

Abstract
Summary
Whole-genome bisulfite sequencing (WGBS) has emerged as the gold-standard technique in genome-scale studies of DNA methylation. Mapping reads from WGBS requires unique considerations that make the process more time-consuming than in other sequencing applications. Typical WGBS data sets contain several hundred million reads, adding to this analysis challenge. We present the WALT tool for mapping WGBS reads. WALT uses a strategy of hashing periodic spaced seeds, which leads to significant speedup compared with the most efficient methods currently available. Although many existing WGBS mappers slow down with read length, WALT improves in speed. Importantly, these speed gains do not sacrifice accuracy.
Availability and implementation
WALT is available under the GPL v3 license, and downloadable from https://github.com/smithlabcode/walt.
Contact
andrewds@usc.edu or tingchen@usc.edu
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is essential for many biological processes and is found altered in a variety of human diseases (Bock, 2012). Technological advances over the past decade have enabled whole-genome bisulfite sequencing (WGBS), which has become the gold-standard approach for studying cytosine (C) methylation due to its ability to quantify methylation levels unambiguously for nearly all Cs in mammalian genomes. Treatment with sodium bisulfite converts each unmethylated C into uracil, which is reported as thymine (T) in sequenced reads. The most computationally intensive step analysis of WGBS data is to map reads to the reference genome. The bisulfite conversion makes mapping more complicated, and each data set typically includes hundreds of millions of reads.
In recent years, several mappers have been developed specifically for reads from bisulfite sequencing, which we refer to here as ‘bisulfite reads’. These mappers use a variety of strategies. For example, Bismark (Krueger and Andrews, 2011) converts every C in the reads and the reference genome to T, then applies Bowtie (Langmead et al., 2009) to map the converted reads to the converted reference genome. BSMAP (Xi and Li, 2009) builds a pre-compiled hash table to minimize the cache-miss latency when searching possible genome positions for k-mers. BSMAP also applies a bitwise masking method to efficiently count the number of mismatches for asymmetric conversion of C→T. Existing tools are not fast enough to map a large WGBS data set in a day using a single processing core. The challenge in mapping bisulfite reads is derived from two related sources: accommodating the asymmetric conversion of C→T, and the reduced entropy of the alphabet (i.e. nearly 50% T). Here we present WALT, a new mapper for bisulfite reads. WALT is faster than existing methods, and has advantages in accuracy.
2 Methods
WALT employs periodic spaced seeds, originally described by Chen et al. (2009), to filter candidate mapping positions for each read. Three spaced seeds (010) *, 0(010) * and 00(010) * are used to guarantee full sensitivity for two mismatches (Supplementary Table S1). The ones (‘1’) in the spaced seeds indicate positions that must match, while the zeros (‘0’) indicate positions that are not required to match. The star (‘*’) indicates repeating of the 010 pattern until the spaced seed reaches the desired weight. Three seeds are generated for a given read, and each seed is obtained by concatenating all positions in the read corresponding to a ‘1’ in the spaced seed.
WALT converts all Cs in both the reads and the reference genome to Ts for mapping. When processing paired-end reads, or in the case of post-bisulfite adaptor ligation, some reads will be complementary to the original converted strand. For those A-rich reads, guanines (G) are converted to adenines (A). WALT builds four hash tables for converted reference genomes: C→T forward and reverse, G→A forward and reverse. For one hash table, the 3k possible k-mers over {A, G, T} or {A, C, T} are used as hash keys. Each hash bucket contains genome positions where the corresponding spaced seed can be found. The genome positions in each bucket are sorted by their spaced seed subsequences (Supplementary Fig. S2). When mapping a read, the first k nucleotides of each seed are used to locate hash buckets and binary search is applied for nucleotides after the first k. WALT validates each candidate position by aligning the whole read against the genome sequence to count the number of mismatches. For a given read, exact matching genomic positions are identified using the first seed, positions with one mismatch are covered by first two seeds, and using all three seeds guarantees full sensitivity for two mismatches (Supplementary Fig. S1).
3 Results
We assessed the performance of WALT in comparison with Bismark, BSMAP and other tools shown in Supplementary Material. All tests were run using identical Intel Xeon processors using a single core (details in Supplementary Material). We used three datasets: (1) SRR1532534, (2) SRR948855 (Fortin et al., 2014) and (3) SRR2296821 (Yong-Villalobos et al., 2015) for this comparison, evaluating both speed and accuracy. All of them are paired-end data by Illumina HiSeq 2000. The read length, reference genome and number of reads in each dataset are shown in Table 1.
Table 1.
Information about data sets used
| Data set | Read length | Genome | No. of reads | |
|---|---|---|---|---|
| (1) | SRR1532534 | 90 | Human (hg19) | 50,000,000 |
| (2) | SRR948855 | 100 | Human (hg19) | 50,000,000 |
| (3) | SRR2296821 | 90 | Arabidopsis (TAIR10) | 21,543,659 |
For the two human datasets, Table 2 shows a comparison of runtimes for WALT, Bismark and BSMAP. WALT is about 36× faster than Bismark and 7× faster than BSMAP in single-end mapping. For paired-end mapping, WALT is approximately 15× faster than Bismark and 7× faster than BSMAP. WALT does a look-up for three seeds when reads have more than one mismatch, but only one seed for exact matches, and two for one or two mismatches. Since In our experiments, on the average 61.8% and 11.1% of reads are matched with 0 and 1 mismatch, respectively, the average number of lookups per read is only 1.49 (Supplementary Table S13). The Arabidopsis thaliana genome is relatively small, leading to fewer candidates in each hash bucket. Even with this small genome, WALT nearly matches the speed of BSMAP and is still approximately 25× faster than Bismark.
Table 2.
Runtime of WALT, Bismark and BSMAP (hours)
| Single-end |
Paired-end |
|||||
|---|---|---|---|---|---|---|
| WALT | Bismark | BSMAP | WALT | Bismark | BSMAP | |
| (1) | 0.71 | 26.85 | 4.14 | 2.43 | 38.72 | 12.32 |
| (2) | 0.85 | 29.13 | 6.09 | 2.61 | 37.10 | 21.55 |
| (3) | 0.09 | 3.11 | 0.09 | 0.32 | 7.46 | 0.22 |
Figure 1 shows the runtime on reads with different lengths generated from SRR948855 (Supplementary Table S12). WALT runs faster on longer reads, while Bismark and BSMAP slow down significantly. WALT and BSMAP both are based on hash table method. The time required to validate each candidate mapping location is proportional to the read length. However, WALT uses heavier seeds for longer reads and gets less false positive candidates, while BSMAP uses fixed seed length. Bismark uses Bowtie to map the reads, which essentially conducts an inexact search in a compressed suffix array, which is also more costly for longer reads.
Fig. 1.
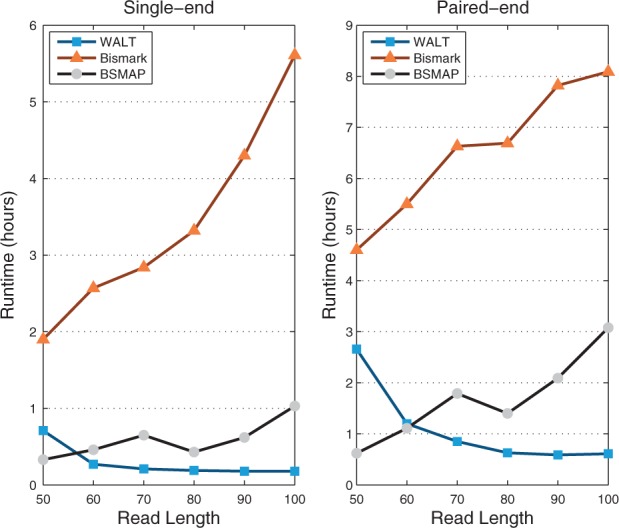
Runtime as a function of read length
Mapping accuracy is essential in WGBS as incorrect mapping may lead to bias in estimates of methylation levels and other epigenomic features in downstream analyses. Ambiguously mapping reads—those for which the best matching genome position is not unique—are excluded from most analyses as their interpretation requires project-specific considerations. We used the program mrsFAST (Hach et al., 2010), with C→T converted reads and reference genome, to obtain all possible mapping positions for all reads; mrsFAST is designed to produce all mappings. This provided a ground truth in measuring mapping accuracy. For a uniquely mappable read (as determined by mrsFAST), if the bisulfite mapper reports it as uniquely mapping, then it counts as true positive (TP), otherwise false negative (FN). For a read found to be ambiguously mapped or unmapped, if the bisulfite mapper reports it as such it counts as TN, otherwise FP. We then compute precision and recall for each mapper:
WALT had the best precision while BSMAP had the best recall on all datasets (Table 3). Bismark had the lowest recall and precision. BSMAP reported 27 and 293% more incorrect uniquely mapped reads than WALT in single- and paired-end mapping, respectively.
Table 3.
Recall (R) and precision (P) of WALT, Bismark and BSMAP
| Single-end |
Paired-end |
|||||
|---|---|---|---|---|---|---|
| WALT | Bismark | BSMAP | WALT | Bismark | BSMAP | |
| (1) R | 0.982 | 0.973 | 0.994 | 0.965 | 0.968 | 0.991 |
| P | 0.991 | 0.976 | 0.988 | 0.984 | 0.943 | 0.967 |
| (2) R | 0.984 | 0.974 | 0.995 | 0.971 | 0.972 | 0.993 |
| P | 0.993 | 0.969 | 0.991 | 0.989 | 0.935 | 0.973 |
| (3) R | 0.987 | 0.974 | 0.995 | 0.986 | 0.959 | 0.994 |
| P | 0.995 | 0.978 | 0.993 | 0.996 | 0.960 | 0.967 |
Supplementary Material
Acknowledgement
We thank Benjamin Decato and Meng Zhou for valuable comments.
Funding
This work was partially supported by National Institutes of Health grant HG006015 (ADS)
Conflict of Interest: none declared.
References
- Bock C. (2012) Analysing and interpreting DNA methylation data. Nat. Rev. Gen., 13, 705–719. [DOI] [PubMed] [Google Scholar]
- Chen Y. et al. (2009) PerM: efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. Bioinformatics, 25, 2514–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J.P. et al. (2014) Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol., 15, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F. et al. (2010) mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods, 7, 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. (2011) Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y., Li W. (2009) BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 10, 232.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong-Villalobos L. et al. (2015) Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc. Natl. Acad. Sci. USA, 112, E7293–E7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.