

Abstract
Methods for displaying large numbers of peptides on solid surfaces are essential for high-throughput characterization of peptide function and binding properties. Here we describe a method for converting the >107 flow cell-bound clusters of identical DNA strands generated by the Illumina DNA sequencing technology into clusters of complementary RNA, and subsequently peptide clusters. In this method, we modify the flow cell-bound primers with ribonucleotides enabling them to be used by poliovirus 3Dpol polymerase. The primers hybridize to the clustered DNA leading to RNA clusters. The RNAs fold into functional protein- or small molecule-binding aptamers. We next used the mRNA display approach to synthesize flow cell-tethered peptides from these RNA clusters. The peptides show selective binding to their cognate antibodies. The methods described here provide an approach for using DNA clusters to template peptide synthesis on an Illumina flow cell, providing new opportunities for massively parallel peptide-based assays.
Keywords: next-generation DNA sequencing, aptamers, mRNA display, peptide display, peptide binding assays, primer-dependent RNA synthesis
Graphical abstract

Introduction
Massively parallel biochemistry assays provides the ability to increase the throughput of biological discovery. Illumina next-generation sequencing (NGS) is one such example.[1] Illumina sequencing requires the formation of >107 DNA clusters containing ~103 identical DNA strands.[1] DNA sequencing is then performed at each cluster. Thus, the Illumina sequencing instrument is a platform for massively parallel, fluorescence-based reactions.[1]
Currently, the Illumina instrument is limited to oligonucleotide-based reactions, but has not been used for peptide-based reactions. Recent studies showed that the strands of DNA within clusters can be transcribed to complementary RNA (cRNA).[2],[3] RNA polymerase binds the DNA and transcribes until it is halted by a DNA-tethered roadblock protein, e.g. streptavidin or Tus. Thus, the product is a DNA-polymerase complex with a noncovalently attached RNA. Using this approach, massively parallel binding assays of fluorescent proteins to tethered RNA targets was described.[2],[3]
Although RNA can be synthesized from the DNA clusters, there are no methods to synthesize peptides encoded by the DNA clusters. Here we describe the synthesis of model peptides directed by the sequence of the DNA clusters. This method utilizes a novel method to synthesize RNA from the DNA clusters such that the cRNA is covalently linked to the Illumina surface. The cRNA can be translated and the synthesized peptide remains attached to the clusters. We synthesize two peptides encoded by the DNA clusters and show that the resulting peptide clusters can be readily detected using epitope-specific antibodies. The methods described here provide an approach to utilize the Illumina platform to generate DNA clusters that encode peptide sequences, and then synthesize and display peptides. These methods could be potentially scaled to enable the sequence information of a DNA cluster to be linked to the encoded peptide, allowing Illumina flow cells to function as an addressable peptide display platform.
Results and Discussion
Strategy for synthesis of RNA on a next generation sequencing platform
To synthesize peptides encoded by DNA clusters, we first wanted to synthesize covalently-linked RNA on the flow cell. The current methods produce RNA that is noncovalently tethered to the DNA clusters via stalled RNA polymerase.[2],[3] These complexes can decompose over time and are assayed within 72 h of formation using loss-of-signal normalization techniques.[2],[3] To overcome this, we sought to generate covalently attached RNA. To do this, we took advantage of the DNA primers that are present on the flow cell surface that are used for creating the DNA clusters.[1] Each DNA cluster is derived from one single-stranded (ss) DNA that is allowed to anneal to a flow cell-bound primer, followed by DNA polymerase-mediated synthesis of a complementary strand of DNA, a process called “bridge amplification.” After several rounds, a DNA cluster is formed with all DNA bound via their 5′ ends. Sequencing is performed by DNA polymerase using a primer and nucleotides with cleavable fluorescent tags.[1]
RNA cannot be synthesized from the DNA clusters using the flow cell-bound primers because the common RNA polymerases do not efficiently covalently attach the newly synthesized RNA to the 3′-OH of the primer. Thus, T7 and other common RNA polymerases are “primer independent.”[4] We reasoned that covalently linked RNA could be generated if a primer-dependent RNA polymerase is used. Although primer-dependent RNA polymerases that use DNA primers and DNA templates have not been described, we considered viral RNA-dependent RNA polymerases, such as 3D polymerase (3Dpol) from poliovirus.[5],[6] Although 3Dpol uses RNA templates in vivo, DNA templates can be used in vitro.[7] Importantly, 3Dpol–synthesized RNA is extended from the 3′-OH of the primer resulting in a covalently attached product (Scheme 1).[8],[5]
Scheme 1.

Principle of covalently-linked RNA synthesis on the Illumina flow cell.
Synthesis of complementary RNA using hybrid RNA-DNA primers
A problem is that the flow cell-bound primers are DNA, while 3Dpol strongly prefers an RNA primer.[8] To overcome this, we reasoned that enzymatic modification of the primers to contain 3′-ribonucleotides might generate primers that are compatible with 3Dpol. To test these DNA-RNA hybrid primers with 3Dpol, we used a “self-priming” DNA template. A self-priming template loops back to hybridize with an internal sequence. The 3′-OH functions as a primer (Figure 1a). We modified the 3′-OH with terminal transferase (TdT) and GTP, which adds 2-3 guanosine ribonucleotides to the DNA 3′-OH.[9] The DNA template contained three cytosines as the first three residues following the primer complimentary domain, such that the TdT-added guanosines would hybridize to these cytosines (Figure 1a).
Figure 1.

3Dpol allows RNA synthesis using a hybrid RNA-DNA primer. (a) Schematic of the self-priming DNA template used to test the suitability of a hybrid RNA-DNA primer. In this template, the 3′ end of the DNA loops back to hybridize with an internal domain in the sequence. TdT is used to add guanosine ribonucleotides that converts the 3′ end into an RNA sequence. Denaturing gel electrophoresis can be used to confirm synthesis of cRNA (red). (b) 3Dpol exhibits RNA synthesis using a hybrid RNA-DNA primer. A self-priming template was incubated with 3Dpol and ribonucleotides to initiate RNA synthesis. Product is only seen if the self-priming template is pretreated with TdT to add guanosine ribonucleotide, indicating that 3Dpol requires a hybrid RNA-DNA primer for efficient synthesis on this template. Reactions were analyzed by denaturing gel electrophoresis and subsequently stained with SYBR Gold to visualize the products. The increased product length is consistent with full-length synthesis of the RNA product.
To synthesize cRNA, the DNA templates were incubated with 3Dpol and ribonucleotides. Only templates that contained ribonucleotides at the 3′ end showed an increased size by denaturing gel electrophoresis (Fig. 1b). The increased size was consistent with covalent elongation of ~23 nt of cRNA. Thus, DNA-RNA hybrid primers can be utilized by 3Dpol to synthesize cRNA off of a DNA template.
To test this approach on flow cells, we used cDNA clusters designed to contain three cytosine residues as the first residues following the primer-complimentary domain, as described above. The cDNA clusters with 3′-end labeled with TdT and FITC-dideoxyadenosine triphosphate (Figure 2a). Importantly, this does not result in labeling of the primers that are on the flow cell because the flow cell primers are 3′-phosphorylated. However, after FITC-labeling of the DNA clusters, the primers were dephosphorylated using T4 polynucleotide kinase, and then converted to DNA-RNA hybrid primers with TdT and GTP. We allowed the DNA-RNA hybrid primers to anneal to the cDNA strands (Scheme 1). Then, transcription was performed using 3Dpol, ribonucleotides, and Cy5-labeled uridine triphosphate (Cy5-UTP). This resulted in Cy5-RNA that colocalized with the FITC-labeled cDNA clusters, demonstrating 3Dpol-mediated RNA synthesis on the Illumina flow cell (Figure 2a).
Figure 2.

RNA-DNA hybrid primers enable cRNA synthesis using cDNA clusters on the Illumina flow cell. (a) DNA-RNA hybrid primers allow RNA synthesis on the Illumina flow cell. In these experiments, a GAIIx flow cell that underwent standard Illumina paired-end clustering of cDNA libraries was used. The resulting double-stranded DNA clusters were denatured with NaOH, which resulted in ssDNA clusters with 3′- OH and primers with 3′-phosphate. Then the ssDNA clusters were 3′-end labeled with FITC-ddATP using TdT-catalyzed chain termination, resulting in FITC-labeled cDNA clusters (middle panel). At this point, the primers were dephosphorylated using PNK to yield 3′-OH. Next, the primer 3′-OH groups were treated with TdT and ribonucleoside GTP to produce DNA-RNA hybrid primers with 2-3 guanosine residues in the 3′-end[9]. The flow cell was heated and then cooled to allow the cDNA clusters to hybridize to the DNA-RNA hybrid primers. RNA was transcribed into cRNA using 3Dpol and Cy5-UTP (red). As can be seen, RNA synthesis was only detected when the primers were modified with TdT and the ribonucleotide guanosine triphosphate. Scale bar = 1 μm. (b) Test of 3Dpol variants for their ability to promote RNA synthesis on the Illumina flow cell. We tested 3Dpol from poliovirus (3Dpol) and coxsackievirus (3Dcox WT), as well a mutant coxsackievirus polymerase (3Dcox I230F), which is an engineered low fidelity variant. Flow cell treatment, primer modification and transcription with the indicated 3Dpol were carried out as described in (a). This shows that all the 3Dpol variants initiated transcription on the cDNA clusters; however the most RNA synthesis was seen with the poliovirus 3Dpol (3Dpol). Scale bar = 1 μm. (c) Quantification of primer-dependent transcription of cDNA clusters on Illumina flow cell. The Cy5-fluorescence in the RNA clusters and FITC-fluorescence in the DNA clusters from (a) and (b) was quantified, and the Cy5/FITC ratio was calculated and normalized to 3Dpol. Quantification shows that 3Dpol was the most efficient polymerase variant, producing ~40% more RNA than coxsackievirus 3Dpol and its I230F variant. n = 50 clusters per condition. Error bars = s.d.
Characterization of RNA synthesis by different 3Dpol variants
We next asked if coxsackievirus 3Dpol and recently engineered low fidelity variants could be used for RNA synthesis.[10] However, the other 3Dpol variants produced markedly less RNA than the poliovirus 3Dpol (Figure 2b and c). Thus, poliovirus 3Dpol was used for all subsequent RNA synthesis experiments.
RNA clusters are full length and covalently linked to the flow cell surface
We next asked if the Cy5-labeled RNA is indeed covalently linked. To test this, we selectively degraded the DNA and the RNA in DNA/RNA clusters using DNase I or RNase A and T respectively. DNase I degrades DNA in DNA/RNA duplexes and has preference for pyrimidine-purine-pyrimidine sequences.[11] This minimizes degradation of the short (21 nt) DNA primer domain in the DNA-RNA hybrid strand which contains only one pyrimidine-purine-pyrimidine sequence.[1] If the RNA were attached via complementary hybridization, DNA degradation would release the RNA. However, DNase I treatment did not reduce the amount of Cy5-RNA, while it removed ~ 60% of the FITC-DNA fluorescence in the clusters (Figure 3a and b). As a control, RNase A and T reduced Cy5 fluorescence by ~90% (Figure 3a and b). Thus, 3Dpol-synthesized RNA is covalently linked to the primer on the flow cell.
Figure 3.

DNA removal and Verification of full length RNA synthesis on the Illumina flow cell. (a) Removal of the DNA and RNA strands after RNA synthesis. After RNA synthesis, a RNA:DNA heteroduplex is created. In these experiments, RNA was synthesized using Cy5-UTP (red) to visualize the RNA. The DNA was visualized using 3′ end labelling with TdT and FITC-ddATP. Following RNA synthesis, treatment with DNase I lead to the degradation of the DNA, leaving single-stranded covalently-linked RNA clusters. Then the RNA was degraded with RNase A and RNAse T, which selectively degrade RNA at C and U residues, or RNA at G residues, respectively. Scale bar = 1 μm. (b) Quantification of percent DNA degradation on the Illumina flow cell following RNA synthesis. Cy5 and FITC fluorescence in the cDNA clusters in (a) was quantified, the background subtracted and the percentage degradation was normalized to untreated clusters. This shows that DNase I removed ~60% of the FITC-DNA fluorescence in the clusters but did not reduce the amount of Cy5-RNA, and the RNAse treatment removed ~90% of the Cy5-RNA fluorescence. n = 50 clusters per condition. Error bars = s.d. (c) Verification of full-length RNA synthesis. The RNA and DNA strands in the RNA/DNA clusters were separately hybridized with FITC-labeled oligonucleotides, which are complimentary to the 3′-end of either the DNA or RNA strands in the clusters. As the FITC-oligonucleotide was targeted to the 3′-end of the RNA this also demonstrates that the synthesized RNA is full length. Scale bar = 1 μm. (d) Quantification of transcription efficiency. FITC fluorescence in the cDNA clusters in (c) was quantified, the background subtracted and normalized to the FITC fluorescence in the DNA clusters. This shows that the RNA/DNA ratio in the RNA/DNA clusters is ~1:1, indicating that the 3Dpol transcription efficiency on the DNA clusters is ~100%. n = 50 clusters per condition. Error bars = s.d.
We confirmed the RNA transcripts were full length using a complementary FITC-labeled oligonucleotide that hybridizes to the 30 nt at the 3′-end of the newly synthesized RNA. In this experiment the DNA clusters were not end-labeled with FITC. Hybridization of the oligonucleotide was readily detectable (Figure 3c), indicating that the synthesized RNA is full length.
To estimate the percent of DNA strands that are transcribed into a cRNA, we hybridized a FITC-labeled oligonucleotide that hybridized to either the RNA or DNA. We separately measured the cluster fluorescence when either the RNA or the DNA was the target of the hybridizing oligonucleotide. Quantification of the FITC fluorescence showed that the RNA/DNA ratio is approximately 1:1 in the RNA/DNA duplex clusters (Figure 3c and d). Thus, 3Dpol enables efficient full-length cRNA synthesis in DNA clusters on the flow cell.
Measurement of protein and fluorophore binding to RNA aptamers displayed in clusters
To further confirm RNA synthesis, we synthesized RNA with measureable biochemical properties. First, we prepared DNA clusters encoding the S1 RNA aptamer. The RNA was then synthesized using 3Dpol as described above. The flow cell was sequentially titrated with increasing concentrations of Cy5-labeled streptavidin to allow quantification of streptavidin binding (Figure 4a and b). This showed that streptavidin binds to the S1 RNA clusters with an affinity of Kd = ~92 nM, which is consistent with the previously reported S1 affinity (Kd = 70 nM).[12]
Figure 4.
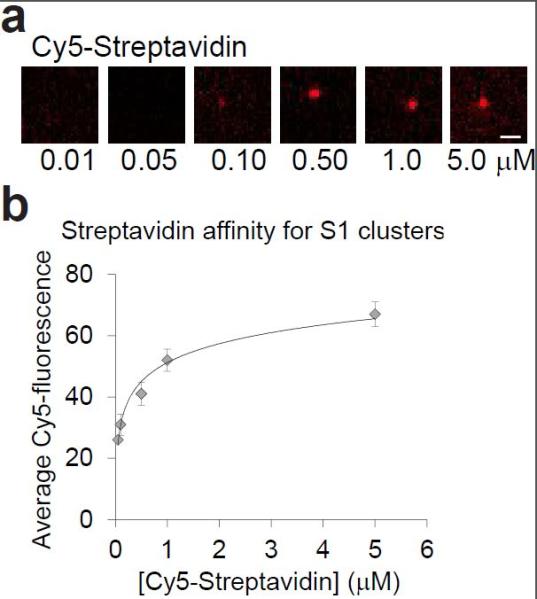
Measurement of the affinity of streptavidin for the S1 aptamer RNA clusters on the Illumina flow cell. (a) Streptavidin binding to S1 RNA aptamer clusters. A flow cells displaying the streptavidin-binding RNA aptamer S1 was generated by clustering DNA oligonucleotides encoding the S1 aptamer at a low density. After DNA clustering, the RNA was synthesized using 3Dpol without Cy5-RNA labelling. Then the flow cell was incubated with increasing amounts of Cy5-labeled streptavidin. The ability of Cy5-labeled streptavidin to bind the aptamer shows that the S1 aptamer is folded and that RNA-binding proteins have access to the RNA clusters. Scale bar = 1 μm. (b) Streptavidin affinity for S1 RNA clusters. The average Cy5 fluorescence in the Cy5-streptavidin/S1 clusters was quantified and plotted versus the streptavidin concentration. This shows that streptavidin binds to the S1 RNA clusters with an affinity of Kd of ~90 nM. n = 50 clusters per condition. Error bars = s.d.
Next, we generated RNA clusters of the Spinach aptamer.[13] Spinach binds and turns on the fluorescence of DFHBI [3,5-difluoro-4-hydroxybenzylidene imidazolinone], which is otherwise non-fluorescent.[13] The flow cells were created with DNA encoding a randomized library of aptamer sequences mixed with 10% Spinach-encoding DNA. As an additional negative control for nonspecific fluorophore association, we an unrelated cDNA library, PhiX.[1] Incubation of the flow cell with DFHBI resulted in fluorescence activation of the Spinach library clusters, but not the PhiX library (Figure S1). These data demonstrate that folded and functional covalently-linked RNA aptamers could be assayed on the flow cell.
Strategy for synthesis of peptides on a next generation sequencing platform
We next sought to use the mRNA display approach[14],[15] to synthesize peptides encoded by the cDNA clusters. In mRNA display, the 3′ end of the mRNA is modified with puromycin.[14],[15] Puromycin can be covalently linked to the RNA by hybridization of puromycin-labeled oligonucleotide.[14],[15] When the ribosome approaches the puromycin, the RNA-bound puromycin is incorporated into the nascent peptide chain. Since the puromycin is attached to the RNA it terminates translation, and the resulting product is a peptide-RNA conjugate (Scheme 2).[14],[15]
Scheme 2.

Principle of translation using mRNA-display on the Illumina flow cell.
For translation, we used bacterial ribosomes, which bind RNA internally at the Shine-Dalgarno sequence.[16] We also included a translation initiation enhancer[17] upstream of the Shine-Dalgarno sequence. Importantly, it is not necessary to change the Illumina pipeline or redesign the input cDNA: in order to add a Shine-Dalgarno sequence, the standard Illumina adapter sequences are modified. [1] Thus, we redesigned Illumina adapter sequences to incorporate the Shine-Dalgarno sequence and the ribosome-binding enhancer sequence[17] (Table S1; translation adapters).
Translation on the Illumina surface using mRNA display
Next we asked if mRNA on the flow cell could be used as a template for in vitro translation. To test this, we clustered cDNA encoding myc- and FLAG-tag peptides followed by a hexaglycine spacer (Table S1). cRNA clusters were synthesized as described above. Then, we treated a flow cell containing clustered FLAG mRNA with in vitro translation mix including [35S] methionine. No puromycin was used in this experiment. As a result, the newly synthesized protein is expected to be released into the reaction solution. As a control, we translated a FLAG-mRNA in solution. Quantification of the resulting 35S-labeled FLAG peptide via gel electrophoresis followed by autoradiography indicated that both the flow-cell bound FLAG mRNA and the FLAG mRNA in solution yielded a peptide product (Figure 5a; lanes 1, 2, and 4). This was blocked by the translation inhibitor hygromycin B (Figure 5a; lanes 3 and 5). Thus, flow cell-bound FLAG mRNA can be used as a template for ribosomes.
Figure 5.

Translation of mRNA clusters into peptides on the Illumina flow cell. (a) RNA on the Illumina flow cell can be used as a template for protein synthesis. mRNA clusters encoding FLAG peptide were generated on an Illumina flow cell as a low density as described above. The FLAG mRNA templates on the Illumina flow cell and in solution were translated using S30 in vitro translation mix supplemented with 35S-labeled methionine. The mRNA template in solution served as a control for translation of a template that is not bound to the glass. Addition of the bacterial ribosome inhibitor hygromycin B was included in a separate flow cell lane as negative controls for translation. The translation mix was recovered from each flow cell lane and analyzed by polyacrylamide gel electrophoresis followed by 35S-autoradiography. This shows that translation of the flow cell 5′-linked FLAG mRNA yielded a product that is comparable to the translation product from mRNA with a free 5′-end. Additionally, the translation inhibitor abolished translation. (b) Translation of mRNA clusters using mRNA-display. The mRNA clusters described in (a) were hybridized with a complementary 3′-puromycin-modified oligonucleotide, according to standard mRNA-display techniques. The puromycin-DNA oligonucleotide included a linker, which was designed to function as a spacer that allows puromycin to enter the ribosome during translation. The puromycin (pur) linker was Cy3-modified in the 5′-end to allow quantification of the RNA-puromycin clusters. The resulting FLAG mRNA-puromycin clusters were treated with in vitro translation mix (“Ribosome”) supplemented with FITC-labeled tRNA-Lysine to enable visualization of the mRNA-puromycin-peptide clusters. Addition of the S30 translation inhibitor, hygromycin B was included in a separate flow cell lane as a negative controls for translation. Co-localization of the FITC-peptide and the Cy3-puromycin-mRNA shows that peptide synthesis occurred on the mRNA-puromycin clusters. The FITC-lysine signal reflects protein synthesis since it was blocked by hygromycin B, a ribosome inhibitor. Scale bar = 1 μm. (c) Selective antibody-binding to peptides translated on Illumina flow cell. A FLAG and myc peptide-displaying flow cell was generated by adding in vitro translation mix (“Ribosome”) as described in (b) without FITC-Lys labeling. Then, the flow cell was simultaneously incubated with a mouse FLAG antibody and a rabbit myc antibody followed by incubation with Cy5-anti-mouse and FITC-anti-rabbit secondary antibodies. The RNA clusters are labeled via the Cy3-puromycin (pur) oligonucleotide linker. Colocalization of the Cy3-puromycin oligonucleotide signal, which indicates mRNA, and the FITC- or Cy5-labeled antibodies shows that the FLAG and myc antibodies selectively bound to the FLAG and myc peptide, respectively. Scale bar = 1 μm.
To retain the peptide on the clusters, we hybridized a 3′-puromycin-modified DNA oligonucleotide (Table S1) to the FLAG-mRNA clusters (Scheme 2).[18] The oligonucleotide was hybridized uniformly over all the clusters since it targets the sequencing primer-complementary domain in the 3′-end of the RNA.[1] As per the mRNA display protocol, the puromycin was linked to the oligonucleotide via a flexible linker to facilitate the entry of puromycin into the ribosome during translation.[18]
To label the synthesized peptide, the translation was carried out with FITC-labeled lysine-tRNA. Additionally, the puromycin-labeled oligonucleotide was further modified with Cy3 at the 5′-end, to visualize the puromycin oligonucleotide-hybridized mRNA clusters. Following addition of the translation mix, FITC-labeled peptide was readily detected and colocalized with the Cy3-labeled puromycin-mRNA clusters (Figure 5b). Thus, peptide synthesis was initiated on the mRNA clusters (Figure 5b). Furthermore, hygromycin B abolished synthesis of FITC-labeled peptide (Figure 5b). Together, these data demonstrate peptide synthesis from mRNA clusters on the Illumina flow cell.
Selective antibody-binding to displayed peptide-mRNA clusters
cDNA-encoded peptide clusters could be useful for various biochemical assays. These assays require that the peptides be accessible to proteins in solution. To test this, we asked if the peptides can bind cognate antibodies. Unlabeled FLAG and myc peptides were synthesized from RNA clusters as described above. Next, the flow cell was incubated with both mouse anti-FLAG and rabbit anti-myc antibodies followed by Cy5-anti-mouse and FITC-anti-rabbit secondary antibodies. This showed that the FLAG and myc antibodies can selectively bind the FLAG and myc peptide, respectively (Figure 5c). Thus the displayed peptides are accessible and could be used in other types of biochemical assays.
Conclusions
The Illumina next-generation sequencing system is a platform for massively parallel high-throughput biochemical reactions. The use of the Illumina platform for biochemical reactions has been limited to RNA biochemistry.[2],[3] Here we demonstrate that the Illumina flow cell can be repurposed for peptide synthesis.
Our method takes advantage of a novel method for synthesis of covalently attached RNA to the Illumina flow cell surface. This is potentially advantageous since the flow cells can be stringently washed and reused. Indeed, our assays illustrate the advantages of covalently attached RNA with experiments that included high-temperature, washes with low salt and high detergent concentrations, as well as enzymatic activity on the RNA. Additionally, our method yields high transcription efficiency compared to previously reported yields.[2],[3]
We demonstrated the versatility of this method by encoding and generating clusters of RNA aptamers with specific measurable properties. Thus we could measure small molecule and protein binding to RNAs, as has been described using the tethered RNA approach.[2],[3] However, here we show for the first time synthesis of cDNA-encoded peptides. These peptides are accessible to solution-phase reagents and thus could participate in a variety of biochemical reactions.
The methods described here can provide the basis for massively parallel presentation and assays of peptide sequences encoded by diverse types of libraries. Peptide presentation is particularly valuable on the Illumina platform since the functional properties of a peptide can be assayed after the sequence of the DNA cluster is determined. Thus, DNA sequencing of a library can be completed, at which point the reagents needed for RNA synthesis and peptide synthesis can be added. The GAIIx platform allows users to program steps for reagent addition after DNA sequencing is completed. Subsequent fluorescent assays, such as the antibody-binding experiments performed here, can be coupled to fluorescent measurements at each cluster. Thus, assays on the encoded protein can be designed to produce fluorescence signals that could then be linked to the specific DNA sequence that encodes the peptide. Thus, massively parallel assays on >107 peptides in each flow cell is the next major direction for this technology.
Experimental Section
Reagents and equipment
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich and sequencing reagents were from Illumina. DFHBI was from Lucerna Technologies (New York, NY). Commercially available reagents were used without further purification. All experiments were carried out with RNase and DNase free H2O. All Illumina flow cell clustering were performed on a cBot (Illumina) at the Epigenomics Core at Weill Cornell Medical College. DNA sequencing and was performed on a Genome Analyzer IIx (GAIIx; Illumina). Fluorescence images were acquired with a CoolSnap HQ2 CCD camera through a 60× oil objective (Plan Apo 1.4 numerical aperture) mounted on a Nikon TE2000 epifluorescence microscope and analyzed with the NIS-Elements software. FITC was imaged with a sputter-coated excitation filter 470/40 nm, dichroic mirror 495 nm (long-pass), and emission filter 525/50 (Chroma Technology), Cy3 was imaged with a sputter coated excitation filter 560/40 nm and emission filter 630/75 nm (Chroma Technology), Cy5 was imaged with a sputter coated excitation filter 620/60 nm and emission filter 700/75 nm (Chroma Technology), and DAPI was imaged with a sputter coated excitation filter 350/50 nm and emission filter 460/50 nm (Chroma Technology). Image analyses were completed with NIS-Elements AR 3.2 (Nikon). PCR was performed in an Eppendorf Mastercycler ep gradient S thermocycler.
Primer modification and transcription of self-priming DNA template in solution
A self-priming DNA template (Table 1; IDT technologies; 10 μM) was modified with 2-3 guanosine moieties by treatment of the 3′-OH with TdT (2 U/μL, 100 μL 1 × TdT Buffer, New England Biolabs) supplemented with ribonucleoside guanosine triphosphate (GTP; 10 μM) for 1 h at 37°C. This resulted in a DNA-RNA hybrid primer with 2-3 RNA moieties in the 3′-end. The DNA-RNA hybrid oligonucleotide was purified using ethanol precipitation. The self-priming DNA-RNA hybrid or the unmodified DNA template (10 μM) were treated with 3Dpol (Poliovirus[19]; 10 μM) in 1 × Pol buffer [50 μL; HEPES pH 7.5 (50 mM), MgCl2 (1.5 mM), MnCl2 (1.5 mM), NaCl (90 mM), dithiothreitol (5mM), ribonucleoside nucleotide triphosphate (200 μM)] for 2 h at 37°C. The reaction mixture was run on a Novex® TBE-Urea Gel, 15% (Life Technologies) according to manufacturer’s protocols.
cDNA library preparation for clustering on the Illumina flow cell
Libraries for clustering on the Illumina flow cells were prepared according to standard Illumina Paired-End GAII library preparation protocols, with the exception of modification of the Illumina adapters. The Illumina adapters for transcription were redesigned to include the three guanosines required for complementarity with the modified RNA-DNA hybrid primers (Table S1). The Illumina adapters for translation were redesigned to include these three guanosines, as well the Shine-Dalgarno sequence, an A/U-translation initiation enhancer, a start codon, and peptide spacers (Table S1). We designed these adapters with 5′-phosphates such that they can be used in Illumina’s standard protocol for library preparation. Note, that no other modifications to Illumina’s standard library preparation, clustering, and paired-end sequencings protocols are necessary for transcription and translation of the cDNA clusters.
Generation of RNA clusters on the Illumina flow cell
To optimize RNA synthesis, we used Paired-End GAIIx flow cells (Illumina) displaying cDNA libraries that were clustered on a cBot (Illumina) according to standard Illumina paired-end sequencing protocols. The resulting flow cells consist of single-stranded primers and double-stranded DNA clusters, in which one strand is attached to the flow cell surface via the 5′-end with a complementary strand hybridized to it. The DNA clusters have 3′-OH and the primers have 3′-phosphates.First, the dsDNA clusters were denatured with NaOH (0.1 M, 300 μL per lane) for 20 min at 60°C. Then, the resulting flow cells consist of 3′-phosphorylated primers as well as ssDNA clusters with 3′-OH, which are covalently linked via the 5′-end to the flow cell.Then, the ssDNA clusters were selectively labeled with FITC via terminal transferase (TdT)-catalyzed 3′ OH labeling by treatment with TdT (2 U/μL, 50 μL 1 × TdT Buffer, New England Biolabs) supplemented with FITC-labeled 2',3'-dideoxyadenosine-5'-triphosphate (ddATP; 10 μM; PerkinElmer) for 1 h at 37°C. This treatment does not label the primers since the primers have a 3′ phosphate. The flow cell was washed with 5 × saline-sodium citrate (SSC) buffer (3 × 500 μL/lane; Corning) and imaged. In experiments without FITC-DNA labeling this step was omitted.Then, the flow cell 3′-phosphorylated primers were dephosphorylated with polynucleotide kinase (PNK; 1 U/μL, 50 μL 1 × T4 PNK Buffer, New England Biolabs) for 1 h at 37°C to yield primers with 3′-OH. The flow cell was washed with 5 × SSC buffer (3 × 500 μL/lane).The resulting flow cell primers 3′-OH were then modified with 2-3 Guanine moieties by treatment of the 3′-OH with TdT (2 U/μL, 50 μL 1 × TdT Buffer, New England Biolabs) supplemented with ribonucleoside guanosine triphosphate (GTP; 10 μM) for 1 h at 37°C. This resulted in a DNA-RNA hybrid primers with 2-3 RNA moieties in the 3′-end. The flow cell was washed with 5 × SSC buffer (3 × 500 μL/lane).Then the DNA clusters were transcribed into complementary RNA using various viral 3Dpol variants (Poliovirus wild-type, Coxsackievirus wild-type and I230F variant; 10 μM) in 1 × Pol buffer [50 μL/lane; HEPES pH 7.5 (50 mM), MgCl2 (1.5 mM), MnCl2 (1.5 mM), NaCl (90 mM), dithiothreitol (5 mM), ribonucleoside nucleotide triphosphate (200 μM)] supplemented with ribonucleoside Cy5-aminoallyl-uridine triphosphate (Cy5-UTP; 66.67 μM; GE Healthcare) for 2 h at 37°C. Cy5-UTP was incorporated during transcription to allow quantification of the RNA clusters. The flow cell was washed with 5 × SSC buffer (3 × 500 μL/lane) and imaged.
Degradation of DNA in DNA/RNA duplex clusters on the Illumina flow cell
Following RNA synthesis the complementary DNA was degraded using DNase I (0.02 U/μL, 50 μL/lane 1 × DNase I Buffer, New England Biolabs) for 10 min at 37°C, which yielded covalently linked ssRNA clusters, which are complementary to the sequenced DNA clusters. The flow cell was washed with 5 × SSC buffer (3 × 500 μL/lane) and imaged. Occasionally, the ssRNA clusters were selectively degraded with RNase A and T (1 U/μL RNase A, 0.5 U/μL RNase T, 50 μL/lane 1 × RNase A/T Buffer, Thermo Scientific) for 2 h at 37°C. The flow cell was washed with 5 × SSC buffer (3 × 500 μL/lane) and imaged.
Generation of aptamer RNA clusters the on Illumina flow cell
cDNA encoding Spinach or S1 aptamers (Table S1) were clustered in separate lanes on a Paired-End GAIIx flow cell (Illumina) using standard Illumina GAIIx protocols on a cBOT clustering instrument (Illumina). Following clustering of the cDNA, the resulting clusters were transcribed into RNA clusters without FITC-ddATP-and Cy5-UTP-labeling and the complementary DNA was degraded as described above.
Imaging of Spinach on the Illumina flow cell
The Spinach aptamer-displaying flow cell was equilibrated with Spinach imaging buffer [500 μL/lane; HEPES pH 7.4 (40 mM), KCl (125 mM), MgCl2 (10 mM), DFHBI (20 μM)] for 1 h and imaged.
Streptavidin binding to S1 aptamer clusters on the Illumina flow cell
The S1 aptamer-displaying flow cell was equilibrated with streptavidin imaging buffer [SIB; 500 μL/lane; Tris-HCl pH 7.4 (40 mM), KCl (100 mM), MgCl2 (10 mM), NaCl (150 mM)] at room temperature. It was then imaged to measure the background intensity. It was then equilibrated successively with increasing concentrations of Cy5-streptavidin (0.010 μM, 0.050 μM, 0.10 μM, 0.5 μM, 1.0 μM, 5.0 μM; LifeTechnologies) in SIB (100 μL/lane) for 30 min at room temperature. The flow cell was washed with SIB (500 μL/lane) and immediately imaged.
Generation of mRNA templates for translation in solution
cDNA encoding myc- and FLAG-tag peptides (Table S1) were amplified by PCR using Phusion High-Fidelity PCR Master Mix (New England Biolabs) and primers [T7 Illumina fwd and Illumina rev; Table S1], which included a complementary 5′ T7 promoter sequence (lowercase) according to manufacturer’s protocol. dsDNA was purified using QIAquick PCR Purification Kit (Qiagen) according to manufacturer’s protocol.This construct was used as templates for in vitro T7 transcription reactions (Epicentre) according to manufacturer’s protocol. The mRNA template was purified using RNaseEasy Purification Kit (Qiagen) according to the manufacturer’s protocol.
Generation of mRNA-puromycin-linker clusters for mRNA display on the Illumina flow cell
cDNA encoding myc- and FLAG-tag peptides (Table S1) were clustered on an Paired-End GAIIx flow cell (Illumina) using standard Illumina GAIIx protocols on a cBOT (Illumina). Following clustering of the myc and FLAG cDNA, the resulting clusters were transcribed into RNA clusters without FITC-ddATP- and Cy5-UTP-labeling and the complementary DNA was degraded as described above. Hereafter, the mRNA in select flow cell lanes was hybridized with a Cy3-labeled DNA-puromycin linker (5 nM; Table S1) in Illumina HT1 buffer (500 μL/lane) at 60°C for 10 min and 40°C for 10 min. The flow cell was immediately washed with 40°C Illumina HT1 (3 × 500 μL/lane) and 37°C phosphate buffered saline supplemented with Tween20 (PBST; 0.1% Tween20; 500 μL/lane) and the flow cell was imaged.
Translation of the mRNA clusters on the Illumina flow cells and mRNA templates in solution
Translation of the FLAG- and myc-mRNA clusters with stop codons on the flow cell was performed with PURExpress in vitro translation kit (New England Biolabs) according to manufacturer’s protocol. Briefly: translation mix [25 μL/lane; Solution A (10 μL), Solution B (7.5 μL), RNaseOut (0.5 μL; Life Technologies), EasyTag™ L-[35S]-Methionine (2 μL; PerkinElmer), H2O (2.0 μL)] was incubated on the FLAG- and myc-mRNA with stop codons clustered flow cell at 37°C for 30 min. Translation in solution was carried out using mRNA template (3.32 nM) in 10 μL translation mix. For translation inhibition the translation mix was supplemented with hygromycin B (50 μg/μL). After translation the flow-through was collected separately from each flow cell lane.
35S-gel electrophoresis
The collected translation mix (10 μL) was run on a Novex 16% Tricine Protein Gel (ThermoFisher) according to the manufacturer’s protocol. The gel was fixed and dried using Novex DryEase Mini-Gel Drying system (ThermoFisher) according to manufacturer’s protocol. The gel was imaged on a Typhoon Triad Phosphorimager using ImageQuant software.
mRNA display on the Illumina surface
Translation of the puromycin-linker/mRNA clusters without stop codons on the flow cell was performed with S30 T7 High-Yield Protein Expression System (Promega, Lot: 0000100529) translation kit according manufacturer’s protocol. Briefly: translation mix [25 μL/lane; S30 Premix Without Amino Acids (10 μL), Amino Acids Mixture Minus Lysine (2.5 μL), S30 Extract (7.5 μL), RNaseOut (0.5 μL; Life Technologies), Lysine (5 μL) or FluoroTect™ Green Lys (5 μL; Promega)] was incubated on the RNA/DNA-puromycin clustered flow cell at 30°C for 60 min. For translation inhibition the translation mix was supplemented with hygromycin B (50 μg/μL). To allow covalent linkage of the puromycin to the polypeptide the flow cell was immediately treated with PBS (500 μL/lane) supplemented with MgCl2 (50 mM) and KCl (500 mM) and incubated for 1 h at room temperature. The flow cell was immediately washed with PBST (3 × 500 μL/lane) and imaged.
Antibody binding to peptide-displaying clusters
All antibody solutions were centrifuged at 10,000 g for 5 min at 4°C prior to addition to the flow cells. The peptide-displaying clusters generated without FITC-Lys labeling were blocked with bovine serum albumin (BSA; 500 ng/μL) in PBST (3 × 250 μL/lane; 5 min each) at room temperature. Following blocking, the flow cell was simultaneously incubated with mouse monoclonal FLAG antibody (Sigma-Aldrich) and rabbit monoclonal myc antibody (Sigma-Aldrich) at 1:1000 dilution in PBST-BSA buffer [PBST with BSA (50 ng/μL)] by adding 100 μL/lane every 3 min for 30 min at room temperature. The flow cell was washed with PBST-BSA (250 μL/lane every 5 min for 15 min) at room temperature. This was followed by incubation with donkey Cy5-anti-mouse DyLight 650 antibody (ThermoFisher) and donkey FITC-anti-rabbit AlexaFluor antibody (Molecular Probes) at 1:1000 dilution in PBST-BSA (100 μL/lane every 3 min for 30 min) at room temperature. The flow cell was washed with PBST (250 μL/lane every 5 min for 15 min) at room temperature and imaged.
Supplementary Material
Acknowledgements
We thank Dr. Wenjiao Song (Weill Cornell Medical College) for synthesis of DFHBI, and Gary Schroth at Illumina for providing Illumina flow cells and sequencing reagents. Supported by NIH grants R01 NS064516, R01 EB010249, and Starr Cancer Consortium grant I9-A9-084 to S.R.J, and R01 AI059130 to O.B.P.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Bentley DR, et al. Nature. 2008;456(7218):53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, Greenleaf WJ. Nat Biotechnol. 2014;32(6):562–568. doi: 10.1038/nbt.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tome JM, Ozer A, Pagano JM, Gheba D, Schroth GP, Lis JT. Nat Methods. 2014;11(6):683–688. doi: 10.1038/nmeth.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sousa R, Chung YJ, Rose JP, Wang BC. Nature. 1993;364(6438):593–599. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- [5].Rodriguez-Wells V, Plotch SJ, DeStefano JJ. Nucleic Acids Res. 2001;29(13):2715–2724. doi: 10.1093/nar/29.13.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Van Dyke TA, Flanegan JB. J Virol. 1980;35(3):732–740. doi: 10.1128/jvi.35.3.732-740.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arnold JJ, Ghosh SK, Cameron CE. J Biol Chem. 1999;274(52):37060–37069. doi: 10.1074/jbc.274.52.37060. [DOI] [PubMed] [Google Scholar]
- [8].Lubinski JM, Ransone LJ, Dasgupta A. J Virol. 1987;61(10):2997–3003. doi: 10.1128/jvi.61.10.2997-3003.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deng G, Wu R. Methods Enzymol. 1983;100:96–116. doi: 10.1016/0076-6879(83)00047-6. [DOI] [PubMed] [Google Scholar]
- [10].Campagnola G, McDonald S, Beaucourt S, Vignuzzi M, Peersen OB. J Virol. 2015;89(1):275–286. doi: 10.1128/JVI.01574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang Z, Fasco MJ, Kaminsky LS. Biotechniques. 1996;20(6):1012–1014. 1016, 1018–1020. doi: 10.2144/96206st02. [DOI] [PubMed] [Google Scholar]
- [12].Srisawat C, Engelke DR. RNA. 2001;7(4):632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Paige JS, Wu KY, Jaffrey SR. Science. 2011;333(6042):642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roberts RW, Szostak JW. Proc Natl Acad Sci U S A. 1997;94(23):12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nemoto N, Miyamoto-Sato E, Husimi Y, Yanagawa H. FEBS Lett. 1997;414(2):405–408. doi: 10.1016/s0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- [16].Shine J, Dalgarno L. Proc Natl Acad Sci U S A. 1974;71(4):1342–1346. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Osterman IA, Evfratov SA, Sergiev PV, Dontsova OA. Nucleic Acids Res. 2013;41(1):474–486. doi: 10.1093/nar/gks989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu R, Barrick JE, Szostak JW, Roberts RW. Methods Enzymol. 2000;318:268–293. doi: 10.1016/s0076-6879(00)18058-9. [DOI] [PubMed] [Google Scholar]
- [19].Thompson AA, Peersen OB. EMBO J. 2004;23(17):3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.