

Abstract
Detection of recent natural selection is a challenging problem in population genetics. Here we introduce the singleton density score (SDS), a method to infer very recent changes in allele frequencies from contemporary genome sequences. Applied to data from the UK10K Project, SDS reflects allele frequency changes in the ancestors of modern Britons during the past ~2000 to 3000 years. We see strong signals of selection at lactase and the major histocompatibility complex, and in favor of blond hair and blue eyes. For polygenic adaptation, we find that recent selection for increased height has driven allele frequency shifts across most of the genome. Moreover, we identify shifts associated with other complex traits, suggesting that polygenic adaptation has played a pervasive role in shaping genotypic and phenotypic variation in modern humans.
Understanding the genetic basis of adaptation is a central goal in evolutionary biology. Most work in humans and other species has focused on identifying signals of strong selection at individual loci (1). In humans, these methods have identified loci involved in adaptations to diet, altitude, and disease resistance, and lighter pigmentation in northern populations (2–4). Early methods for studying selection focused on detecting hard sweeps, in which new mutations under strong positive selection sweep through a population toward fixation (5). But selection often acts on preexisting variants, either in partial sweeps at individual loci (6, 7), or through polygenic adaptation acting simultaneously on many variants across the genome (8, 9).
However, it is difficult to measure recent selection on standing variation. In some cases this may be done by comparing closely related populations with divergent selective pressures (4, 10), or by using ancient DNA when suitable ancestral samples are available (11, 12), but a generally applicable method is lacking.
To tackle this challenge, we introduce the singleton density score (SDS). SDS uses whole-genome sequence data from contemporary samples to infer recent allele frequency changes at single-nucleotide polymorphisms (SNPs). Recent selection distorts the ancestral genealogy of sampled haplotypes, resulting in shorter terminal (tip) branches for the favored allele. Hence, haplotypes carrying the favored allele tend to carry fewer singleton mutations (Fig. 1, A to C, and fig. S1) (13). Following this intuition, we calculate the distance between the nearest singletons on either side of a test SNP as a summary statistic for each individual (Fig. 1D). The distributions of distances for the three genotypes at the test SNP are then used to compute a maximum likelihood estimate of the log-ratio of mean tip-branch lengths for the derived versus ancestral alleles (Fig. 1, E and F) (13). The two alleles act as natural controls for each other, correcting for local variation in mutation and recombination rates, or in the detection of singletons. The predictions are normalized within bins of derived allele frequency to have mean 0 and variance 1, where SDS > 0 corresponds to an increased frequency of the derived allele. In neutral simulations, SDS follows a standard normal distribution, even when considering complex scenarios with recent admixture (figs. S2 and S3).
Fig. 1. Illustration of the SDS method.
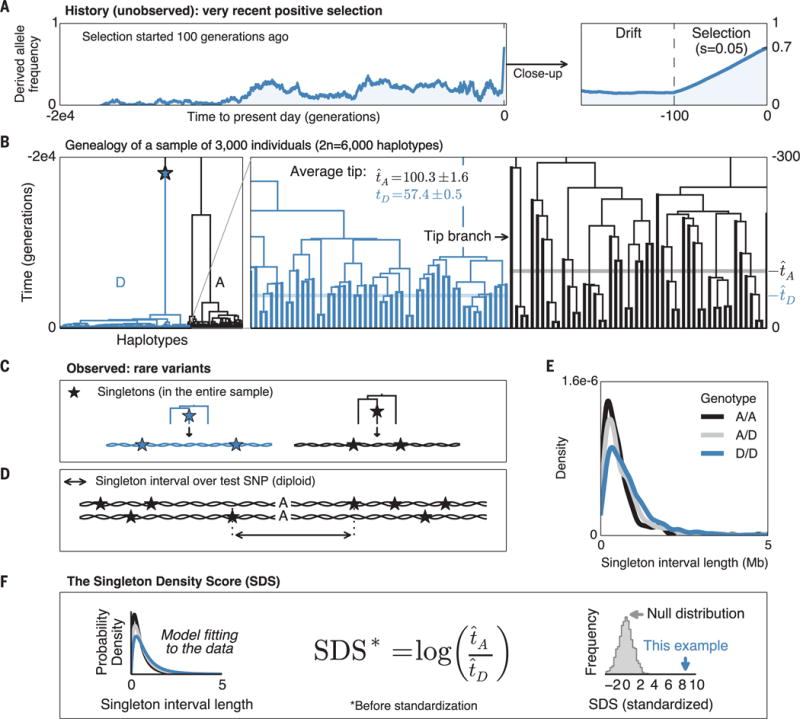
(A) Simulated frequency trajectory for a derived allele that was selected from standing variation starting 100 generations ago. (B) Corresponding genealogy of 3000 present-day genomes. Lineages carrying the derived allele (D) are in blue; ancestral (A) are in black. Enlargement of the genealogy illustrates that tip branches carrying the favored allele (blue) are on average shorter than those carrying the disfavored allele (black). (C) Because favored alleles (blue) tend to have shorter tip branches, their haplotypes tend to have lower singleton density. (D) For each individual, we compute the distance between nearest singletons around the test SNP. (E) Distribution of singleton distances as a function of genotype at the simulated test SNP. (F) Mean tip length t is estimated for each allele from a likelihood model. Unstandardized SDS is a log-ratio of estimated tip lengths; this is standardized to mean 0, variance 1 within bins of derived allele frequency. In this simulated example, SDS is highly significant (P = 1 × 10−17 in favor of the derived allele; relative to neutral simulations). Compare with illustration of drift simulation (fig. S1).
Because SDS measures changes in tip-branch lengths of the genealogy, it detects selection roughly within the timeframe of average tip lengths. For samples of 3000 individuals, this is ~75 generations, according to one recent demographic model (14) (Fig. 2A). At this sample size, SDS is powered to detect ~2% selection, with similar power for selection on standing variation and hard sweeps (Fig. 2, B and C, and fig. S4). Notably, SDS detects little or no signal for selection that stopped >100 generations before present (figs. S4 to S6). The time scale examined by SDS is roughly an order of magnitude shorter than the limits of sensitivity for previous methods that study hard sweeps (13). For example, the integrated haplotype score (iHS)—a commonly used test for hard sweeps (5)—integrates signal over >1000 generations, is generally less powerful, and has no specificity for recent selection (Fig. 2C and figs. S4 and S5).
Fig. 2. Properties of SDS.
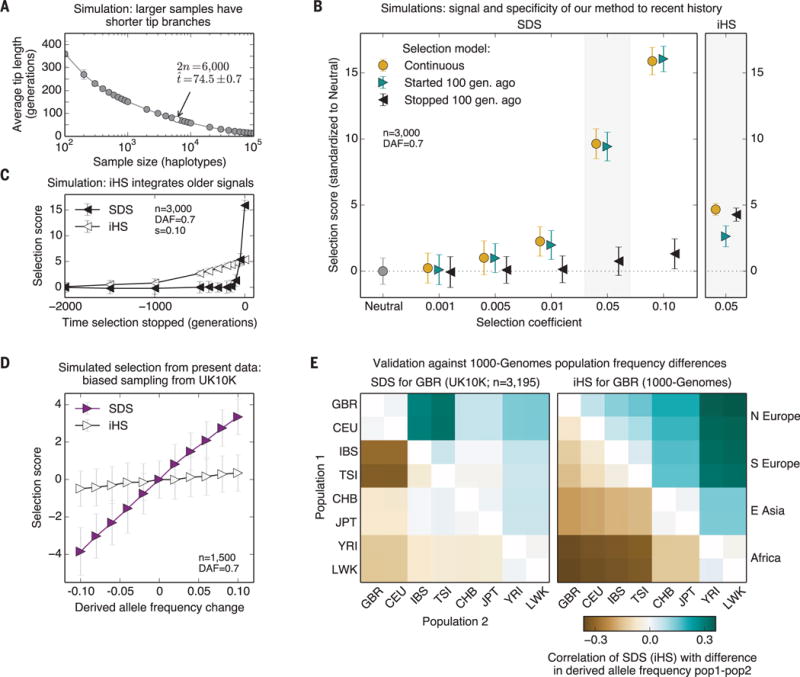
(A) Mean tip length as a function of sample size, for a demographic model with strong recent growth (14) (additional models shown in fig. S10). (B) Power simulations for SDS (mean ± SD) under three models of selection with current derived allele frequency of 0.7: continuous hard sweep (orange); selection starting 100 generations ago (cyan); and hard sweep that stopped 100 generations ago (black). Right panel: corresponding simulations for iHS. (C) SDS and iHS, for sweeps that stopped in the past (s = 0.10), followed by neutral drift. (D) Power to detect simulated selection from present variation using half of the UK10K data. We biasedly sampled 1500 genomes out of 3195 without replacement so as to change the frequencies at randomly chosen SNPs. (E) Allele frequency differences between extant populations (1000-Genomes) versus SDS or iHS. SDS is most correlated with the difference between northern and southern Europe, whereas iHS reflects Europe versus Africa divergence. GBR, British; CEU, Utah residents (northwest European ancestry); IBS, Iberians (Spain); TSI, Tuscans (Italy); CHB, Han (China); JPT, Japanese; YRI, Yoruba (Nigeria); LWK, Luhya (Kenya).
To validate performance in real data, we analyzed 3195 individuals from the UK10K project (15) (fig. S7) (13). To model strong instantaneous selection, we performed biased subsampling of 1500 individuals (without replacement) to change allele frequencies at target SNPs by amounts ranging from 1% to 10% (Fig. 2D and figs. S8 and S9). Although iHS has no power in this test, each 1% change in allele frequency changes the mean SDS by ~0.3 to 0.4 standard deviations. Thus, we expect to have power to detect recent strong selection at individual loci, or weaker signals distributed across many alleles.
We used the set of 3195 genomes to compute SDS for 4.5 million autosomal SNPs with minor allele frequency >5%. We estimate the mean tip length to be 2000 to 3000 years [fig. S10; (13)]. Reassuringly, SDS predictions are correlated with allele-frequency differences between populations (Fig. 2E and fig. S11), and most strongly between southern and northern Europe (Spearman’s ρ = 0.32 ± 0.005). In contrast, iHS measured in a British sample is most correlated with African-European differences. This provides empirical evidence that SDS captures historical frequency changes for times more recent than iHS.
Genome-wide, the largest values of SDS cluster at the lactase locus, a well-known target of selection in Europeans (2, 12) (P = 1 × 10−23; Fig. 3, A and B, and figs. S12 and S13). Based on the magnitude of the signal, we infer that the selection almost certainly persisted into the last 2000 years (fig. S14) (13). The MHC (major histocompatibility complex) region, which has been subject to long-term balancing selection (16, 17), includes the second-highest cluster, with high SDS values across most of the extended MHC region, and at least three independent signals (maximum SDS = 7.9; P = 2 × 10−15; figs. S15 and S16) (13). Curiously, SDS does not support the strongest hit reported from a study of European ancient DNA (12), suggesting complex dynamics of selection in this region (fig. S13). SNPs in the neighborhood of WDFY4 also cross genome-wide significance (P = 3 × 10−11) (fig. S17), but the nature of selection is unclear (13).
Fig. 3. Overview of signals.
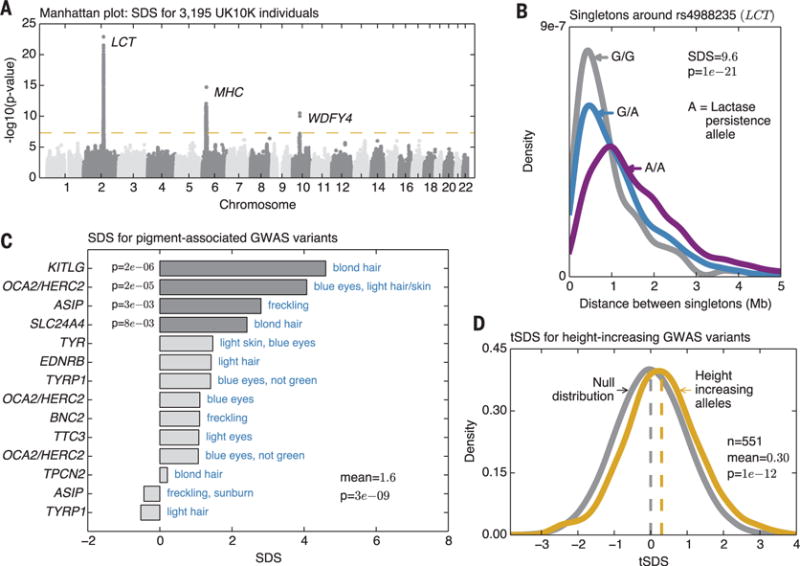
(A) Manhattan plot of SDS P values indicates regions of genome-wide significance (P < 5 × 10−8; P values are two-sided tail probabilities of standard normal). (B) Distributions of singleton distances at the lactase locus, partitioned by genotypes at the causal site. Compare to simulated signals (Fig. 1E). (C) SDS signals for a curated set of segregating variants with known effects on pigmentation shows overall increase in derived allele frequencies (one-sided P values). (D) Distribution of tSDS scores at 551 height-associated SNPs. tSDS is polarized so that tSDS > 0 implies increased frequency of the “tall” allele.
We next considered GWAS-associated variants from the genome-wide association study (GWAS) catalog (18). Overall, these have significantly inflated SDS variance (P = 5 × 10−7 excluding lactase and MHC) (fig. S18) (13). Examining categories of related variants, we found a strong enrichment for variants associated with pigmentation (Fig. 3C and figs. S19 to S21) (13). Although the major determinants of light skin pigmentation in Europe are near fixation and thus not testable by SDS, there is a strong overall enrichment of selection in favor of derived variants associated with lighter pigmentation, especially of hair and eye color (P = 3 × 10−9 for mean SDS > 0).
It has been proposed that another major mechanism of adaptation may be through polygenic selection on complex traits (8). Polygenic adaptation can potentially change phenotypes rapidly, through small, directed allele frequency shifts at many loci, yet leave only weak signals at individual loci. Currently, the best candidate for poly-genic selection in humans is height, as northern Europeans have come to possess more “tall” alleles than southern Europeans over the past ~5000 years (9, 12, 19, 20).
We thus examined SDS for height-associated SNPs from a recent meta-analysis (21). To aid our analysis of height and other traits, we reset the sign of SDS scores for each trait such that positive values indicate increased frequency of the trait-increasing allele instead of the derived allele. We call these new metrics trait-SDS (tSDS) scores. The mean tSDS for 551 height-associated SNPs is significantly positive (mean = 0.30; P = 4 × 10−11), indicating that indeed, on average, “tall” alleles have been increasing in frequency within the past ~2000 to 3000 years in the ancestors of the British (Fig. 3D).
Because most complex-trait heritability is due to SNPs that do not reach GWAS significance (22), we hypothesized that we could increase power by including all SNPs, not just genome-wide significant hits. We thus used all SNPs to test for genome-wide rank correlation between tSDS and GWAS Z score [block-jackknife was used to account for linkage disequilibrium (LD)] (13). Notably, when testing height, mean tSDS is positive across nearly the entire range of P values, and the correlation is extremely significant (Spearman ρ = 0.078; P = 9 × 10−74; fig. S22A) (13). This is not an artifact of uncontrolled population structure in the GWAS, as the correlation is even stronger for a smaller family-based GWAS that provides stringent structure control (20) (Spearman ρ = 0.094; P = 9 × 10−163; Fig. 4A).
Fig. 4. Signals of polygenic adaptation.
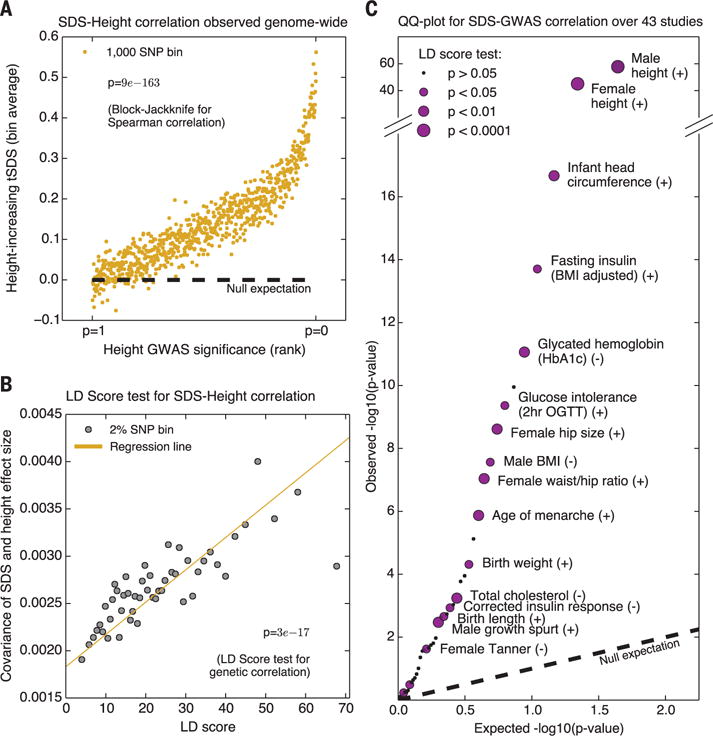
(A) Mean tSDS of SNPs, where tSDS > 0 implies increased frequency of the “tall” allele in a recent family-based study (20). The x axis is ordered from least significant SNPs (P ~ 1) to most significant (P ~ 0), and SNPs are placed into bins of 1000 consecutive SNPs for easier visualization. (B) Covariance of height Z score and SDS, as a function of LD score, provides evidence that selection on height is truly polygenic (P = 2 × 10−11; LD score regression). (C) QQ-plot testing for a correlation between GWAS Z score and tSDS for 43 traits. tSDS > 0 implies increased frequency of the “trait-increasing” allele. Significant traits that are also nominally significant by LD score regression (P < 0.05, one-sided test) are labeled.
Our observation that the signal is stronger in the smaller family-based study may indicate that standard GWAS methods have overcorrected for population structure that pervasively correlates with the phenotypic signal. Further, it may seem counterintuitive that, in aggregate, even non-significant SNPs could have a detectable association with tSDS. However, we estimate that 85% of SNPs in this data set are associated with nonzero effects on height (including through LD tagging) and that the direction of effect is estimated correctly for 68% of all SNPs (23) (fig. S23) (13). Thus, our results indicate that polygenic selection on height has affected allele frequencies across most of the genome.
Aside from height and body mass index (BMI) (19), evidence for selection on other complex traits has generally been weak [e.g., (12, 19)]. We expanded our test to consider 43 traits for which genome-wide GWAS data are available (tables S1 and S2). Notably, many traits show highly significant associations between SDS and GWAS effect sizes (Fig. 4C). Because large-scale family studies are not available for most traits, we used LD score regression to verify these correlations (24, 25). This method uses the property that the covariance between two correlated polygenic signals should increase with the amount of LD if they share an underlying genetic basis, but should be nearly independent of LD for spurious associations resulting from stratification (13). Indeed for height, LD score regression is highly significant (P = 3 × 10−17, family data; P = 2 × 10−11, meta-analysis; Fig. 4B and fig. S22B). Notably, most of the other significant traits are also nominally significant by this stringent test and persist in multiple genomic contexts (Fig. 4C and figs. S24 to S27).
Although height has the strongest signal, we also see signals for increased infant head circumference and birth weight, and increases in female hip size; as well as on variants underlying metabolic traits; male-specific signal for decreased BMI; and in favor of later sexual maturation in women, but not in men. Multiple regression analysis indicates that none of the examined traits, including height, uniquely underlies the top associations (fig. S28) (13). Although these signals are highly intriguing, and some match known phenotypes of modern Britons (13), the confounding role—if any—of population structure in contributing to these signals remains to be fully determined.
In this study, we have introduced a method for inferring very recent changes in allele frequencies that is widely applicable across human populations and other species. We found that human adaptation continued well into historical times, with polygenic adaptation being an important force shaping both genotypic and phenotypic variation.
Supplementary Material
Acknowledgments
We thank G. Coop, R. Durbin, H. Fraser, N. Patterson, J. Pickrell, M. Przeworski, G. Sella, M. Robinson, P. Visscher, and the anonymous reviewers for comments; and A. Bhaskar for technical assistance. This work was supported by NIH grants ES025009, 5T32HG000044-19, and MH101825 and by the Howard Hughes Medical Institute. SDS scores and software are available through the Dryad Digital Repository at http://datadryad.org/resource/doi:10.5061/dryad.kd58f and GitHub at https://github.com/yairf/SDS, as well as through the authors’ website at http://pritchardlab.stanford.edu.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/354/6313/760/suppl/DC1
Materials and Methods
References (26–63)
REFERENCES AND NOTES
- 1.Vitti JJ, Grossman SR, Sabeti PC. Annu Rev Genet. 2013;47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- 2.Bersaglieri T, et al. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamason RL, et al. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 4.Yi X, et al. Science. 2010;329:75–78. [Google Scholar]
- 5.Voight BF, Kudaravalli S, Wen X, Pritchard JK. PLOS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hermisson J, Pennings PS. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Przeworski M, Coop G, Wall JD. Evolution. 2005;59:2312–2323. [PubMed] [Google Scholar]
- 8.Pritchard JK, Pickrell JK, Coop G. Curr Biol. 2010;20:R208–R215. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turchin MC, et al. Nat Genet. 2012;44:1015–1019. doi: 10.1038/ng.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatia G, et al. Am J Hum Genet. 2011;89:368–381. doi: 10.1016/j.ajhg.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilde S, et al. Proc Natl Acad Sci USA. 2014;111:4832–4837. doi: 10.1073/pnas.1316513111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathieson I, et al. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Information on materials and methods is available on Science Online.
- 14.Tennessen JA, et al. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter K, et al. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bakker PIW, Raychaudhuri S. Hum Mol Genet. 2012;21(R1):R29–R36. doi: 10.1093/hmg/dds384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leffler EM, et al. Science. 2013;339:1578–1582. doi: 10.1126/science.1234070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welter D, et al. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg JJ, Coop G. PLOS Genet. 2014;10:e1004412. doi: 10.1371/journal.pgen.1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MR, et al. Nat Genet. 2015;47:1357–1362. doi: 10.1038/ng.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood AR, et al. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, et al. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephens M. bioRxiv. 2016 [Google Scholar]
- 24.Bulik-Sullivan BK, et al. Nat Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulik-Sullivan B, et al. Nat Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.