

SUMMARY
Specific interactions between proteins govern essential physiological processes including signaling. Many enzymes, especially the family of serine/threonine phosphatases (PSPs: PP1, PP2A and PP2B/calcineurin/CN), recruit substrates and regulatory proteins by binding Short Linear Motifs (SLiMs), short sequences found within intrinsically disordered regions (IDRs) that mediate specific protein:protein interactions. While tremendous progress had been made in identifying where and how SLiMs bind PSPs, especially PP1 and CN, essentially nothing is known about how SLiMs bind PP2A, a validated cancer drug target. Here we describe three structures of PP2A:SLiM interaction (B56: pS-RepoMan, B56:pS-BubR1 and B56:pSpS-BubR1), show that this PP2A-specific SLiM is defined as LSPIxE and then use this data to discover scores of likely PP2A regulators and substrates. Together, these data not only provide a powerful approach for dissecting PP2A interaction networks in cells but also for targeting PP2A diseases, such as cancer.
Keywords: PP2A, B56, small linear motif (SLiM), BubR1, RepoMan, mitosis, phosphorylation
IN BRIEF
Wang et al. report that BubR1 and RepoMan bind directly to PP2A-B56 using an LSPIxE short linear motif (SLiM), where phosphorylation of the Ser residue enhances binding. Using this SLiM motif, the authors identify more than 100 other potential PP2A targeting proteins and/or substrates.
INTRODUCTION
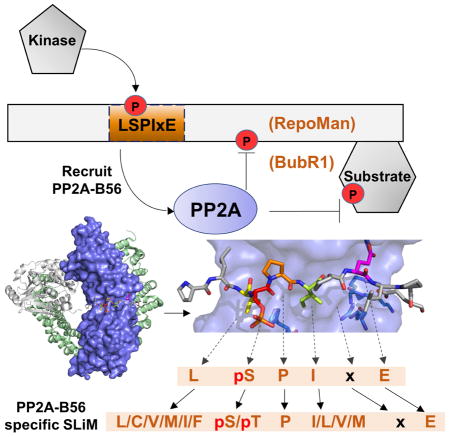
The serine/threonine Protein Phosphatase 2A (PP2A) recruits two distinct regulatory subunits (an A and a B subunit, the latter being either PR55 [B], B56 [PR61, B′], PR72 [B″] or PR93/PR110 [B‴]) to form highly selective holoenzymes (Shi, 2009). However, how these PP2A holoenzymes recruit substrates or are targeted to substrate-containing compartments is poorly understood. The serine/threonine Protein Phosphatases 1 and 2B (PP1; PP2B or Calcineurin/CN) bind substrates and regulatory proteins using short functional sequences known as Short Linear Motifs (SLiMs) (Peti and Page, 2015; Roy and Cyert, 2009). These SLiMs are found within intrinsically disordered regions (IDRs) of substrates and regulatory proteins and mediate specific protein:protein interactions (Ragusa et al., 2010). Sequence differences in and around the SLiM alter the binding affinity for a single anchoring site on binding partners, e.g. kinases or phosphatases (Nygren and Scott, 2015). Furthermore, SLiMs are also regulated by posttranslational modifications, e.g. phosphorylation, which alters their ability to bind their cognate binding partner (Kim et al., 2003). Together, these differences allow for a highly dynamic regulation of signaling pathways via SLiM interactions.
SLiMs have been identified in the substrates and regulatory proteins of PP1 (RVxF, ΦΦ, SILK) and calcineurin (LxVP, PxIxIT, AID). Furthermore, subsequent structural studies have revealed how and where these SLiMs bind their respective phosphatases (Choy et al., 2014; Grigoriu et al., 2013; Hurley et al., 2007; Li et al., 2007; Terrak et al., 2004). It is now established that these SLiM interactions are essential for PSP function and signal fidelity. Indeed, it was recently shown that the blockbuster immunosuppressant drugs FK506 and cyclosporin A inhibit CN activity by binding the LxVP SLiM interaction pocket; i.e., they block substrate binding and, as a consequence, dephosphorylation (Grigoriu et al., 2013; Jin and Harrison, 2002). Thus, SLiM interaction sites are compelling drug targets as it is the interaction of folded enzymes and unfolded SLiMs that control many signaling pathways in the cell.
Recently, the first SLiM specific for PP2A was identified in the kinase BubR1 and the nuclear scaffolding protein RepoMan (Figure S1A) (Kruse et al., 2013; Qian et al., 2013; Suijkerbuijk et al., 2012; Xu et al., 2013). These studies demonstrated that BubR1 residues 662–682 and RepoMan residues 581–599, each of which includes a short LSPI motif surrounding a Cdk1 phosphorylation site (S670BR1; S591RM), are necessary for recruiting PP2A-B56. Although phosphorylation of these serines enhances PP2A-B56 recruitment by both proteins, there is conflicting data regarding the role of additional BubR1 phosphorylation sites (S676BubR1; T680BubR1) for B56 binding, as these sites are not conserved in RepoMan. Because mutations that disrupt B56 engagement leads to abnormal chromosome congression and segregation (BubR1) (Xu et al., 2013) and mitotic chromosome targeting (RepoMan) (Qian et al., 2013), it is now clear that this PP2A-specific SLiM interaction is critical for cell cycle regulation. However, where this SLiM binds B56 and the role, if any, of residues outside the LSPI sequence in B56 binding is unknown.
Here we address these outstanding questions by determining the structures of three B56:LSPI complexes. The structures identify the LSPI binding pocket on B56 and explain why serine phosphorylation is essential for binding. They also reveal that the SLiM is longer: LSPIxE. By using this expanded SLiM definition, we identified nearly 100 PP2A:B56 interactors, substantially expanding the B56 and, in parallel, the PP2A interactome. As the SLiMs binding pockets on phosphatases have been successfully targeted by FDA approved drugs (i.e., FK506 and cyclosporin A, which target CN), a detailed molecular understanding of this site on PP2A-B56 will provide opportunities for the development of PP2A specific therapeutics for cancer.
RESULTS
BubR1 and RepoMan LSPI-containing peptides bind directly to B56
To determine how BubR1 and RepoMan recruit PP2A:B56, we determined three crystal structures (Figure 1A; Table 1): (1) B56:pS-BubR1 (2.79 Å, mono-phosphorylated BubR1, 663TLSIKKL-pS-PIIEDDREADH681; D are S/T→D mutations that mimic phosphorylation and residues underlined are observed in the electron density and thus modeled), (2) B56:pSpS-BubR1 (2.82 Å, dually-phosphorylated BubR1, 668KL-pS-PIIED- pS676) and (3) B56:pS-RepoMan (2.85 Å, mono-phosphorylated RepoMan, 581RDIASKKPLL-pS-PIPELPEVPE601). The SLiM-containing peptides bind B56 with KD’s of 0.6, 2.0 and 0.1 μM, respectively (isothermal titration calorimetry, ITC; Figure 1B, Table S1). Electron density (Figure 1C) for the bound peptides was evident in the initial molecular replacement maps. The structures of B56 in all three complexes are nearly identical to the free (Magnusdottir et al., 2009) and holoenzyme bound (Xu et al., 2009) structures (RMSDs of ~0.5 Å and ~0.8 Å, respectively; Figure S1B). The largest differences are observed in aa 51–64, which adopt a distinct conformation in the three B56:peptide complexes due to crystal contacts. Consistent with other B56 structures, no electron density was observed for B56 residues 110–127 (Magnusdottir et al., 2009). Finally, as expected, the BubR1/RepoMan binding pocket on B56 is fully accessible within the context of the PP2A-B56 holoenzyme (Figure 1D).
Figure 1. Structure of the B56:BubR1 complex.

(A) pS-RepoMan (orange), pS-BubR1 (light blue) and pSpS-BubR1 (magenta) bind B56 (surface colored according to sequence conservation).
(B) Binding isotherms of pS-RepoMan and pS-BubR1 with B56.
(C) Electron density of the pS-RepoMan, pS-BubR1 and pSpS-BubR1 peptides (2Fo-Fc, σ = 1.0).
(D) B56:pS-RepoMan complex (teal:orange) is superimposed on the B56 subunit in PP2A (PDBID 3FGA; catalytic domain, grey; A subunit, white; B56, dark grey); the distance between the PP2A catalytic center (Mn2+ ions in magenta) and the pS residue (red) in the LSPIxE motif is illustrated.
(E) BubR1/RepoMan peptides (orange surface; sticks colored as in (C)) bind B56 between heat repeats 3 and 4 (lavender and blue). B56 heat repeat schematic, with the corresponding helices numbered.
See also Figures S1 and S2 and Tables S1 and S2.
Table 1.
Data collection and refinement statistics
| B56:pS-RepoMana,b | B56:pS-BUBR1a,c | B56:pSpS-BUBR1a,d | |
|---|---|---|---|
| Data collection | |||
| Space group | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Cell dimensions | |||
| a, b, c (Å) | 53.6, 109.0, 120.0 | 53.2, 107.8, 118.0 | 53.3, 107.4, 117.7 |
| A, β, γ(°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 38.21 - 2.85 (3.00 - 2.85)* | 39.52 - 2.79 (2.94 - 2.79)* | 39.51 - 2.82 (2.97 - 2.82)* |
| Rmerge | 0.097 (0.885) | 0.072 (1.382) | 0.075 (0.894) |
| Mean I /σI | 13.0 (2.0) | 16.8 (1.7) | 13.5 (1.8) |
| Completeness (%) | 99.3 (97.3) | 98.4 (93.2) | 98.7 (93.8) |
| Multiplicity | 5.5 (5.6) | 6.6 (6.7) | 5.3 (5.3) |
| CC1/2 | 0.998 (0.686) | 0.999 (0.625) | 0.998 (0.686) |
| Refinement | |||
| Resolution (Å) | 38.21 – 2.85 | 39.52 – 2.79 | 39.52 – 2.82 |
| No. reflections | 16968 | 17130 | 16683 |
| Rwork / Rfree | 0.198/0.227 | 0.198/0.227 | 0.208/0.231 |
| No. atoms | |||
| Protein | 2768 | 2771 | 2738 |
| Water | 23 | 10 | 13 |
| B-factors | |||
| Protein | 66.4 | 86.7 | 75.5 |
| Water | 60.4 | 78.5 | 75.2 |
| RMS deviations | |||
| Bond lengths (Å) | 0.002 | 0.002 | 0.002 |
| Bond angles (°) | 0.651 | 0.690 | 0.730 |
| Ramachandran | |||
| Outliers (%) | 1.2 | 0.3 | 0.6 |
| Allowed (%) | 2.4 | 3.3 | 4.0 |
| Favored (%) | 96.4 | 96.4 | 95.4 |
| Clashscore | 1.63 | 1.45 | 2.20 |
| PDBID | 5SW9 | 5K6S | 5SWF |
Data was collected from a single crystal
pS-RepoMan, 581RDIASKKPLL(pS)PIPELPEVPE601
pS-BUBR1, 663TLSIKKL(pS)PIIEDDREADH681
pSpS-BUBR1, 668KL(pS)PIIED(pS)676
Values in parentheses are for highest-resolution shell.
BubR1 and RepoMan bind B56 in a highly conserved pocket between the 3rd and 4th heat repeats
Both BubR1 and RepoMan bind B56 in an extended conformation at the center of the concave surface defined by the C-terminal helices of the B56 HEAT repeats 3 and 4 (Figure 1E). The interaction is extensive, burying ~1200 Å2 of solvent accessible surface area in the three complexes, ~25% larger than that observed for other well-established SLiM interactions (Grigoriu et al., 2013). The BubR1/RepoMan residues that interact most extensively with B56 are L669BR1/L590RM, pS670BR1/pS591RM, I672BR1/I593RM and E674BR1/E595RM (Table S2). Notably, the B56 residues that interact directly with BubR1/RepoMan are conserved among all B56 isoforms (Figures 1A and S1C), explaining why both proteins bind equally effectively to all B56 isoforms in vivo (Qian et al., 2013; Suijkerbuijk et al., 2012; Xu et al., 2013).
The complexes show that B56 binds BubR1 and RepoMan using both hydrophobic (Figure 2A and S3) and electrostatic/polar interactions (Figures 2B, 2C and S3). LSPI residues L669BR1/L590RM and I672BR1/I593RM bind into two adjacent hydrophobic pockets. The ‘Leu’ binding pocket is defined by K183B56, T184B56, H187B56, R188B56, E226B56 and I227B56 (Figure 2A, lower panel) while the ‘Ile’ binding pocket, which is deeper and ideally shaped to bind Ile residues, is defined by H187B56, Y190B56, I227B56 and I231B56 (Figure 2A, middle panel). These interactions are essential for binding as mutating these residues to alanine (ASPA; or LSPA) in either BubR1 or RepoMan abolishes B56 binding (Kruse et al., 2013; Qian et al., 2013).
Figure 2. The PP2A:B56 specific SLiM is LSPIxE.

(A) The B56 binding pocket (lavender) with pS-RepoMan (orange), pS-BubR1 (light blue) and pSpS-BubR1 (magenta); LSPIxE residues in dark pink and the RepoMan residues that make additional B56 contacts in orange; residues not conserved between the peptides are written as RepoMan/BubR1. The B56 residues (blue) that constitute the LSPIxE ‘L’, ‘I’ and ‘E’ pockets are shown in the left panels. The peptide sequences used for complex formation are shown below: grey, not modeled due to lack of density; bold and shaded, modeled residues; dark pink, residues conserved between the peptides; yellow, RepoMan residues that define the E598 pocket (see (C)); italics, side chains not modeled; underline, phosphomimetic mutations.
(B) Electrostatic and H-bond interactions between the ‘pS’ residues and the ‘E’ residues of the LSPIxE motif; colored as in (A). Electrostatic and H-bond interactions are shown as dashes (black, ≤ 4.0 Å; grey, ≤ 5.0 Å) with the distances in Å provided.
(C) The E598RM binding pocket that is specific to RepoMan; colored as in (A). Electrostatic and H-bond interactions as in (B).
See also Figure S3 and Tables S3–S5.
These hydrophobic interactions optimally position the phosphorylated serines of the three peptides to project away from the surface of B56 and form bidentate salt bridges with R188B56 (Figures 2A and 2B, lower panel). This allows H187B56 to form hydrogen bonds with the pS670BR1/pS591RM carbonyl, restraining the bound peptides. Although the P671BR1/P592RM sidechains point up away from B56, they play an important structural role by directing the BubR1/RepoMan peptide chains back towards B56, allowing I672BR1/I593RM to bind into the deep ‘Ile’ hydrophobic pocket. P671BR1/P592RM are also required for the efficient phosphorylation of S670BubR1/S591RM by CDK1 (its kinase motif is S/T-P), an event that strongly enhances the binding of this SLiM to B56. Finally, R197B56 further restrains the orientation of the BubR1/RepoMan peptides by forming a hydrogen bond with the carbonyl of Ile673BubR1 (Figure 2C).
Unexpectedly, we also observed a third salt bridge between E674BR1/E595RM and both K240B56 and H243B56 (Figure 2B, upper panel) This interaction allows E674BR1/E595RM to bind a third deep pocket in B56 defined by F235B56, K240B56 and H243B56 (Figure 2A, upper panel). As a consequence, E674BR1/E595RM are two of the most buried peptide residues in the B56 complexes (Table S2), suggesting that this interaction is critical for B56 binding. This was confirmed by peptide binding studies which showed mutating this residue to an alanine weakens the affinity of BubR1 for B56 to levels nearly identical to that observed by mutating either the ‘L’ or ‘I’ in the LSPI motif to an alanine (Kruse et al., 2013). Together, these data demonstrate that the PP2A-B56 specific SLiM corresponds to an LSPIxE motif, with phosphorylation of the ‘S’ residue enhancing binding via key electrostatic interactions with B56.
RepoMan binds to B56 using an extended LSPIxE motif (LSPIxExPE)
Our structures revealed multiple, prominent electrostatic interactions between BubR1/RepoMan and B56. In order to understand the importance of the electrostatic interactions on binding, we used ITC (ITC data summarized in Table S1, Figure S2). We first measured the affinity between BubR1 and B56 at different salt concentrations (300 mM to 150 mM; increasing concentrations of salt weaken the strength of electrostatic interactions due to ionic shielding). The binding affinity of BubR1 668KLpSPIIEDE676 for B56 increases by ~3.6 fold as the salt concentration is reduced, confirming the importance of electrostatics for overall binding strength. We then tested the role of specific ionic interactions. The most prominent interaction is that mediated by pS670BR1/pS591RM, which binds H187B56 and R188B56. Previous work showed that in vivo substitution of the anchoring pS residue with a D is not phosphomimicking (Qian et al., 2015). Our structures reveal why this is the case: an Asp is too short to effectively reach the R188B56 side chain. However, the structures suggested that a E would also not be phosphomimicking. This is because three of the four pS670 oxygens mediate ionic interactions, something not possible with either a Glu or Asp (Figure 2B, lower panel). As hypothesized, substituting E for pS (668KLpSPIIEDE676 vs 668KLEPIIEDE676) weakens the affinity for B56 by 2.4-fold. This demonstrates that although D and E are often used to mimic phosphorylated residues, neither are suitable mimics for the pS residue in the PP2A-B56 specific LSPIxE motif. A recent interesting study confirms that a S670EBR1 substitution does not function as a phosphorylation mimic for defining this interaction (Wang et al., 2016).
We then tested the role of residues outside the LSPIxE SLiM for binding. Phosphorylation of a BubR1 residue C-terminal to the LSPIxE motif, S676BR1, has been suggested to be important for B56 binding even thought this serine is not conserved in RepoMan (the corresponding residue is P677RM). Our structures show that neither the S676DBR1 (pS-BubR1) nor the pS676BR1 (pSpS-BubR1) residue makes significant contacts with B56, suggesting this residue does not significantly contribute to binding. We tested this using ITC (Table S1; Fig. S2). The data showed that, unlike pS670BR1, pS676BR1 can be substituted by Glu without affecting binding affinity for B56 (668KLpSPIIEDE676 vs 668KLpSPIIEDpS676). Furthermore, removing this residue altogether only modestly weakens binding, reducing it by ~1.7-fold (668KLpSPIIED675).
The peptides used for these ITC measurements constitute the structurally determined core LSPIxE motif. However, we also performed ITC measurements with a longer peptide (Table S1). This peptide exhibited ~3 fold stronger binding. This affinity increase is due largely to a ~35% reduction in entropy, likely due to the different amino acid composition and/or the different chain length of the peptide. We then repeated the ITC with the longer phosphorylated RepoMan LSPIxE peptide. This peptide bound B56 ~5-fold more strongly than the longer BubR1 peptide. This increase in affinity was due, in part, to a large reduction in entropy compared to BubR1. However, it was also due to direct interactions between B56 and RepoMan not observed in either of the B56:BubR1 complexes. Namely, RepoMan, but not BubR1, forms an additional bidentate salt bridge between E598RM and R201B56 (Figure 2C). Together, these data demonstrate the importance of both enthalpic and entropic contributions to the overall binding energies between IDP SLiMs and their folded protein binding partners. They also demonstrate how residues outside the core LSPIxE SLiM of distinct B56 regulators and substrates function to fine tune their affinities for B56.
Using the LpSPIxE SLiM to identify B56 interactors
Our structures of the three B56:LSPIxE complexes, in conjunction with appropriate secondary filters, can now be leveraged to define the appropriate PP2A SLiM motif(s) that will successfully identify PP2A interacting proteins. This is complementary to a sequence conservation and computational prediction approach that was recently used to identify LSPI sequences (Hertz et al., 2016). By using these structures as a ‘molecular ruler’ to define the LSPIxE SLiM, we focus on identifying the most stringent B56 interactors.
Using the structurally-determined LSPIxE SLiM and ScanProsite (de Castro et al., 2006), we identified 13 instances (sites) of this motif in 13 different human proteins, ~90% fewer than using the LSPI definition alone (Figure 3A; Tables S3, S4). Because the LSPIxE motif in both BubR1 and RepoMan are present in IDRs (required for the extended binding observed in the B56:BubR1 complex) and because phosphorylation significantly enhances binding, we applied two additional secondary filters: multiple disorder prediction algorithms to ensure the identified sites are in IDRs and (2) NetPhos (Blom et al., 1999) to ensure sites are phosphorylated. Applying these filters identifies 10 LSPIxE sites in 10 distinct proteins (Figure 3B; Table S3). Thus, we predict that these 10 proteins bind directly to PP2A:B56 and do so in a manner identical to that observed for BubR1. Interestingly, as observed for BuBR1 and RepoMan, 60% of the identified potential B56 interacting proteins are present in the nucleus, correlating well with the essential role of PP2A during the cell cycle.
Figure 3. Potential PP2A:B56 LSPIxE interactors.

(A) Bar graph illustrating the number of motifs identified in the human UniProt database using listed motifs. Blue, total sites identified; orange, ‘likely interactors’, i.e., sequences that are (i) predicted to be in IDRs and (ii) predicted to be phosphorylated.
(B) Gene names and schematics of the 10 human proteins with ‘LSPIxE’ sites.
(C) Cellular localization of the likely interactors of B56.
(D) Models of the PP2A-B56:BubR1 and PP2A-B56:RepoMan complexes; S893 is dephosphorylated by PP2A-B56.
It is immediately apparent, however, that this strict definition of the LSPIxE SLiM will fail to detect PP2A interactors that have similar, yet also binding compatible sequences. We analyzed the B56:BubR1 structure to identify amino acids that can be accommodated in the Leu- and Ile-binding pockets; i.e. we used the B56 structures as a rigid geometric ruler. This approach is valid as these pockets are unchanged between the three B56 complexes (Figure 1A) and in the structures of B56 alone and the PP2A:B56 holoenzyme (Figures S1B). The Leu-binding pocket is larger and less well-defined than the Ile-binding pocket (Figure 2A, lower panel) and thus can accommodate a Val, Met or Cys and, with some small B56 sidechain rearrangements, an Ile or a Phe. Trp and Tyr residues are excluded as they are too bulky to fit into the pocket. Similarly, polar and charged residues are excluded as there are no amino acids in the pocket positioned to mediate compensatory polar interactions. Expanding the motif to include these additional residues modestly increases the number of sites and proteins identified ([LCVMIF]-SPIxE, 20 sites in 19 proteins; Figure 3A; Table S4). Interestingly, RepoMan, a protein with a confirmed B56-specific LSPIxE SLiM, is predicted to have two LSPIxE sites using this expanded motif (590LSPIpE595, confirmed; 935MSPIkE940, discovered in this analysis). This gives rise to the intriguing possibility that these distal sites might be activated by phosphorylation via distinct kinases at different times during the cell cycle and thereby differentially regulate PP2A targeting during mitosis.
In contrast to the open Leu-binding pocket, the Ile-binding pocket is more constrained (Figure 2A, middle panel). An analysis of the binding pocket suggests that a Leu, Met and Val can also be accommodated at this site; as with the Leu-binding pocket, larger amino acids and charged/polar amino acids are too bulky or charged, respectively, to effectively bind this pocket without significant structural rearrangements, an observation consistent with previous mutagenesis data (Qian et al., 2013). Combining our expanded definition of the Leu binding site with the Ile binding site results in a much greater number of sites identified: 75 [LCVMIF]-SP-[ILVM]-xE sites (Figure 3A; Table S4). Allowing the phosphorylated residue to be either a Ser or Thr further increases the number of sites identified by ~30% to 104 [LCVMIF]-[ST]-P-[ILVM]-xE sites (Figure 3A; Tables S4, S5). Although the application of these strict geometric rules and filters may not capture all interactors, it identifies those most likely bind in the B56 LSPIxE SLiM binding pocket.
Strikingly, as for the 10 sites identified using the strict definition of the motif (LSPIxE), nearly half of the sites using the expanded definition are in nuclear proteins (Figure 3C). Several of the identified proteins have already been associated with PP2A in vivo or shown to have roles in cell division, a process where PP2A activity is vital (Table S5). For example, Kif4A interacts directly with PP2A B56 subunits (Bastos et al., 2014). Our analysis identified a 1224CSPIeE1229 SLiM sequence near the C-terminus of Kif4A (1226CSPIeE1231 in Kif4B). Furthermore, studies have shown that Ser1125Kif4A is phosphorylated during mitosis (Dephoure et al., 2008). Our analysis strongly suggests that Kif4A binds directly to PP2A:B56 via this LSPIxE SLiM. We also discovered that well-known mitotic proteins also contain LSPIxE sites, and thus likely also target PP2A during the cell cycle. For example, RepoMan has two LSPIxE sequences; given that Ser936RM has already been identified to be phosphorylated (Rigbolt et al., 2011), it is likely that 935MSPIkE940 also binds and targets PP2A. Most intriguing in relation to BubR1 is the identification of LSPIxE sites in both Bub1 (654FSPIqE659) and KNL1 (1041LTPLeE1046). This is of interest as KNL1 is a scaffold for both Bub1 and BubR1 and our findings suggests that all three proteins may function to target PP2A to kinetochores during mitosis.
DISCUSSION
In contrast to protein tyrosine phosphatases (PTPs), identifying PSP substrates, especially those of PP1 and PP2A, has been comparatively difficult. This is because the approaches for identifying substrates of PTPs, including non-covalent substrate trapping mutants (Flint et al., 1997) and selective inhibitors (Honkanen et al., 1994), are much less developed for PP1 and PP2A. It is for these reasons that it has been especially challenging to identify bona fide substrates of PP2A, PP1 and CN and, as a consequence, to fully elucidate the roles of PSPs in distinct signaling cascades. The identification of SLiMs that are specific for these PSPs provide the first steps to overcome this difficulty.
Our structures reveal that LSPIxE defines the PP2A-specific SLiM, with all five residues in the SLiM mediating key interactions with B56. In particular, the ‘E’ in the LSPIxE SLiM interacts extensively with H243B56. This is of special interest as the B56 H243P mutation has been identified in embryonal carcinoma tumors (Nobumori et al., 2013). The structure also explains why the phosphorylation of the ‘S’ residue enhances binding to B56; it mediates key electrostatic interactions with H187B56 and R188B56 in B56. This differs from PP1-specific SLiMs (RVxF, SILK, among others), where phosphorylation reduces PP1 binding (Kim et al., 2003). The differential response of PSPs to the phosphorylation state of their specific SLiMs allows PSPs to associate with discrete regulators at different times to direct distinct biological outcomes.
Most importantly, the discovery that the PP2A-specific SLiM is LSPIxE also led to the identification of 104 instances of this motif in 98 human proteins that arepotential PP2A interactors (Figure 3A; Table S5). Only a handful of these proteins were previously identified as PP2A targeting proteins, as they require specific cellular conditions, i.e. phosphorylation, in order to effectively engage PP2A. Whether the identified proteins function solely as targeting proteins, like BubR1, or also substrates, like RepoMan, remains to be determined (Figure 3D). Finally, these B56:SLiM structures also have important implications for the development of novel compounds that target PP2A. PSPs were generally considered to be undruggable because natural product inhibitors of PSP active sites are poorly selective and highly toxic. However, recent structural and functional studies are leading to the emerging view that PSP SLiM binding sites are highly targetable. For example, the LxVP SLiM binding pocket, which is used by scores of CN substrates to bind CN, is the binding site of the immunosuppressants FK506 and CSA; i.e., they potently inhibit CN by simply blocking the phosphatase from binding its substrates. It is now clear that a similar strategy can be used to target PP2A, thereby providing a powerful approach for both dissecting PP2A signaling pathways and targeting PP2A diseases, such as cancer.
EXPERIMENTAL PROCEDURES
Detailed protein expression, purification, crystallization, structure determination and ITC methods can be found in the Supplemental Procedures.
Structure determination
B56γ31–380 was purified using his-tag, TEV cleavage and size exclusion chromatography and incubated with BubR1/RM peptides in a 1:5 molar ratio. Crystals of the complexes grew in 0.1 M HEPES pH 7.75, 0.8 M LiCl and 8% PEG6K (B56:pS-BubR1) or 8% PEG8K (B56:pSpS-BubR1, B56:pS-RepoMan) using hanging drop vapor diffusion at RT. Data were collected at SSRL beamline 12.2 at 100 K and a wavelength of 0.98 Å using a Pilatus 6M PAD detector. The data were processed and the structures refined as described in Supplemental Procedures. Data collection and refinement details are provided in Table 1.
Bioinformatics
ScanProsite (de Castro et al., 2006) was used to identify additional PP2A interacting proteins that contain an LSPIxE SLiM. Definition of the search sequences were based on the experimental 3-dimensional B56:pS-RepoMan, B56:pS-BuBR1 and B56:pSpS-BuBR1 complex structures. The probability of these proteins containing functional LSPIxE motifs (i.e., ones that bind B56 in the B56 LSPIxE-motif binding grooves) was further evaluated disorder prediction (method used described in detail in Supplemental Procedures) as it is well recognized that SLiM are only identified in IDRs. Furthermore, as phosphorylation was experimentally shown to significantly enhance binding, NetPhos was used to select only hits that have a high likelihood to be phosphorylated (NetPhos >0.5) (Blom et al., 1999).
Supplementary Material
HIGHLIGHTS.
Crystal structures of PP2A B56 in complex with phosphorylated RepoMan and BubR1
RepoMan and BubR1 bind B56 using both hydrophobic and electrostatic interactions
The PP2A-B56 specific short linear motif (SLiM) is L-pS-P-I-x-E
The identification of more than 100 proteins that likely bind PP2A via this motif
Acknowledgments
The work was supported by NINDS (1R01NS091336; W.P.) NIGMS (R01GM098482, R.P.), the Fund for Scientific Research-Flanders (G.0473.12, G.0482.12; M.B.) and a Flemish Concerted Research Action (GOA 15/016, M.B.). Some data was obtained at the BU Structural Biology Core Facility (BioMed, Brown University). Crystallographic data were collected at SSRL. Use of SSRL is supported by the U.S. DOE, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH-NIGMS (P41GM103393).
Footnotes
ACCESSION NUMBERS
The accession number for the reported crystal structures are 5K6S (B56:pS-BubR1), 5SW9 (B56:pS-RepoMan) and 5SWF (B56:pSpS-BubR1).
Supplemental information includes three figures, five tables and supplemental experimental procedures.
CONTRIBUTIONS
X.W, M.B, W.P. and R.P conceived experiments and wrote the manuscript. X.W., R.B., W.P. and R.P performed and analyzed experiments. X.W, R.B. W.P., M.B and R.P. discussed the data and manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bastos RN, Cundell MJ, Barr FA. KIF4A and PP2A-B56 form a spatially restricted feedback loop opposing Aurora B at the anaphase central spindle. J Cell Biol. 2014;207:683–693. doi: 10.1083/jcb.201409129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Choy MS, Hieke M, Kumar GS, Lewis GR, Gonzalez-DeWhitt KR, Kessler RP, Stein BJ, Hessenberger M, Nairn AC, Peti W, et al. Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc Natl Acad Sci U S A. 2014;111:4097–4102. doi: 10.1073/pnas.1317395111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, Peti W. The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS Biol. 2013;11:e1001492. doi: 10.1371/journal.pbio.1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz EP, Kruse T, Davey NE, Lopez-Mendez B, Sigurethsson JO, Montoya G, Olsen JV, Nilsson J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.06.024. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Codispoti BA, Tse K, Boynton AL, Honkanan RE. Characterization of natural toxins with inhibitory activity against serine/threonine protein phosphatases. Toxicon. 1994;32:339–350. doi: 10.1016/0041-0101(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Hurley TD, Yang J, Zhang L, Goodwin KD, Zou Q, Cortese M, Dunker AK, DePaoli-Roach AA. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J Biol Chem. 2007;282:28874–28883. doi: 10.1074/jbc.M703472200. [DOI] [PubMed] [Google Scholar]
- Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci U S A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Watanabe T, Allen PB, Kim YM, Lee SJ, Greengard P, Nairn AC, Kwon YG. PNUTS, a protein phosphatase 1 (PP1) nuclear targeting subunit. Characterization of its PP1- and RNA-binding domains and regulation by phosphorylation. J Biol Chem. 2003;278:13819–13828. doi: 10.1074/jbc.M209621200. [DOI] [PubMed] [Google Scholar]
- Kruse T, Zhang G, Larsen MS, Lischetti T, Streicher W, Kragh Nielsen T, Bjorn SP, Nilsson J. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J Cell Sci. 2013;126:1086–1092. doi: 10.1242/jcs.122481. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang L, Rao A, Harrison SC, Hogan PG. Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. J Mol Biol. 2007;369:1296–1306. doi: 10.1016/j.jmb.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Magnusdottir A, Stenmark P, Flodin S, Nyman T, Kotenyova T, Graslund S, Ogg D, Nordlund P. The structure of the PP2A regulatory subunit B56 gamma: the remaining piece of the PP2A jigsaw puzzle. Proteins. 2009;74:212–221. doi: 10.1002/prot.22150. [DOI] [PubMed] [Google Scholar]
- Nobumori Y, Shouse GP, Wu Y, Lee KJ, Shen B, Liu X. B56gamma tumor-associated mutations provide new mechanisms for B56gamma-PP2A tumor suppressor activity. Mol Cancer Res. 2013;11:995–1003. doi: 10.1158/1541-7786.MCR-12-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren PJ, Scott JD. Therapeutic strategies for anchored kinases and phosphatases: exploiting short linear motifs and intrinsic disorder. Front Pharmacol. 2015;6:158. doi: 10.3389/fphar.2015.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W, Page R. Strategies to make protein serine/threonine (PP1, calcineurin) and tyrosine phosphatases (PTP1B) druggable: achieving specificity by targeting substrate and regulatory protein interaction sites. Bioorg Med Chem. 2015;23:2781–2785. doi: 10.1016/j.bmc.2015.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Beullens M, Huang J, De Munter S, Lesage B, Bollen M. Cdk1 orders mitotic events through coordination of a chromosome-associated phosphatase switch. Nat Commun. 2015;6:10215. doi: 10.1038/ncomms10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Beullens M, Lesage B, Bollen M. Aurora B defines its own chromosomal targeting by opposing the recruitment of the phosphatase scaffold Repo-Man. Curr Biol. 2013;23:1136–1143. doi: 10.1016/j.cub.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat Struct Mol Biol. 2010;17:459–464. doi: 10.1038/nsmb.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B. Systemwide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4:rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- Roy J, Cyert MS. Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal. 2009;2:re9. doi: 10.1126/scisignal.2100re9. [DOI] [PubMed] [Google Scholar]
- Shi Y. Assembly and structure of protein phosphatase 2A. Sci China C Life Sci. 2009;52:135–146. doi: 10.1007/s11427-009-0018-3. [DOI] [PubMed] [Google Scholar]
- Suijkerbuijk SJ, Vleugel M, Teixeira A, Kops GJ. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev Cell. 2012;23:745–755. doi: 10.1016/j.devcel.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R. Structural basis of protein phosphatase 1 regulation. Nature. 2004;429:780–784. doi: 10.1038/nature02582. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang Z, Yu T, Yang H, Virshup DM, Kops GJ, Lee SH, Zhou W, Li X, Xu W, et al. Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell. 2016;7:516–526. doi: 10.1007/s13238-016-0283-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Raetz EA, Kitagawa M, Virshup DM, Lee SH. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol Open. 2013;2:479–486. doi: 10.1242/bio.20134051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Cetin B, Anger M, Cho US, Helmhart W, Nasmyth K, Xu W. Structure and function of the PP2A-shugoshin interaction. Mol Cell. 2009;35:426–441. doi: 10.1016/j.molcel.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.