

Abstract
Background
Comprehensive genomic sequencing (CGS) has the potential to revolutionize precision medicine for cancer patients across the globe. However, to date large-scale genomic sequencing of cancer patients has been limited to Western populations. In order to understand possible ethnic and geographic differences and to explore the broader application of CGS to other populations, we sequenced a panel of 415 important cancer genes to characterize clinically actionable genomic driver events in 201 Japanese patients with colorectal cancer (CRC).
Methods
Using next-generation sequencing methods, we examined all exons of 415 known cancer genes in Japanese CRC patients (n = 201) and evaluated for concordance among independent data obtained from US patients with CRC (n = 108) and from The Cancer Genome Atlas-CRC whole exome sequencing (WES) database (n = 224). Mutation data from non-hypermutated Japanese CRC patients were extracted and clustered by gene mutation patterns. Two different sets of genes from the 415-gene panel were used for clustering: 61 genes with frequent alteration in CRC and 26 genes that are clinically actionable in CRC.
Results
The 415-gene panel is able to identify all of the critical mutations in tumor samples as well as WES, including identifying hypermutated tumors. Although the overall mutation spectrum of the Japanese patients is similar to that of the Western population, we found significant differences in the frequencies of mutations in ERBB2 and BRAF. We show that the 415-gene panel identifies a number of clinically actionable mutations in KRAS, NRAS, and BRAF that are not detected by hot-spot testing. We also discovered that 26% of cases have mutations in genes involved in DNA double-strand break repair pathway. Unsupervised clustering revealed that a panel of 26 genes can be used to classify the patients into eight different categories, each of which can optimally be treated with a particular combination therapy.
Conclusions
Use of a panel of 415 genes can reliably identify all of the critical mutations in CRC patients and this information of CGS can be used to determine the most optimal treatment for patients of all ethnicities.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-016-0387-8) contains supplementary material, which is available to authorized users.
Keywords: Colorectal cancer, Precision medicine, Ethnicity, Japanese, Comprehensive genomic sequencing, Actionable driver mutation, Hypermutation
Background
Cancer remains the leading cause of death worldwide with colorectal cancer (CRC) among the most common indications, accounting for 700,000 deaths per year [1]. Utilizing next-generation sequencing technology, projects such as The Cancer Genome Atlas (TCGA) and others have profiled genomic changes in several cancer types including CRC [2–9]. The ultimate goal of cancer genome profiling is to enable precision medicine, the tailoring of treatments based on unique genomic changes of each patient’s individual tumor. For instance, the importance of genomic evaluation of RAS and RAF for advanced CRC patients has been widely accepted, since it has been revealed that tumors with RAS or RAF mutations show resistance to anti-EGFR therapies [10]. Initially, mutations in these genes were found to occur in “hot-spots” (i.e. KRAS codon 12, 13, or BRAF V600E) [11–13], however, whole exome sequencing (WES) has revealed that mutations outside of hot-spots can also influence therapeutic responses [14, 15]. Yet, WES may not be practical in the clinical setting due to its high cost, shallow sequencing depth, and excessive information about variants/genes of unknown significance [16, 17]. Although sequencing studies of CRC have been reported [4, 18–20], tumors from Asian populations have not been the subject of comprehensive evaluation. We now report the results from the analysis of 201 Japanese CRC patients.
Since all of the reported studies examined the mutational spectrum using WES, and WES is clinically expensive and time-consuming, we hypothesized that sequencing a panel of cancer-associated genes would identify essentially all actionable genomic driver mutations and further determine mutational burden in CRC, both of which can enable development of personalized treatment strategies. In the current study, we tested this hypothesis utilizing a 415-gene panel designed for solid tumors at a very high depth of coverage (~500×) in Japanese patients (n = 201 tumors) and evaluated for concordance among independent data obtained from US patients with colon cancer (n = 108 tumors) (J-CRC and US-CRC, respectively) and from the TCGA-CRC WES database (n = 224 tumors). Here, we report that comprehensive genomic sequencing (CGS) with a 415-gene panel can accurately determine high mutation burden (somatic mutation rate) and that there are differences in the frequency of mutations in ERBB2 and BRAF. Hierarchical clustering of clinical data revealed that a subset of 26 genes can classify all of the CRC patients into eight categories, each of which can be effectively treated with available drugs or drugs in development.
Methods
Patient cohorts and sample inclusion criteria
Japanese cohort
A total of 201 patients diagnosed with stage I–IV CRC according to AJCC 7th edition [21] who had curative surgery between 2009 and 2015 at Niigata University Medical and Dental Hospital or Niigata Cancer Center Hospital were enrolled (Additional file 1: Table S4). Patients with familial adenomatous polyposis, inflammatory bowel disease, or synchronous multiple CRCs were excluded.
US cohort
A total of 108 patients with histologically confirmed diagnosis of primary colorectal adenocarcinoma (stage I–IV) between 2014 and 2016 submitted for CGS as part of routine medical examination were included in this study. All tumor samples that had > 50% tumor content after macrodissection, as determined through routine hematoxylin and eosin (H&E) stain by an independent pathologist, were included. A full waiver of authorization under the Health Insurance Portability and Accountability Act (HIPAA) was granted to enable retrospective analyses for samples obtained without prior consent. All data were de-identified prior to inclusion in this study.
Sequencing library preparation
For Japanese and US patient samples, archival tissue in the form of formalin-fixed, paraffin embedded (FFPE) tumor or unstained tissue sections obtained during routine biopsy and/or resection were used for analysis. An independent pathologist evaluated tumor content on H&E stained slides for each study sample to ensure > 50% tumor content was present. Where applicable, unstained slides were macro-dissected to enrich for tumor content and genomic DNA (gDNA) was extracted using BiOstic FFPE Tissue DNA Isolation Kit (Mo Bio Laboratories, Inc.). All sample prep, CGS, and analytics were performed in a CLIA/CAP-accredited laboratory (KEW Inc; Cambridge, MA, USA).
Comprehensive genomic sequencing
FFPE gDNA (50–150 ng) was converted into libraries and enriched for the 415 genes with CANCERPLEX (KEW Inc.; Cambridge, MA, USA). CANCERPLEX is a clinically validated 415-gene panel enriched for coding regions and selected introns of genes with known association in cancer. Sequencing was performed on the Illumina MiSeq and NextSeq platforms with average 500× sequencing depth. Genomic data were then processed through a proprietary bioinformatics platform and knowledge base to identify multiple classes of genomic abnormalities including single nucleotide substitutions (SNPs), small insertions/deletions (indels), copy number variations (CNV), and translocations in ALK, RET, and ROS1. A threshold of 10% allelic fraction was used for SNPs and indels and thresholds of >2.5-fold (gains) and 0.5-fold (loss) were used. To assess somatic status of mutations in a tumor-only setting, we employed a filtering strategy similar to one recently published [22] with minor differences. In short, variants were deprioritized if they were present in a combination of dbSNP, 1000 Genomes, and ExAC databases (at AF > 1%). Next, allele frequencies for each mutation were used to fit a model to determine whether the variant is likely germline heterozygous or somatic. Finally, results underwent manual molecular pathologist review validating somatic versus possible germline status of a variant. Based on published and our experience, this approach allows the correct discrimination between germline and somatic variants in more than 99% of cases. Mutated burden was determined by non-synonymous SNPs present in the tumor that have population frequency of < 1% dbSNP and 1000 Genomes databases.
Downsampling TCGA mutation data
COAD-READ mutation data for the TCGA-CRC samples (n = 224 samples) were downloaded from the Broad GDAC Firehose website (https://gdac.broadinstitute.org/). Similar to the 415-gene panel bioinformatics pipeline, silent mutations that were not protein altering were removed from the dataset. To compare mutation burden of the 415-gene panel to TCGA WES data, the dataset of SNPs was downsampled to the 415 genes in the panel and the mutation rate determined in the panel was calculated as mutations/Mb. To produce receiver operating characteristic (ROC) curves, genes were selected randomly to produce panels of 400, 300, 200, 100, and 50 genes. Mutation burden was calculated using only CGS panel genes and individual ROC curves were used to evaluate how well mutation burden predicted hypermutated samples. This process was repeated 100 times and average ROC curves were produced at each panel size. In addition, individual ROC curves were produced using all genes and only those genes in KEW’s CANCERPLEX panel.
Mutation signature
Each single nucleotide variant (SNV) was classified in a matrix of the 96 possible substitutions based on the sequence context comprising the nucleotides 5′and 3′ to the position of the mutation. Mutational signatures were extracted using non-negative matrix factorization analysis with the SomaticSignatures R package [23] and plotted with ggplots R package (http://ggplot2.org/). This analysis identified complex signatures, different between hypermutated and non-hypermutated cases. Deconvolution of the complex profiles in order to identify components matching to COSMIC mutational signatures was done using deconstruct Sigs R package [24].
Mismatch repair immunohistochemistry (MMR-IHC)
Immunohistochemistry (IHC) staining was performed on the 40 samples of Japanese CRC with highest mutation rates. Slides were stained for four mismatch repair (MMR) proteins, MLH1 (clone G168-15), MSH2 (clone FE11) MSH2 (clone BC/44), and PMS2 (clone A16-4), and were scored by two pathologists. For US clinical cases, clinical records were reviewed and results of MMR studies were recorded when available.
Mutation analysis and visualization
Genomic data for Japanese (n = 201) and US patients (n = 108) obtained from CGS were mined in OncoPrinter (www.cbioportal.org). Pathway genes were selected based on previously published TCGA data [4] that are included in the 415-gene panel. For TCGA analyses, genomic profiles were selected in cBioPortal for mutations and putative copy-number alterations from GISTIC for which tumor sequence data are available (n = 224). For each pathway, the number of total uniquely altered cases was determined. Statistical significance was determined by Fisher’s exact two-tail test with a 95% confidence interval. For dsDNA break repair pathway analysis, the statistical significance of Japanese and US datasets was determined as compared to TCGA.
To align mutations with their protein domains, genomic data for Japanese, US, and TCGA datasets were analyzed in Mutation Mapper (www.cbioportal.org). Lollipop figures were generated for select genes implicated in colorectal adenocarcinoma. For BRAF and KRAS, data were further segregated by hypermutation status (hypermutated versus non-hypermutated).
Gene clustering analysis
Mutation data from non-hypermutated J-CRC patients (n = 184 tumors) were extracted and clustered by gene mutation patterns. Two different sets of genes from the 415-gene panel were used for clustering: (1) 61 genes with frequent alteration in CRC; and (2) 26 genes that are clinically actionable in CRC. For this analysis, KRAS and NRAS were integrated into one gene as a RAS.
The number of common mutated genes related to donors i and j was presented as an element cij of an N × N matrix, where N is the number of non-hypermutated donors. In order to normalize the elements of this N dimension symmetric matrix into values ranging from 0 to 1, the original element was replaced by 1 / (cij + 1) that indicated the level of similarity between donors i and j. Because of this normalization, donors with more common mutated genes would more possibly come from a relatively close group. Consequently, a matrix with the normalized values between all donors was created. Hierarchical clustering of the matrix was performed for classifying donor groups with different mutated-gene patterns by Euclidean distance and Ward’s clustering. For the 26-gene set, donors were divided into eight groups based on the hierarchical clustered dendrogram, which clearly distinguished donors by the different mutated-gene patterns. On the other hand, for the 61-gene set, donors were divided into 17 groups. These clusterings were performed by software R (https://www.r-project.org/).
Model selection of clustering
Clustering stability was evaluated by R package clValid for statistical and biological validation of clustering results (https://cran.r-project.org/web/packages/clValid/index.html). This method would produce the results of four stability measures called APN (average portion of non-overlap), AD (average distance), ADM (average distance between means), and FOM (figure of merit). For each index, a lower value means higher stability. We attempted clustering stabilities for combinations of different numbers of clusters obtained by cutting a dendrogram (2–12 for the 26-gene set and 2–24 for the 61-gene set) with different distance methods (“Euclidean,” “maximum,” “manhattan,” “canberra,” and “minkowski”) and clustering methods (“ward.D,” “ward.D2,” “single,” “complete,” “average,” “mcquitty,” “median,” and “centroid”). All combinations of these three parameters were evaluated and the parameters with the lowest values of each stability index were extracted. Of these, the common parameter sets with relatively lower values among the four stability indices were selected. The most appropriate cluster number, distance method, and clustering method were determined from the resulted parameter settings, taking into account that the number of donors presented in clusters (>5 donors) would be maximized as possible and the primary mutated genes would be clear. The final selected parameter settings were the Euclidean distance method and ward.D clustering in both sets and eight clusters for the 26-gene set and 17 clusters for the 61-gene set.
Statistical analysis of clinical information
To estimate associations between mutated-gene patterns and clinical information such as sex, rectum/colon, and left/right, a two-tailed Fisher’s exact test was applied in each cluster. Additionally, in order to explore associations between mutated-gene patterns and tumor aggressiveness, seven clinical variables were dichotomized into less or more aggressive factors for colon cancer onsets in the following manner: lymphatic invasion (absence/presence), vascular invasion (absence/presence), histopathological grade (G1/G2 or G3), size of primary tumor (T1/T2 or T3/T4), spread to regional lymph node (N0 or N1/N2), distant metastasis (M0 or M1), and tumor stage (I/II or III/IV). In each cluster, two-tailed Fisher’s exact test was applied to all clinical categories by comparing the distribution in a cluster group to that of all the donors in the other groups. Note that in the case of statistical signature for 17 hypermutated donors, two-tailed Fisher’s exact test was conducted against 184 non-hypermutated donors as a reference set.
Patients were followed every 1–6 months at outpatient clinics. Medical records and survival data were obtained for all 104 Stage IV CRC patients. Among them, 46 patients received anti-EGFR therapies. Seven out of the 46 patients with surgical resection were excluded and 39 patients were included for the analysis of clinical outcomes. Tumor assessments at baseline included a computed tomography (CT) scan of the abdomen as well as of other relevant sites of the disease. Follow-up scans to assess response were obtained after cycles 1 and 2 and every two cycles thereafter. Responses were determined using RECIST 1.0. Six patients who showed progression disease before the first assessment for RECIST were excluded and 33 patients were included for waterfall plot analysis. The best calculated responses on the basis of measurable lesions were analyzed by waterfall plot.
The follow-up period for progression-free survival was defined as the interval between the date of diagnosis of metastatic disease and that of progression disease. Survival curves were constructed using the Kaplan–Meier method and differences in survival were evaluated using the log-rank test. Three out of 39 patients were excluded for Kaplan–Meier analysis based on the clustering, since each one of three patients was classified into each different subtype alone. All statistical evaluations were performed using the SPSS 22 software package (SPSS Japan Inc., Tokyo, Japan). All tests were two-sided and a P value < 0.05 was considered statistically significant.
While conducting the two-tailed Fisher’s exact test as above, the statistical powers of the tests were also estimated by R package statmod (https://cran.r-project.org/web/packages/statmod/index.html). Some clinical categories showing significant differences (p < 0.05) were at insufficient power levels (power < 0.8). It is known that power is related to sample size and, in other words, the power of tests could be promoted by adjusting the effect size of samples [25]. Therefore, for these significant but low-power contingency tables, we made a prediction of the number of donors that could meet a sufficient power level under the premise that the hypothetical cross-tabulations had the same cell percentages as that of 184 non-hypermutated donors. The prediction was performed for sample sizes in the range of 20–500 with increments of ten donors for each step and both P value and power of Fisher’s exact test were calculated for assumed contingency table at each step. By this means, a minimum effect non-hypermutated donor number was obtained and this sample size could become a reference in future studies. The statistical power calculation and prediction for the above-mentioned Fisher’s exact test were simulated 1000 times for each cross-tabulation.
Gene-based statistical analysis
To estimate associations between genes and tumor aggressiveness, we performed Fisher’s exact test for each gene in seven clinical categories. Subsequently, significant genes with at least one clinical category (p < 0.05) were extracted. A matrix between the genes and the clinical categories were created based on log odds ratio for the extracted genes. Finally, the matrix was clustered by Euclidean distance and Ward’s method. In this clustering, positive and negative infinity values are replaced by 4 and −4 as pseudonumbers, respectively.
Results
Genomic alterations in cancer signaling pathways
Utilizing the CGS platform (Additional file 1: Table S1), we assessed the genes and pathways most frequently altered in the test samples (Fig. 1). We found that the same sets of alterations were generally detected by both WES and CGS. Genomic alterations in oncogenic pathways involving cell cycle, RAS/RAF, PI3K, and WNT were comparable (Fig. 1) [4]. However, we found statistically significant differences in ERBB2 (p < 0.05), APC (p < 0.001), TP53 (p < 0.001), CDKN2A (p < 0.05), and NRAS (p < 0.05) mutations in Japanese patients as compared with US patients (Fig. 1a–c), which may reflect epidemiological differences between the two populations [26, 27].
Fig. 1.
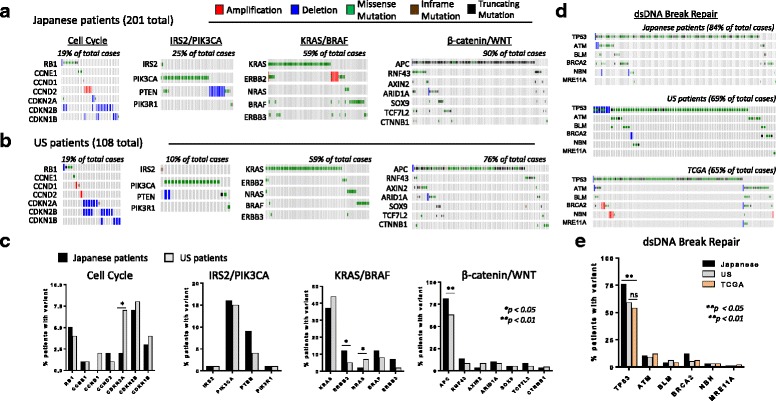
Genetic aberrations across common oncogenic pathways in CRC. Japanese patients (a) and US patients (b) were evaluated for gene alterations in the key cancer pathways. Amplification (red), deletion (blue), missense point mutations (green), or frameshift mutations (brown). Altered cases are defined as the total number of unique samples with a genetic aberration in each pathway. c Percent of patients with a variation for each given gene. Statistical significance was determined using Fisher’s exact test. d J-CRC, US-CRC, and TCGA sample data were evaluated for gene alterations in the dsDNA break repair pathway in the 415-gene panel. e Percent of patients with a variation for each given gene. Statistical significance was determined using Fisher’s exact test
Given the recent recognition that tumors with DNA double-strand break repair defects (most notably BRCA1/2 mutations) are more sensitive to PARP inhibitors [28] and the recent approval of olaparib for advanced ovarian cancer, we undertook a comprehensive analysis of the DNA double-strand break repair pathway. Currently BRCA1/2 mutation status alone is used to identify patients for olaparib treatment; however, mutations in other genes can lead to DNA double-strand break repair defects [28, 29]. Therefore, those genes may also be useful in determining olaparib sensitivity. Excluding TP53, which is not used for selection of PARP inhibitors, we analyzed the five DNA repair pathway genes that are most commonly mutated in Japanese and US patients and compared with TCGA samples (Fig. 1d and e). We found genomic alterations in all five DNA repair genes, including BRCA2, which represent a significant proportion of CRC patients (26% of Japanese, 21% of US, and 19% of TCGA samples).
Mutation rates detected by targeted sequencing with cancer gene panel
The clinical significance of identifying hypermutated tumors has recently been demonstrated in several studies correlating mutation burden with the development of neo-antigens and clinical response to immunotherapy drugs [4, 30–33]. We found hypermutated tumors as identified by CGS: 17 (8%) in J-CRC and two (3%) in US-CRC (Fig. 2a and b), generally correlated with DNA mismatch repair deficiency (MMR-D) as detected by standard clinical IHC evaluation for MMR proteins (MLH1, MSH2, MSH6, and PMS2). For Lynch syndrome genes, both somatic and potentially germline pathogenic mutations were included in the analysis (see “Methods”). One patient showed loss of MSH2 expression by IHC supported by genetic loss for MSH2 gene without a hypermutated phenotype while conversely two patients with the highest mutation burdens were MMR-intact and microsatellite stable but had POLE mutations, demonstrating that although often useful in predicting hypermutation status, neither MMR-D nor MSI-H alone can fully predict all hypermutated tumors. Similar analysis of US-CRC clinical cases confirmed the ability of CGS to detect hypermutated tumors, although the clinical bias to perform CGS on advanced cases of MSS-CRC in US community oncology practice may explain the low percentage of hypermutated tumors found in these samples. To further validate utility of CGS in identifying hypermutated tumors, we downsampled the TCGA WES data (n = 224 tumors) [4] to the subset of 415 genes in the CGS platform. This analysis not only accurately identified the hypermutated tumors (both MMR-deficient and MMR-intact) but also showed strong correlation in mutation rates between the 415-gene panel and WES (Fig. 2c). The average mutation rate detected by CGS was higher than that detected by WES reflecting the fact that the panel content was in part selected to include genes more frequently mutated in cancer. We further downsampled the TCGA data to random gene panels of descending size (400, 300, 200, 100, and 50) and determined that panels smaller than 300 genes lacked sufficient statistical power to accurately identify hypermutated cases (Fig. 2d), thus demonstrating that the CGS platform (roughly 1/2000th of the genome) is comparable to WES in generating mutation rates and to distinguish hypermutated and non-hypermutated tumors.
Fig. 2.
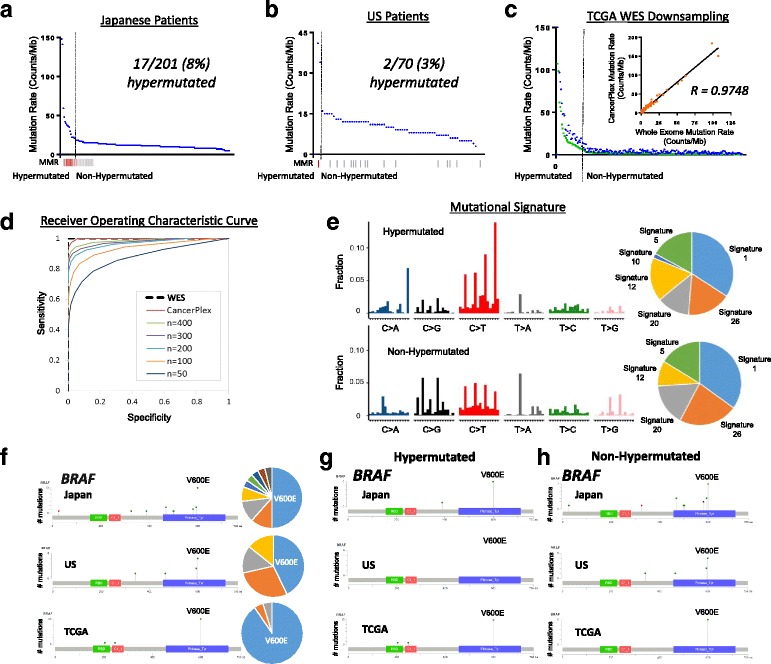
Mutation rates in Japanese and US CRC patients. Mutation rates from Japanese patients (a) and US patients (b) were determined by the number of non-synonymous SNVs in the 415-gene panel. Hypermutated and non-hypermutated cancers separated by the dashed line. Red, MMR-deficient; gray, MMR-intact; white, no data. c Data from TCGA CRC cases (green) were downsampled to the content of the 415-gene CGS platform (blue; non-synonymous SNPs). Correlation between mutation rates determined by CGS and WES (insert). d ROC analysis using the 415-gene CGS platform, WES, and random sets of 400, 300, 200, 100, and 50 genes as predictors of hypermutated samples (TCGA dataset). e Aggregated mutational signature profiles for hypermutated (top) and non-hypermutated cases (bottom). The pie charts represent inferred contribution of COSMIC signatures to corresponding profiles. f Mutations in BRAF for Japanese patients (n = 201), US patients (n = 108), and TCGA samples (n = 224) were aligned to protein domains. The number of mutations at each given amino acid were plotted in corresponding pie graphs. As shown, BRAF V600E was the highest frequency mutations in each protein. Patient samples were further plotted by mutation status: (g) BRAF-hypermutated, (h) BRAF-non-hypermutated
We further explored the utility of CGS to provide clinically meaningful patterns of mutational signatures [34] from the J-CRC cohort (Fig. 2e). Based upon the signatures described in COSMIC (http://cancer.sanger.ac.uk/cosmic)), we found that Signatures 20 and 26 contributed the largest proportion of total somatic SNVs and were similar to previous findings. Both signatures were associated with defective DNA repair [34]. Interestingly, in the hypermutated-cases only we identified Signature 10 (C > A SNVs at TpCpT context), previously shown to correlate with altered activity of DNA polymerase epsilon [34] (termed “ultra-hypermutators” by COSMIC). Indeed, we determined that the two cases with the highest mutation burdens were MMR-intact with mutations in their POLE gene: V411L in the exonuclease (proofreading) domain in one case and P286R in the polymerase domain in the other demonstrating the capacity of CGS in identifying clinically useful mutational signatures.
Genomic evaluation of key driver genes
Recent updates in clinical guidelines, in both Japan and in the US, have made the genomic evaluation of KRAS, NRAS, and BRAF essential for treatment planning. Most mutations in these genes cluster in “hot-spots” (i.e. KRAS codon 12, 13; NRAS codon 61; BRAF codon 600); however, data from large full-gene sequencing projects have identified additional mutations outside these hot-spots (e.g. KRAS codon 22, 33, 59, etc.). We compared the distribution of somatic mutation across these key genes between Japanese and US cohorts and with the TCGA (Fig. 2f–h, Additional file 1: Figure S1). While the KRAS mutation patterns in different cohorts appeared similar, BRAF mutation patterns presented key differences. BRAF mutations present in TCGA-CRC samples were predominantly represented by V600E which is often restricted to hypermutated tumors and agrees with previous reports [35–37]. The TCGA database shows that BRAF mutations in non-hypermutated tumors were also significantly more frequent in right-sided tumors. In contrast to previous studies, both Japanese and US-CRC cases had a wide range of non-V600E mutations inside and outside the kinase domain including D594G, a kinase-dead BRAF that can drive tumor progression through interactions with CRAF [38]. In addition, BRAF mutations were found in both left-sided and right-sided tumors (Additional file 1: Table S2). This finding may suggest unique therapeutic strategies for not only right-sided, but also left-sided tumors that were enriched for alternate BRAF mutations. Consistent with previous findings in TCGA-CRC cases [39], we found APC and RNF43 truncating mutations mutually exclusive in J-CRC and in US-CRC (Fig. 1) with significant enrichment of RNF43 alterations, particularly G659 mutations, in MMR-deficient tumors (Additional file 1: Figure S2). Analysis of additional key driver genes showed similar patterns of mutation between Japanese, US, and TCGA cohorts (Additional file 1: Figure S1). Similar to TCGA results, no gene fusions were found in well-characterized driver genes ALK, RET, or ROS1.
Genomic alterations and tumor aggressiveness
Unlike earlier genomic profiling studies, this study also included clinical outcomes data that was used to determine the relationship between mutation profile and patient outcomes. CRC is a clinically diverse disease and it has been long considered that genomic heterogeneity is vital to understanding this diversity. Tumors can be classified by degree of lymphatic invasion, vascular invasion, histopathological grade, TNM classifications, and tumor stage [21]. We therefore examined the association between gene alterations and clinical features. Among the 415 genes, we found that genes significantly enriched in at least one certain category (p < 0.05) were distinctly classified into more aggressive or less aggressive groups (Additional file 1: Figure S3 and Table S3). For example, mutations in genes such as PTEN, SMAD2, TGFB2, and SRC implicated in epithelial-mesenchymal transition, metastasis, and cancer progression [40, 41], were enriched in more aggressive groups while the other genes clustered in the less aggressive groups.
Cluster analysis for Japanese CRC mutations
Several approaches to identify genomic subtypes have been proposed to correlate genomic landscape with clinical features in CRC. Despite differing methods of classification, the hypermutated subtype has commonly emerged across various genomic profiling efforts. In agreement with these findings, we identified a subgroup of 17 Japanese patients with hypermutated tumors as characterized by CGS (Fig. 1). We therefore performed hierarchical clustering of mutations in a subset of genes frequently altered in CRC (n = 61 genes) in the Japanese cohort of non-hypermutated patients (n = 184 tumors) to further assess the association between gene alterations and clinical features in CRC (Additional file 1: Figure S4). We identified that all patients can be classified into 12 typical clusters (Additional file 1: Figure S4). We further examined associations between each of these clusters with clinicopathological features, such as sex, tumor location, and pathologic stage (Additional file 1: Figure S4B). Of note, patients in Cluster 7 (n = 49 tumors) with primary mutated genes APC and TP53 significantly associated with the location of left side (p < 0.01), less lymph node metastasis (p < 0.05), and less distant metastasis (p < 0.05) compared with patients in all other clusters (Additional file 1: Figure S4B). These findings suggest that there are clear associations between mutation spectrum and clinical characteristics of Japanese CRC patients.
Additional cluster analysis on a subset of 26 genes associated with targeted therapies either already approved or in late-phase development in Japan (Phase II or III) (Fig. 3, Additional file 1: Figure S5A) identified seven clusters with mutated genes and a single cluster with no mutated genes. Patients with KRAS mutations (Clusters 6–8; n = 75 tumors) were classified into three clusters, while patients without KRAS mutations were classified into either Cluster 1 (n = 49 tumors) with “all wild-type” genes or Clusters 2–5 (n = 60 tumors) with mutations in actionable driver genes including ERBB2, PIK3CA, RNF43, BRAF, and PTEN. Patients in Cluster 1 were associated with tumors in the left side (p < 0.01), while patients in Cluster 7 (n = 17 tumors) with RAS and PIK3CA mutations were associated with tumors on the right side (p < 0.05), consistent with previous reports [42]. Interestingly, patients in Cluster 2 (n = 8 tumors) with ERBB2 mutations were associated with smallest tumor size, significantly less lymphatic invasion (p < 0.01) and early stage (p < 0.05), while patients in Cluster 5 (n = 29 tumors) harboring PTEN mutations exhibited significantly more lymphatic (p < 0.05) and vascular invasion (p < 0.01) with more metastasis.
Fig. 3.
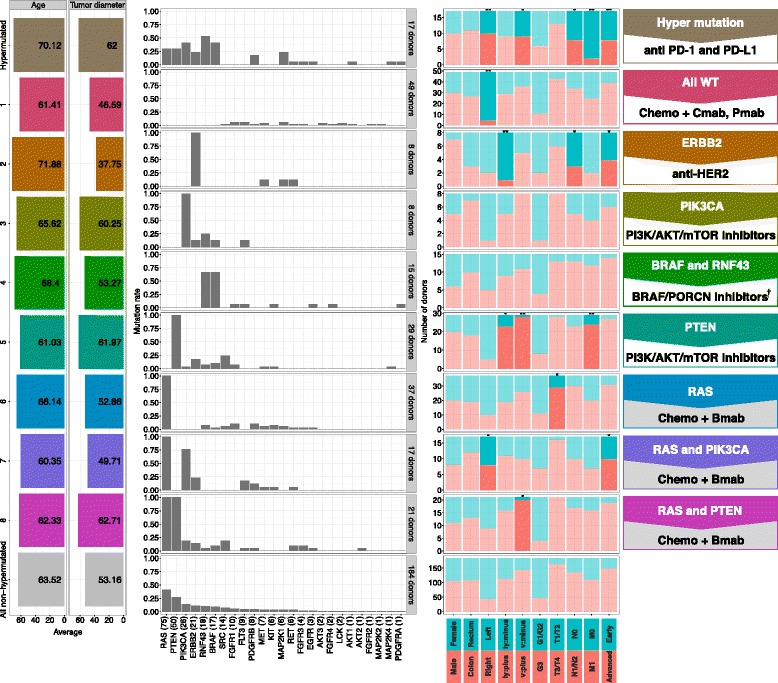
Cluster of 26-gene co-mutation patterns. Cluster analysis was performed on non-hypermutated Japanese CRC samples (n = 184 tumors) by using Euclidean distance and Ward’s clustering method and co-mutation patterns of the 26-gene subset with statistical analysis are shown. Mutation rate in each group is shown as a bar graph in the middle panel. Group-based mean values for age and tumor diameter are shown (left) with cluster colors and fraction for clinical information (right). Dark bars indicate significant difference (p < 0.05, two-tailed Fisher’s exact test) to the distribution of all other non-hypermutated donors, light bars are non-significant (*p < 0.05, **p < 0.01). Chemo chemotherapy; Cmab Cetuximab; Pmab Panitumumab; Bmab Bevacizumab. †Combination therapy with other inhibitors (e.g. anti-EGFR, MEK inhibitors) will be recommended
Outcome of Stage IV CRC patients and clinical potential of cluster analysis based on CGS platform
Next, we examined clinical outcomes of Stage IV CRC patients to explore the clinical potential of cluster analysis based on CGS for Japanese CRC patients. Kaplan–Meier analysis for patients with Stage IV CRC (n = 102, excluded two hypermutated cases) revealed that overall survival rates were significantly different among the subtypes based on cluster analysis on a subset of the 26 genes associated with targeted therapies (Fig. 3, Additional file 1: Figure S5B). The 26 genes included RTK and RAS pathway, such as KRAS, BRAF, NRAS, and ERBB2, which have known associations with resistance to anti-EGFR targeted therapies in CRC patients [10]. We therefore hypothesized that the cluster analysis based on the 26 genes estimates the effect of anti-EGFR therapies. Waterfall plot analysis demonstrated the best calculated responses on the basis of measurable lesions in 33 patients treated with anti-EGFR therapies and revealed that all the three patients with progressive disease belong to subgroups with actionable driver mutations (RNF43 and BRAF; Cluster 4 and RAS; Cluster 6), but not subgroup of “all wild-type” without actionable mutations (Cluster 1) (Fig. 4a). Moreover, in agreement with previous findings [43], swimmers plot and Kaplan–Meier analysis demonstrated that patients in subgroup of “all wild-type” showed significantly better progression-free survival as compared to patients in subgroups of “mutated” (Clusters 2–6 and hyper-mutated subgroup) (p = 0.009) (Fig. 4b and c). Moreover, Kaplan–Meier analysis further demonstrated a significant difference among subgroups when the subgroups with actionable mutations were stratified based on the clustering (p = 0.001) (Fig. 4d). These findings indicate clinical potential of clustering based on the 415-gene CGS platform with its ability to estimate the survival of patients with Stage IV CRC treated with targeted therapies.
Fig. 4.
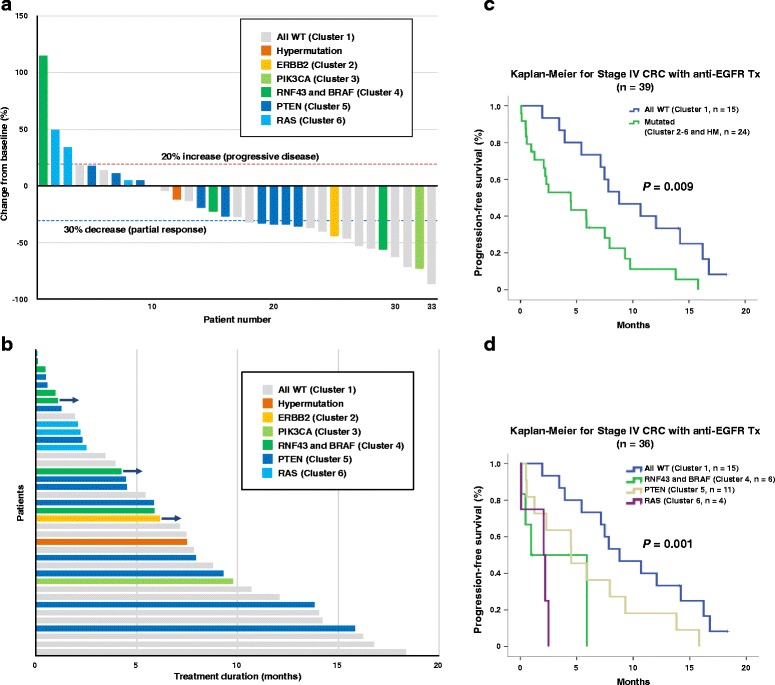
Clinical outcomes of Stage IV patients treated with anti-EGFR therapies. a Waterfall plot for 33 patients with Stage IV CRC after anti-EGFR targeted therapy in addition to cytotoxic chemotherapy. The vertical axis shows the best calculated responses on the basis of measurable lesions in each individual patient. b Swimmers plot for 39 patients with Stage IV CRC treated with anti-EGFR therapies. The horizontal axis shows progression-free survival for each patient. c, d Kaplan–Meier survival estimates according to genomic subgroups. c Progression-free survival was analyzed in 39 patients with Stage IV CRC treated with anti-EGFR therapies. The patients were divided to “All WT (wild type)” (Cluster 1; n = 15) or “Mutated” (Clusters 2–8; n = 24) based on the cluster analysis with targeted therapy-related 26 genes. d Progression-free survival was analyzed for 36 patients with Stage IV CRC treated with anti-EGFR therapies based on subgroups (All WT, cluster 1; RNF and BRAF, cluster 4; PTEN, cluster 5; RAS, cluster 6) by clustering with the 26 genes
Discussion
In the current study, we performed CGS sequencing with a 415-gene panel to probe actionable driver mutations at a very high depth of coverage in the largest series of Japanese patients (n = 201 tumors) and evaluated for concordance among independent data obtained from US patients with colon cancer (n = 108 tumors) and from the TCGA-CRC WES database (n = 224 tumors). We identified overall similarities and some distinct population differences in detecting clinically actionable oncogenic driver events. We correlated mutation burden with DNA mismatch repair status, obtained clear genomic mutational signatures, and identified genomic alteration patterns in Japanese and the US-CRC patients similar to those previously identified by WES by the TCGA. We also found statically significant increases in ERBB2 APC, TP53, and NRAS mutations in Japanese patients as compared with US patients, which may reflect epidemiological differences between the two populations. Interestingly, we found that 11 of 24 BRAF mutations occurred outside the hot-spot V600E. Since mutations other than V600E are known to be activating, our results underscore the importance of sequencing all BRAF exons to assess the optimal therapeutic approach. Moreover, we report here a novel, significant correlation between APC and TP53 mutations with tumors presented on the left side, emphasizing the utility of CGS sequencing as an invaluable resource for better understanding the genomic landscape of CRC.
To explore the clinical potential of CGS, we performed cluster analysis with the set of clinically actionable genes in CRC (n = 26 genes) related to targeted therapies either approved or in late-phase development in Japan and obtained eight typical subgroups in addition to the “hypermutated” subgroup. CRC patients in the “hypermutated” subgroup are expected to benefit most from treatment with immune checkpoint inhibitors. Patients in the “all wild-type” cluster (Cluster 1) may respond best to anti-EGFR therapies, such as Cetuximab and Panitumumab given the lack of contraindicated KRAS mutations. However, patients in Clusters 2–5 had driver mutations downstream of the EGFR pathway, suggesting resistance to anti-EGFR therapies and hence better response to therapies targeting PIK3CA, ERBB2, RNF43/BRAF, or PTEN. Patients in Clusters 6–8 had KRAS mutations and therefore may benefit from chemotherapy + Bevacizumab given their expected resistance to anti-EGFR therapy. Thus, these findings underscore the clinical potential of examining a smaller (26 gene) panel, by which we could identify suitable targeted therapies based on the clustering of actionable gene mutations.
Given the clinical significance of hot-spot KRAS mutations (codons 12 and 13) in patients with advanced CRC to anti-EGFR therapy resistance, KRAS mutation testing has become mandatory testing in Japanese patients before administering anti-EGFR therapy [44]. Indeed, most of the patients treated with anti-EGFR therapies in this study had been identified not to have hot-spot KRAS mutations (codons 12 and 13) and thus considered as KRAS wild-type, except for a few patients who had been treated before testing became required. Recent studies have identified alterations in genes downstream of EGFR (RTKs and RAS pathway) in addition to hot-spot KRAS mutations as likely indicators of primary and secondary resistance to anti-EGFR antibody therapies [10]. We therefore probed the clinical relevance of gene alterations in RTKs and RAS pathway in addition to KRAS mutations as identified by CGS in Japanese CRC patients. Interestingly, there were three patients with progressive disease on anti-EGFR therapy and CGS revealed that two out of the three patients had previously unidentified mutations downstream of EGFR emphasizing that hot-spot testing alone is inadequate in guiding therapeutic strategies. Moreover, Kaplan–Meier analysis demonstrated that patients in the subgroup without alterations in RTKs and RAS pathway showed significantly better progression-free survival than patients in subgroups with mutations, although most of the patients had been previously considered as KRAS wild-type. Taken together, we have demonstrated that CGS captures broad actionable genomic driver mutations in Japanese patients with advanced CRC satisfying a currently unmet critical need to better guide personalized therapeutic approaches in Japan.
Conclusions
We demonstrate concordance of CGS between Japanese and US patients with CRC and with WES in the TCGA database. We further illustrate how CGS testing captures broad actionable genomic driver mutations as well as high mutational burden and highlight its potential to impact clinical outcomes of patients. These findings emphasize the clinical potential of CGS for patients with CRC in Japan and warrant further clinical investigation through prospective randomized clinical trials to confirm the application.
Acknowledgements
We thank our colleagues Karen Dresser, Yosuke Tajima, Takuma Okamura, Hitoshi Nogami, Satoshi Maruyama, Yasumasa Takii, Takashi Kawasaki, and Keiichi Homma.
Funding
This project was supported by funding from Denka Co., Ltd. M. Nagahashi is supported by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research Grant Number 15H05676 and 15 K15471, the Uehara Memorial Foundation, Nakayama Cancer Research Institute, Takeda Science Foundation, and Tsukada Medical Foundation. T. Wakai is supported by the JSPS Grant-in-Aid for Scientific Research Grant Number 15H04927 and 16 K15610. S. Okuda is supported by the JSPS Grant-in-Aid for Scientific Research Grant Number 26700029. K. Takabe is supported by NIH/NCI grant R01CA160688 and Susan G. Komen Investigator Initiated Research Grant IIR12222224. S. Lyle is supported by a grant from the Massachusetts Life Sciences Center.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are not publicly available due to data and privacy protection considerations but may be available on justified request. The raw data (COAD-READ) for the 224 TCGA samples were downloaded from the Broad GDAC Firehose website (https://gdac.broadinstitute.org/).
Authors’ contributions
M. Nagahashi, TW, SO, and SL were project leaders. M. Nagahashi, TW, YS, SO, JER, KT, AP, and SL wrote the manuscript. H. Ichikawa, HK, TK, JS, RY, NS, YK, HU, KY, EO, and SK analyzed clinical data. H. Izutsu, KK, M. Nakada, JT, MR, JH, WP, RS, JER, AP, YL, SO, and SL examined genomic alterations and pathways. YL and SO performed bioinformatics analysis. All authors read and approved the final manuscript.
Competing interests
H. Izutsu, KK, and M. Nakada are employees of Denka Co., Ltd. M. Nakada holds stock in Denka Co., Ltd. JT, MR, JH, RS, JER, AP, and SL are employees of KEW Inc., who have been granted stock options by KEW Inc. The remaining authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Collection and use of all specimens in this study were approved by the Institutional Review Boards of Niigata University Graduate School of Medical and Dental Sciences (#772) and Niigata Cancer Center Hospital (#642). The protocol for this study was approved by Western Institutional Review Board in US. Informed consent was obtained from all participants.
Abbreviations
- CGS
Comprehensive genomic sequencing
- CNV
Copy number variation
- FFPE
Formalin-fixed, paraffin embedded
- MMR-D
Mismatch repair deficiency
- SNV
Single nucleotide variant
- TCGA
The Cancer Genome Atlas
- WES
Whole exome sequencing
Additional file
Location of genetic aberrations for Japanese and US patients, and TCGA samples. Mutations in (A) APC, (B) ERBB2, (C) TP53, (D) NRAS, and (E) KRAS for Japanese patients (n = 201), US patients (n = 108), and TCGA samples (n = 224) were aligned to protein domains. The number of mutations at each given amino acid were plotted in corresponding pie graphs. As shown, KRAS G12 were the highest frequency mutations. Patient samples were further plotted by mutation status (F) KRAS-hypermutated and (G) KRAS-non-hypermutated. Figure S2. Correlation of RNF43 mutations with MMR. (A) The frequencies of APC and RNF43 mutations were determined by MMR phenotype. Statistical significance was determined by Fisher’s exact test. (B) Mutation mapper analysis identified G659 as most frequently altered in MMR-D cases. Figure S3. Gene-based statistical analysis for clinical information. Genes were filtered based on Fisher’s exact test (p < 0.05). Cell values are log odds ratios colored from blue to red. Dendrograms were created by Euclidean distance and Ward’s method. Less (blue) or more (red) aggressive factors of seven clinical variables are shown: lymphatic invasion (ly), vascular invasion (v), histopathological grade (G), TNM classifications (T, N, and M), and tumor stage. Figure S4. Cluster of 61-gene co-mutation patterns. (A) Cluster analysis was performed on non-hypermutated Japanese CRC samples (n = 184 tumors) by using Euclidean distance and Ward’s clustering method (closest distance to common mutated genes are colored yellow to blue). (B) Co-mutated gene patterns of the 61-gene set with statistical analysis. Mutation rate in each group is shown as a bar graph in the middle panel. Group-based mean values for age and tumor diameter are shown (left) with cluster colors and fraction for clinical information (right). Dark bars indicate significant difference (p < 0.05, two-tailed Fisher’s exact test) to the distribution of all other non-hypermutated donors, light bars are non-significant (**p < 0.01, *p < 0.05). Figure S5. Data complementary to Fig. 3. (A) Cluster analysis was performed on non-hypermutated Japanese CRC samples (n = 184 tumors) by using Euclidean distance and Ward’s clustering method (closest distance to common mutated genes are colored yellow to blue). (B) Kaplan–Meier survival estimates according to genomic subgroups. Overall survival was analyzed in 102 patients with Stage IV CRC treated with anti-EGFR therapies. The patients were divided to “All WT (wild type)” (Cluster 1; n = 25) or “Mutated” (Clusters 2–8; n = 77) based on the cluster analysis with targeted therapy-related 26 genes. Table S1. The 415-gene list for the CGS platform. Table S2. BRAF mutation and tumor location (J-CRC, n = 201). Table S3. Raw data for gene-based statistical analysis for clinical information. Table S4. Clinicopathological characteristics of 201 CRC patients. (PDF 1435 kb)
Contributor Information
Toshifumi Wakai, Email: wakait@med.niigata-u.ac.jp.
Shujiro Okuda, Email: okd@med.niigata-u.ac.jp.
Stephen Lyle, Email: slyle@kewinc.com.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464:993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263–7. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–27. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- 12.Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–8. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.French AJ, Sargent DJ, Burgart LJ, Foster NR, Kabat BF, Goldberg R, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res. 2008;14:3408–15. doi: 10.1158/1078-0432.CCR-07-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris VK, Lucas FA, Overman MJ, Eng C, Morelli MP, Jiang ZQ, et al. Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Ann Oncol. 2014;25:2008–14. doi: 10.1093/annonc/mdu252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chevrier S, Arnould L, Ghiringhelli F, Coudert B, Fumoleau P, Boidot R. Next-generation sequencing analysis of lung and colon carcinomas reveals a variety of genetic alterations. Int J Oncol. 2014;45:1167–74. doi: 10.3892/ijo.2014.2528. [DOI] [PubMed] [Google Scholar]
- 16.Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schell MJ, Yang M, Teer JK, Lo FY, Madan A, Coppola D, et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat Commun. 2016;7:11743. doi: 10.1038/ncomms11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454. doi: 10.1186/s13059-014-0454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edge SB, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC cancer staging manual. 7. New York: Springer; 2010. [Google Scholar]
- 22.Garofalo A, Sholl L, Reardon B, Taylor-Weiner A, Amin-Mansour A, Miao D, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8:79. doi: 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015;31:3673–5. doi: 10.1093/bioinformatics/btv408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Indira V, Vasanthakumari R, Jegadeeshwaran R, Sugumaran V. Determination of minimum sample size for fault diagnosis of automobile hydraulic brake system using power analysis. Eng Sci Technol Int J. 2015;18:59–69. doi: 10.1016/j.jestch.2014.09.007. [DOI] [Google Scholar]
- 26.Inra JA, Steyerberg EW, Grover S, McFarland A, Syngal S, Kastrinos F. Racial variation in frequency and phenotypes of APC and MUTYH mutations in 6,169 individuals undergoing genetic testing. Genet Med. 2015;17:815–21. doi: 10.1038/gim.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang K, Lamprecht SA, Shinozaki H, Fan K, Yang W, Newmark HL, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J Nutr. 2008;138:1658–63. doi: 10.3945/jn.108.090985. [DOI] [PubMed] [Google Scholar]
- 28.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–70. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 29.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 30.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 31.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TM, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013;155:858–68. doi: 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brim H, Mokarram P, Naghibalhossaini F, Saberi-Firoozi M, Al-Mandhari M, Al-Mawaly K, et al. Impact of BRAF, MLH1 on the incidence of microsatellite instability high colorectal cancer in populations based study. Mol Cancer. 2008;7:68. doi: 10.1186/1476-4598-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasuck CG, Leite SM, Komatsuzaki F, Ferreira AC, Oliveira VC, Gomes KB. Association between methylation in mismatch repair genes, V600E BRAF mutation and microsatellite instability in colorectal cancer patients. Mol Biol Rep. 2012;39:2553–60. doi: 10.1007/s11033-011-1007-8. [DOI] [PubMed] [Google Scholar]
- 37.Patil DT, Shadrach BL, Rybicki LA, Leach BH, Pai RK. Proximal colon cancers and the serrated pathway: a systematic analysis of precursor histology and BRAF mutation status. Mod Pathol. 2012;25:1423–31. doi: 10.1038/modpathol.2012.98. [DOI] [PubMed] [Google Scholar]
- 38.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46:1264–6. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsey S, Langhans SA. Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition. Front Oncol. 2014;4:358. doi: 10.3389/fonc.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhat AA, Pope JL, Smith JJ, Ahmad R, Chen X, Washington MK, et al. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene. 2015;34:4570–80. doi: 10.1038/onc.2014.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol. 2010;28:466–74. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 43.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 44.Yoshino T, Muro K, Yamaguchi K, Nishina T, Denda T, Kudo T, et al. Clinical validation of a multiplex kit for RAS mutations in colorectal cancer: Results of the RASKET (RAS KEy Testing) Prospective, Multicenter Study. EBioMed. 2015;2:317–23. doi: 10.1016/j.ebiom.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available due to data and privacy protection considerations but may be available on justified request. The raw data (COAD-READ) for the 224 TCGA samples were downloaded from the Broad GDAC Firehose website (https://gdac.broadinstitute.org/).