

Abstract
Nitric oxide (NO•) is thought to protect against the damaging effects of myocardial ischemia–reperfusion injury, whereas xanthine oxidoreductase (XOR) normally causes damage through the generation of reactive oxygen species. In the heart, inorganic nitrite 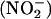 has the potential to act as an endogenous store of NO•, liberated specifically during ischemia. Using a detection method that we developed, we report that under ischemic conditions both rat and human homogenized myocardium and the isolated perfused rat heart (Langendorff preparation) generate NO• from
has the potential to act as an endogenous store of NO•, liberated specifically during ischemia. Using a detection method that we developed, we report that under ischemic conditions both rat and human homogenized myocardium and the isolated perfused rat heart (Langendorff preparation) generate NO• from 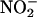 in a reaction that depends on XOR activity. Functional studies of rat hearts in the Langendorff apparatus showed that nitrite (10 and 100 μM) reduced infarct size from 47.3 ± 2.8% (mean percent of control ± SEM) to 17.9 ± 4.2% and 17.4 ± 1.0%, respectively (P < 0.001), and was associated with comparable improvements in recovery of left ventricular function. This protective effect was completely blocked by the NO• scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide (carboxy-PTIO). In summary, the generation of NO• from
in a reaction that depends on XOR activity. Functional studies of rat hearts in the Langendorff apparatus showed that nitrite (10 and 100 μM) reduced infarct size from 47.3 ± 2.8% (mean percent of control ± SEM) to 17.9 ± 4.2% and 17.4 ± 1.0%, respectively (P < 0.001), and was associated with comparable improvements in recovery of left ventricular function. This protective effect was completely blocked by the NO• scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide (carboxy-PTIO). In summary, the generation of NO• from 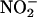 , by XOR, protects the myocardium from ischemia–reperfusion injury. Hence, if XOR is presented with
, by XOR, protects the myocardium from ischemia–reperfusion injury. Hence, if XOR is presented with 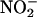 as an alternative substrate, the resultant effects of its activity may be protective, by means of its production of NO•, rather than damaging.
as an alternative substrate, the resultant effects of its activity may be protective, by means of its production of NO•, rather than damaging.
Nitric oxide (NO•) is an important regulator of numerous diverse physiological and pathological processes that is produced by most cell types. It is the metabolism of l-arginine by NO• synthase (NOS) (1), to produce NO• and citrulline, that is widely accepted as the primary source of NO• from biological tissues. However, a growing body of evidence supports alternative NOS-independent mechanisms of NO• synthesis that might operate in situations in which conventional NO• production is impaired (2–6). Inorganic nitrite 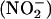 is an endogenous substance produced by the oxidation of NO• in aerobic conditions (2). Conversely, in acidic conditions,
is an endogenous substance produced by the oxidation of NO• in aerobic conditions (2). Conversely, in acidic conditions, 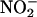 can be chemically reduced back to NO•. This latter reaction seems to be a phenomenon that can be evoked in biological systems and was originally described in the acidic conditions of the human stomach, in which
can be chemically reduced back to NO•. This latter reaction seems to be a phenomenon that can be evoked in biological systems and was originally described in the acidic conditions of the human stomach, in which 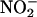 , derived from the sequential reduction of dietary nitrate
, derived from the sequential reduction of dietary nitrate 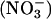 , was reduced to NO• (3, 7). This reaction recently was shown to have important functional effects whereby human saliva rich in
, was reduced to NO• (3, 7). This reaction recently was shown to have important functional effects whereby human saliva rich in 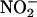 improves gastric mucosal blood flow and mucus thickness when applied to the rat stomach (4). Similarly, in the blood,
improves gastric mucosal blood flow and mucus thickness when applied to the rat stomach (4). Similarly, in the blood, 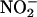 -derived NO•, thought to be generated by means of deoxyhemoglobin, has vasodilator activity (5).
-derived NO•, thought to be generated by means of deoxyhemoglobin, has vasodilator activity (5).
This alternative mechanism of NO• synthesis may be particularly important in ischemic conditions, because the generation of NO• from l-arginine by NOS enzymes depends on oxygen, which is rapidly depleted in ischemia. Studies using rat myocardium clearly demonstrate NOS-independent generation of NO•, detected with EPR spectroscopy (6). This NO• generation was attributed to the chemical reduction of endogenous 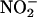 under ischemic conditions (physiological levels of blood and tissue
under ischemic conditions (physiological levels of blood and tissue 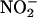 estimates are 0.15–1.0 μM and 5–40 μM, respectively) (2, 6, 8, 9). Ferrari et al. (10) and Gabel et al. (11) suggest that the reduction of
estimates are 0.15–1.0 μM and 5–40 μM, respectively) (2, 6, 8, 9). Ferrari et al. (10) and Gabel et al. (11) suggest that the reduction of 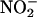 to NO• is derived from a simple acidification reaction; ischemia-induced acidosis is associated with a drop in pH to ≈5.5 in the isolated rat heart preparation after 20 min of global ischemia. However, evidence now suggests that this reaction also may depend on enzymatic catalysis. Purified xanthine oxidoreductase (XOR), under hypoxic conditions, catalyzes the reduction of
to NO• is derived from a simple acidification reaction; ischemia-induced acidosis is associated with a drop in pH to ≈5.5 in the isolated rat heart preparation after 20 min of global ischemia. However, evidence now suggests that this reaction also may depend on enzymatic catalysis. Purified xanthine oxidoreductase (XOR), under hypoxic conditions, catalyzes the reduction of 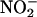 to NO• (12–15). Conventional XOR activity is in purine catabolism, catalyzing the hydroxylation of hypoxanthine to xanthine and xanthine to urate (16). However, provided that the environmental conditions are appropriate, it may be possible that XOR acts as a salvage pathway to maintain levels of NO• in situations where conventional constitutive NOS activity may be compromised. Such situations would include inflammatory cardiovascular conditions (atherosclerosis) with associated endothelial dysfunction and particularly myocardial infarction (15). Indeed, there is good evidence that XOR activity is up-regulated during hypoxia (13, 17–19), with increasing acidosis (14), and with atherosclerosis. In patients with coronary artery disease, endothelium-bound XOR activity is increased by >200% (20). However, whether XOR might determine
to NO• (12–15). Conventional XOR activity is in purine catabolism, catalyzing the hydroxylation of hypoxanthine to xanthine and xanthine to urate (16). However, provided that the environmental conditions are appropriate, it may be possible that XOR acts as a salvage pathway to maintain levels of NO• in situations where conventional constitutive NOS activity may be compromised. Such situations would include inflammatory cardiovascular conditions (atherosclerosis) with associated endothelial dysfunction and particularly myocardial infarction (15). Indeed, there is good evidence that XOR activity is up-regulated during hypoxia (13, 17–19), with increasing acidosis (14), and with atherosclerosis. In patients with coronary artery disease, endothelium-bound XOR activity is increased by >200% (20). However, whether XOR might determine 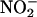 -dependent NO• generation in the heart is unknown.
-dependent NO• generation in the heart is unknown.
Mainly, NO• is considered to be protective in the heart (21). NO• donors protect against ischemia–reperfusion (I/R) damage in in vitro models of infarction (22–24). In addition, knockout of endothelial NOS renders the hearts of mice more sensitive to ischemic insults, with increased infarct size and diminished cardiac function compared with wild type (25, 26). Because 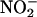 is thought to release NO•, as indicated above, one would expect
is thought to release NO•, as indicated above, one would expect 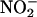 to display cardioprotection in models of I/R injury. However the literature is contradictory. Although acidified
to display cardioprotection in models of I/R injury. However the literature is contradictory. Although acidified 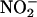 seems to protect against myocardial infarction in cats in vivo (27,28), other studies suggest that
seems to protect against myocardial infarction in cats in vivo (27,28), other studies suggest that 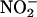 -derived NO• may be contributing to damage (6), and, thus, the effects of NO• derived from
-derived NO• may be contributing to damage (6), and, thus, the effects of NO• derived from 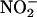 in myocardial injury are uncertain (23, 24).
in myocardial injury are uncertain (23, 24).
We now demonstrate that XOR, during myocardial ischemia, catalyzes the formation of NO• from 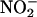 in a pH- and concentration-dependent manner and that this NO• protects the isolated rat heart against the damaging effects of I/R injury. Moreover, we demonstrate that this
in a pH- and concentration-dependent manner and that this NO• protects the isolated rat heart against the damaging effects of I/R injury. Moreover, we demonstrate that this 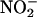 -reducing capacity of XOR is evident in human as well as rat myocardium.
-reducing capacity of XOR is evident in human as well as rat myocardium.
Methods
NO• Production by Rat Heart Homogenates. All experiments were conducted according to The Animals (Scientific Procedures) Act 1986, United Kingdom. The hearts were removed from freshly killed male Wistar rats (Tuck, Rayleigh, U.K.) and immediately frozen in liquid nitrogen and kept at –80°C until processing. The tissue was homogenized in a Dowex chamber with 2 ml of phosphate-buffered solution, containing proteinase inhibitors (1 μM pepstatin A/200 μM PMSF/50 μM leupeptin/1 μg/ml aprotinin). The homogenate was then centrifuged at 13,000 rpm in a Techne genofuge (Jencons-PLS, East Sussex, U.K.) for 5 min, and a Bradford protein assay was performed on the supernatant by using BSA for the standard curve and measuring absorbance by using a spectrophotometric plate reader (MRX-TC Revelation, Dynex Technologies, West Sussex, U.K.).
For the measurement of NO• production, experiments were performed in a sealed 10-ml glass reaction chamber containing citric acid/Na2HPO4 buffer (McIlvaine buffer) at pH 6.0–5.0 and sodium nitrite (10–100 μM) in a total volume of 1 ml. This solution was bubbled with nitrogen gas (100%) to simulate ischemia or room air to create an oxygenated environment, both by means of a NO• scrubbing zero air filter (Sievers, Boulder, CO). The headspace NO• concentration was measured in parts per billion by using continuous sampling with ozone chemiluminescence (Sievers 280A nitric oxide analyzer). We determined the impact of biological tissue on NO• production from 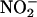 by the addition of heart supernatant (300 μg of protein) and measurement of NO• over 2 min, calculating the rate of NO• production (nmol per g of tissue per s) from the area under the curve.
by the addition of heart supernatant (300 μg of protein) and measurement of NO• over 2 min, calculating the rate of NO• production (nmol per g of tissue per s) from the area under the curve.
To determine whether 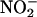 -derived NO• production depended on enzymatic activity, the sample was denatured by boiling before addition to the reaction chamber. To determine the nature of the enzyme involved in NO• production, a range of inhibitors were tested: XOR inhibitors allopurinol (100 μM) and the levorotatory stereoisomer of sodium 8-(3-methoxy-4-phenylsulfinylphenyl)pyrazolo[1,5-α]-1,3,5-triazine-4-olate monohydrate [(–)BOF-4272, 10 μM] (13, 29) and the NOS inhibitor Nω-nitro-l-arginine methyl ester (L-NAME) (300 μM). These inhibitors were incubated with the tissue sample for 30 min before addition to the NO• sampling chamber. In these experiments, NO• production was assessed after a 10-min incubation period in the chamber. After this time, sampling was initiated and the rate of NO• production (nmol per g of tissue per s) was calculated from the area under the curve measured during the initial 18 s of sampling. This method of incubating results in an ≈10-fold lesser rate in the absolute measured NO• production compared with continuous measurement but enables comparison between the inhibitors.
-derived NO• production depended on enzymatic activity, the sample was denatured by boiling before addition to the reaction chamber. To determine the nature of the enzyme involved in NO• production, a range of inhibitors were tested: XOR inhibitors allopurinol (100 μM) and the levorotatory stereoisomer of sodium 8-(3-methoxy-4-phenylsulfinylphenyl)pyrazolo[1,5-α]-1,3,5-triazine-4-olate monohydrate [(–)BOF-4272, 10 μM] (13, 29) and the NOS inhibitor Nω-nitro-l-arginine methyl ester (L-NAME) (300 μM). These inhibitors were incubated with the tissue sample for 30 min before addition to the NO• sampling chamber. In these experiments, NO• production was assessed after a 10-min incubation period in the chamber. After this time, sampling was initiated and the rate of NO• production (nmol per g of tissue per s) was calculated from the area under the curve measured during the initial 18 s of sampling. This method of incubating results in an ≈10-fold lesser rate in the absolute measured NO• production compared with continuous measurement but enables comparison between the inhibitors.
NO• Production by Human Heart Homogenates. These experiments were performed with the approval of the local ethics committee. After informed consent, small amounts of myocardium were obtained from six patients undergoing mitral valve replacement surgery. The tissues were processed to generate heart supernatant samples by using the same methods as for rat heart tissue. The capacity for human heart supernatants to generate NO• from 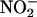 was tested under conditions of pH 5.5 with
was tested under conditions of pH 5.5 with 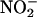 (10–100 μM). In addition, some samples were boiled or treated with the inhibitors allopurinol (100 μM) or L-NAME (300 μM).
(10–100 μM). In addition, some samples were boiled or treated with the inhibitors allopurinol (100 μM) or L-NAME (300 μM).
NO• Production by the Isolated Rat Heart Preparation. Male Wistar rats (Tuck) weighing 260–340 g were anesthetized with i.p. pentobarbital (45 mg/kg) and anticoagulated with heparin (1,000 units/kg) and then killed by cervical dislocation. The hearts were excised, and the aortas were cannulated and perfused with a modified Krebs bicarbonate buffer (118.5 mM sodium chloride/4.8 mM potassium chloride/1.2 mM magnesium sulfate heptahydrate/12 mM glucose/25 mM sodium bicarbonate/1.2 mM potassium dihydrogen orthophosphate/1.7 mM calcium chloride) (30) at a constant flow rate of 40 ml/min per kg of rat. All studies assessing NO• production were conducted in the presence of L-NAME (300 μM) to fully inhibit NO• production from NOS (31).
After 15 min of equilibration and 15 min of recording, saline (control) or 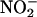 (10–1,000 μM) was infused (0.1 ml/min) via a side arm in the perfusion line for 15 min. This infusion was followed by either the continuation of normal perfusion (non-I/R control) or 60 min of global normothermic ischemia followed by a 30-min reperfusion.
(10–1,000 μM) was infused (0.1 ml/min) via a side arm in the perfusion line for 15 min. This infusion was followed by either the continuation of normal perfusion (non-I/R control) or 60 min of global normothermic ischemia followed by a 30-min reperfusion. 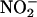 or saline control infusions were continued during ischemia; however, their concentrations were proportionally reduced to achieve equivalent final concentrations in the heart. The
or saline control infusions were continued during ischemia; however, their concentrations were proportionally reduced to achieve equivalent final concentrations in the heart. The 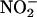 and saline solutions were deoxygenated for the period of global ischemia by bubbling with 95% N2/5% CO2.
and saline solutions were deoxygenated for the period of global ischemia by bubbling with 95% N2/5% CO2.
After the 15 min of equilibration, specially adapted sealable 50-ml chambers were placed around the hearts to collect the NO• gas. These chambers comprised a drawstring plastic membrane over the top to produce a reversible seal around the cannulated aorta, a drainhole near the base for the effluent, creating a “water-seal,” and a resealable port in the middle to allow sampling of the “gas space” around the heart. Gas samples were aspirated into a 50-ml syringe every 15 min, and the NO• concentration was measured with ozone chemiluminescence (Fig. 1).
Fig. 1.

Diagram of the NO•-collecting chamber placed around the isolated perfused heart to collect free NO• gas.
To determine whether XOR is involved in NO• production, hearts were treated with 100 μM allopurinol (13) or 10 μM (–)BOF-4272 (13, 29) (30-min pretreatment for both and then continuously thereafter). To determine whether the reduction of 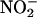 to NO• involved activity at the level of the endothelial cell, the endothelium was removed from the Langendorff mounted heart preparation by bolus injection of 60 μl of 1% Triton X-100 detergent. The efficacy of this method was confirmed by the loss of coronary vasodilatation to bolus doses of the endotheliumdependent vasodilator bradykinin (3–30 pmol).
to NO• involved activity at the level of the endothelial cell, the endothelium was removed from the Langendorff mounted heart preparation by bolus injection of 60 μl of 1% Triton X-100 detergent. The efficacy of this method was confirmed by the loss of coronary vasodilatation to bolus doses of the endotheliumdependent vasodilator bradykinin (3–30 pmol).
Functional Studies. For measurement of left ventricular function and infarct size, the protocol was as described above, except without the NO•-collecting chambers or L-NAME. The left ventricular developed pressure and the rate of change of pressure during diastole (–dP/dt) were measured throughout by using an in-line pressure transducer (Gould, Cleveland) attached to a latex balloon inserted into the left ventricle and inflated to achieve an end diastolic pressure of 8–10 mmHg (1 mmHg = 133 Pa). The transducers were connected to a personal computer for online recording by using chart software. Saline (control) or 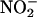 (10 and 100 μM) was infused according to the protocol described in the previous section. Global no-flow ischemia was induced for 30 min, followed by 120-min reperfusion. To determine whether any functional effects of
(10 and 100 μM) was infused according to the protocol described in the previous section. Global no-flow ischemia was induced for 30 min, followed by 120-min reperfusion. To determine whether any functional effects of 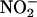 were due to its conversion to NO•, 30 μM NO• scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide (carboxy-PTIO) (32) was infused during the 30-min ischemic period. At the end of reperfusion, the hearts were cut into small pieces and incubated with p-nitroblue tetrazolium (0.5 mg·ml–1 for 20 min at 37°C). In the presence of intact dehydrogenase enzyme systems (normal myocardium), p-nitroblue tetrazolium forms a dark blue formazan, whereas areas of necrosis lack dehydrogenase activity and therefore do not stain (33, 34). Infarct size is expressed as the percentage of the dry weight of the infarcted pieces over the total weight of the heart.
were due to its conversion to NO•, 30 μM NO• scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide (carboxy-PTIO) (32) was infused during the 30-min ischemic period. At the end of reperfusion, the hearts were cut into small pieces and incubated with p-nitroblue tetrazolium (0.5 mg·ml–1 for 20 min at 37°C). In the presence of intact dehydrogenase enzyme systems (normal myocardium), p-nitroblue tetrazolium forms a dark blue formazan, whereas areas of necrosis lack dehydrogenase activity and therefore do not stain (33, 34). Infarct size is expressed as the percentage of the dry weight of the infarcted pieces over the total weight of the heart.
Chemicals. The following chemicals and drugs were used: pepstatin A, PMSF, leupeptin, aprotinin, BSA, sodium nitrite, allopurinol, L-NAME, citric acid, Triton X-100, and p-nitroblue tetrazolium (all from Sigma–Aldrich), Bradford protein assay (Bio-Rad), Na2HPO4 (BDH/AnalaR, Poole, U.K.), carboxy-PTIO (Cayman Chemical, Ann Arbor, MI), bradykinin (Bachem), and (–)BOF-4272 (a generous gift from Otsuka Pharmaceutical Factory, Tokushima, Japan).
Data Analysis and Statistics. All data are expressed as mean ± SEM, where n = the number of animals or humans. Two-way ANOVA was used for two-group comparisons, and one-way ANOVA with Bonferroni's correction was used for multiple group comparisons.
Results
NO• Is Generated from 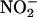 by Rat Heart Homogenates in an XOR-Dependent Reaction. To mimic the acidic conditions typical during myocardial ischemia, rat heart homogenates were incubated at pH 6, 5.5, and 5.0 under “anoxic” (bubbling with N2) and “normoxic” (bubbling with room air) conditions. Addition of physiological concentrations of sodium nitrite (
by Rat Heart Homogenates in an XOR-Dependent Reaction. To mimic the acidic conditions typical during myocardial ischemia, rat heart homogenates were incubated at pH 6, 5.5, and 5.0 under “anoxic” (bubbling with N2) and “normoxic” (bubbling with room air) conditions. Addition of physiological concentrations of sodium nitrite (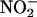 , 10–100 μM), in the absence of heart supernatant, resulted in a small but significant concentration- and pH-dependent yield of NO• gas (n = 3) (Fig. 2). However, the addition of heart supernatant in anoxic conditions led to large increases in the rate of production of NO•; for example, at pH 5.5, this production rate was 4- to 11-fold above that achieved by simple acidification alone (n = 3) (Fig. 2). Heart supernatant also significantly increased NO• production in oxygenated conditions, but by a smaller amount of 1.5- to 2.5-fold at pH 5.5, the impact of heart supernatant being 3.3- to 6.3-fold greater in anoxic than in oxygenated conditions. In addition to anoxia, NO• production in the presence of heart supernatant also depended on increasing
, 10–100 μM), in the absence of heart supernatant, resulted in a small but significant concentration- and pH-dependent yield of NO• gas (n = 3) (Fig. 2). However, the addition of heart supernatant in anoxic conditions led to large increases in the rate of production of NO•; for example, at pH 5.5, this production rate was 4- to 11-fold above that achieved by simple acidification alone (n = 3) (Fig. 2). Heart supernatant also significantly increased NO• production in oxygenated conditions, but by a smaller amount of 1.5- to 2.5-fold at pH 5.5, the impact of heart supernatant being 3.3- to 6.3-fold greater in anoxic than in oxygenated conditions. In addition to anoxia, NO• production in the presence of heart supernatant also depended on increasing 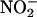 concentration and decreasing pH.
concentration and decreasing pH.
Fig. 2.

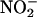 -derived NO• production from rat heart homogenates measured by using ozone chemiluminescence. (a) Typical traces of continuous sampling of NO• production [measured in parts per billion (ppb)] after addition of
-derived NO• production from rat heart homogenates measured by using ozone chemiluminescence. (a) Typical traces of continuous sampling of NO• production [measured in parts per billion (ppb)] after addition of 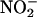 (100 μM) and then heart supernatant at pH 5 under aerobic (O2) and anaerobic conditions (N2). (b–d) Effect of
(100 μM) and then heart supernatant at pH 5 under aerobic (O2) and anaerobic conditions (N2). (b–d) Effect of 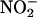 concentration, pH, and aerobic (O2) or anaerobic conditions (N2) on NO• production in the absence (–Heart) and presence (+Heart) of heart supernatant [n = 3 for each data point; *, P < 0.05: +Heart (N2) vs. +Heart (O2); #, P < 0.05: +Heart (N2) vs. –Heart (N2); †, P < 0.05: +Heart (O2) vs. –Heart (O2)].
concentration, pH, and aerobic (O2) or anaerobic conditions (N2) on NO• production in the absence (–Heart) and presence (+Heart) of heart supernatant [n = 3 for each data point; *, P < 0.05: +Heart (N2) vs. +Heart (O2); #, P < 0.05: +Heart (N2) vs. –Heart (N2); †, P < 0.05: +Heart (O2) vs. –Heart (O2)].
NO• production from 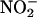 was unaffected by the NOS inhibitor L-NAME (n = 4) (Fig. 3 a–c). However, boiling of the supernatant before incubation with
was unaffected by the NOS inhibitor L-NAME (n = 4) (Fig. 3 a–c). However, boiling of the supernatant before incubation with 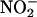 reduced the levels of measured NO• to those achieved in the absence of added tissue supernatant (i.e., simple chemical acidification) (n = 6) (Fig. 3 d–f), implicating a protein, possibly an enzyme. Because there is evidence implicating XOR in the reduction of
reduced the levels of measured NO• to those achieved in the absence of added tissue supernatant (i.e., simple chemical acidification) (n = 6) (Fig. 3 d–f), implicating a protein, possibly an enzyme. Because there is evidence implicating XOR in the reduction of 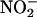 (12–15), we investigated the effect of XOR inhibitors allopurinol and (–)BOF-4272 (13, 29). Both agents inhibited NO• production from
(12–15), we investigated the effect of XOR inhibitors allopurinol and (–)BOF-4272 (13, 29). Both agents inhibited NO• production from 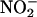 (10–100 μM) at pH 5.5 and 5 and the highest concentration of
(10–100 μM) at pH 5.5 and 5 and the highest concentration of 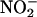 at pH 6 by an amount ≈50% of that because of boiling.
at pH 6 by an amount ≈50% of that because of boiling.
Fig. 3.

Mechanisms of 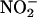 -derived NO• production from rat heart homogenates at pH 6, 5.5, and 5. The effect of L-NAME (a–c) (n = 4 for each data point), and boiled heart, allopurinol (100 μM), and (–)BOF-4272 (10 μM) (d–f) (n = 6 for each data point; *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with control; ###, P < 0.001 boiled vs. allopurinol and (–)BOF-4272).
-derived NO• production from rat heart homogenates at pH 6, 5.5, and 5. The effect of L-NAME (a–c) (n = 4 for each data point), and boiled heart, allopurinol (100 μM), and (–)BOF-4272 (10 μM) (d–f) (n = 6 for each data point; *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with control; ###, P < 0.001 boiled vs. allopurinol and (–)BOF-4272).
Human Myocardium Generates NO• from 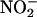 by XOR. Human myocardium also generated NO• from
by XOR. Human myocardium also generated NO• from 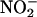 (10 and 100 μM) (i.e., 0.189 ± 0.016 and 0.891 ± 0.181 nmol per g of tissue per s, respectively), its activity being ≈42% of rat myocardium (i.e., 0.429 ± 0.022 and 2.204 ± 0.497 nmol per g of tissue per s, respectively) (n = 3) (Fig. 4a), and this activity was greatly enhanced in anoxic compared with oxygenated conditions. As with the rat tissue, whereas L-NAME had no effect (n = 3) (Fig. 4b), both boiling and pretreatment with allopurinol attenuated NO• production (n = 6) (Fig. 4c).
(10 and 100 μM) (i.e., 0.189 ± 0.016 and 0.891 ± 0.181 nmol per g of tissue per s, respectively), its activity being ≈42% of rat myocardium (i.e., 0.429 ± 0.022 and 2.204 ± 0.497 nmol per g of tissue per s, respectively) (n = 3) (Fig. 4a), and this activity was greatly enhanced in anoxic compared with oxygenated conditions. As with the rat tissue, whereas L-NAME had no effect (n = 3) (Fig. 4b), both boiling and pretreatment with allopurinol attenuated NO• production (n = 6) (Fig. 4c).
Fig. 4.

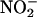 -derived NO• production from human heart homogenates at pH 5.5. (a) The effect of aerobic (O2) and anaerobic (N2) conditions and the absence/presence of heart supernatant [n = 3 for each data point; **, P < 0.01: +Heart (N2) vs. +Heart (O2); ##, P < 0.01: +Heart (N2) vs. –Heart (N2)]. (b) L-NAME (n = 3 for each data point). (c) Boiled heart and allopurinol (n = 6 for each data point; *, P < 0.05; **, P < 0.01 compared with control; and #, P < 0.05 boiled vs. allopurinol).
-derived NO• production from human heart homogenates at pH 5.5. (a) The effect of aerobic (O2) and anaerobic (N2) conditions and the absence/presence of heart supernatant [n = 3 for each data point; **, P < 0.01: +Heart (N2) vs. +Heart (O2); ##, P < 0.01: +Heart (N2) vs. –Heart (N2)]. (b) L-NAME (n = 3 for each data point). (c) Boiled heart and allopurinol (n = 6 for each data point; *, P < 0.05; **, P < 0.01 compared with control; and #, P < 0.05 boiled vs. allopurinol).
The Isolated Rat Heart Preparation Generates NO• from 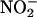 During Ischemia by XOR. Sampling of the gas space around the heart (Fig. 1) demonstrated that infusion of
During Ischemia by XOR. Sampling of the gas space around the heart (Fig. 1) demonstrated that infusion of 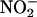 (10–1,000 μM) caused a concentration- and time-dependent increased production of NO• in ischemic conditions, which returned to preischemic levels by 30-min reperfusion (Fig. 5a). In contrast, no elevation in NO• was measured from hearts receiving normal flow throughout, i.e., no I/R insult, despite infusion with
(10–1,000 μM) caused a concentration- and time-dependent increased production of NO• in ischemic conditions, which returned to preischemic levels by 30-min reperfusion (Fig. 5a). In contrast, no elevation in NO• was measured from hearts receiving normal flow throughout, i.e., no I/R insult, despite infusion with 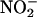 (1,000 μM; n = 4) (data not shown).
(1,000 μM; n = 4) (data not shown).
Fig. 5.

NO• production [in parts per billion (ppb)] from the isolated perfused heart by using the NO• collection chamber. (a) Concentration-dependent production of NO• during ischemia from 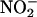 (10, 100, and 1,000 μM; n = 4, 6, and 6, respectively; **, P < 0.01; ***, P < 0.001 compared with saline; n = 4). (b) The effect of allopurinol [100 μM; n = 5; *, P < 0.05 compared with control (with
(10, 100, and 1,000 μM; n = 4, 6, and 6, respectively; **, P < 0.01; ***, P < 0.001 compared with saline; n = 4). (b) The effect of allopurinol [100 μM; n = 5; *, P < 0.05 compared with control (with 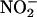 100 μM, n = 6)]. (c) Total NO• production from
100 μM, n = 6)]. (c) Total NO• production from 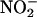 during 1-h ischemia (compared with baseline values) with Triton X-100 (n = 6), allopurinol (100 μM; n = 5), or (–)BOF-4272 (10 μM; n = 5) [*, P < 0.05; **, P < 0.01 compared with control (with
during 1-h ischemia (compared with baseline values) with Triton X-100 (n = 6), allopurinol (100 μM; n = 5), or (–)BOF-4272 (10 μM; n = 5) [*, P < 0.05; **, P < 0.01 compared with control (with 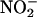 100 μM; n = 18)].
100 μM; n = 18)].
Throughout this series of experiments, L-NAME was included in the Krebs solution to eliminate NOS-derived NO• production and thereby permit study of NO• production from 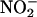 in isolation. Both allopurinol (n = 5) and (–)BOF-4272 (n = 5) suppressed
in isolation. Both allopurinol (n = 5) and (–)BOF-4272 (n = 5) suppressed 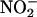 (100 μM)-derived NO• production during ischemia (Fig. 5 b and c), as did removal of the endothelium (n = 6) (Fig. 5c).
(100 μM)-derived NO• production during ischemia (Fig. 5 b and c), as did removal of the endothelium (n = 6) (Fig. 5c).
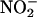 -Derived NO• Is Cardioprotective. In control untreated hearts, after a 30-min ischemic insult, left ventricular developed pressure was profoundly suppressed with recovery of only 51% of preischemic values at 120-min reperfusion (n = 8) (Fig. 6a). In contrast, whereas
-Derived NO• Is Cardioprotective. In control untreated hearts, after a 30-min ischemic insult, left ventricular developed pressure was profoundly suppressed with recovery of only 51% of preischemic values at 120-min reperfusion (n = 8) (Fig. 6a). In contrast, whereas 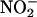 (10 and 100 μM; both n = 6) had no impact on heart function before ischemia, its infusion during ischemia resulted in a recovery of the left ventricular developed pressure (Fig. 6a). Although not damaging per se, scavenging of NO• by using carboxy-PTIO blocked the protective effect of
(10 and 100 μM; both n = 6) had no impact on heart function before ischemia, its infusion during ischemia resulted in a recovery of the left ventricular developed pressure (Fig. 6a). Although not damaging per se, scavenging of NO• by using carboxy-PTIO blocked the protective effect of 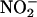 (10 μM) on left ventricular developed pressure (both n = 6) (Fig. 6a).
(10 μM) on left ventricular developed pressure (both n = 6) (Fig. 6a).
Fig. 6.

The protective effects of 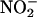 in I/R. (a) The effect of
in I/R. (a) The effect of 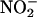 (10 or 100 μM; n = 6 each) compared with saline control (n = 8) on left ventricular developed pressure (LVDP) during I/R. The effect of
(10 or 100 μM; n = 6 each) compared with saline control (n = 8) on left ventricular developed pressure (LVDP) during I/R. The effect of 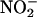 (10 μM) was reversed by the NO• scavenger carboxy-PTIO (
(10 μM) was reversed by the NO• scavenger carboxy-PTIO (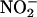 + C-PTIO, n = 6); carboxy-PTIO alone (C-PTIO, 30 μM; n = 6) had no significant effect compared with saline control. (b) The effect of
+ C-PTIO, n = 6); carboxy-PTIO alone (C-PTIO, 30 μM; n = 6) had no significant effect compared with saline control. (b) The effect of 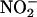 on infarct size compared with saline control in the absence and presence of carboxy-PTIO. **, P < 0.01 compared with saline control; ##, P < 0.01 compared with carboxy-PTIO +
on infarct size compared with saline control in the absence and presence of carboxy-PTIO. **, P < 0.01 compared with saline control; ##, P < 0.01 compared with carboxy-PTIO + 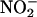 .
.
Diastolic function also was improved by 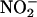 compared with vehicle control. The –dP/dt, expressed as the percentage recovery at the end of reperfusion compared with preischemic levels, was 52.7 ± 1.1% in the control group (n = 8), but in the presence of
compared with vehicle control. The –dP/dt, expressed as the percentage recovery at the end of reperfusion compared with preischemic levels, was 52.7 ± 1.1% in the control group (n = 8), but in the presence of 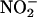 (10 and 100 μM; both n = 6) gave values of 79.6 ± 0.9% and 86.9 ± 2.3%, respectively (both P < 0.001). This protective effect of
(10 and 100 μM; both n = 6) gave values of 79.6 ± 0.9% and 86.9 ± 2.3%, respectively (both P < 0.001). This protective effect of 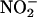 (10 μM) on –dP/dt was reversed by coinfusion of carboxy-PTIO, 39.7 ± 1.2% (n = 6).
(10 μM) on –dP/dt was reversed by coinfusion of carboxy-PTIO, 39.7 ± 1.2% (n = 6).
Likewise, 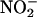 (10 and 100 μM) profoundly suppressed infarct size, an effect reversed by coinfusion with carboxy-PTIO (control, n = 8; other groups, n = 6) (Fig. 6b).
(10 and 100 μM) profoundly suppressed infarct size, an effect reversed by coinfusion with carboxy-PTIO (control, n = 8; other groups, n = 6) (Fig. 6b).
Discussion
Here we demonstrate that 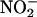 is reduced to NO• during ischemia and protects against I/R damage in the rat heart. Furthermore, by using rat and human heart homogenates and ozone chemiluminescence, we have demonstrated that the quantity of NO• generated increases with increasing
is reduced to NO• during ischemia and protects against I/R damage in the rat heart. Furthermore, by using rat and human heart homogenates and ozone chemiluminescence, we have demonstrated that the quantity of NO• generated increases with increasing 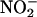 concentration, acidosis (decreasing pH), and hypoxia. This elevation in NO• production is the consequence of two distinct processes: simple chemical acidification,
concentration, acidosis (decreasing pH), and hypoxia. This elevation in NO• production is the consequence of two distinct processes: simple chemical acidification,
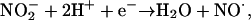 |
[1] |
and enzymatic conversion. This latter component is independent of the principal enzymes responsible for NO• production, the NOS enzymes, but is profoundly suppressed by inhibitors of XOR activity. The capacity for such NO• production was considerable with 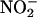 (10 μM) producing ≈108-fold more than that from rat heart homogenates (35).
(10 μM) producing ≈108-fold more than that from rat heart homogenates (35).
XOR was a likely candidate enzyme because, in its purified state, it catalyzes the reduction of 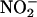 to NO• (12–15, 36). XOR is a complex molybdoflavoenzyme found in the highest concentrations in humans in breast milk, liver, and gut and in plasma from patients with inflammation (16). Its well established function is catalyzing the hydroxylation of hypoxanthine to xanthine and xanthine to urate (16). The activity of XOR is up-regulated with increasing acidosis (14) and during hypoxia (18) by processes involving phosphorylation (13, 17–19), indicating that this enzyme may play an important role in ischemic conditions. Our own studies now clearly demonstrate that, in both rat and human myocardial tissue, XOR does catalyze the reduction of
to NO• (12–15, 36). XOR is a complex molybdoflavoenzyme found in the highest concentrations in humans in breast milk, liver, and gut and in plasma from patients with inflammation (16). Its well established function is catalyzing the hydroxylation of hypoxanthine to xanthine and xanthine to urate (16). The activity of XOR is up-regulated with increasing acidosis (14) and during hypoxia (18) by processes involving phosphorylation (13, 17–19), indicating that this enzyme may play an important role in ischemic conditions. Our own studies now clearly demonstrate that, in both rat and human myocardial tissue, XOR does catalyze the reduction of 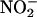 to NO• in biological systems, because inhibitors of XOR activity, both allopurinol and (–)BOF-4272 (which inhibit the molybdenum site of XOR) (29), profoundly suppress NO• production. Furthermore, the concentration of allopurinol (100 μM) used is therapeutically relevant: The upper limit of the recommended concentration of oxypurinol (the single active metabolite of allopurinol) is 100 μM for a sample collected 6–9 h after an oral allopurinol dose in humans (37). Also, BOF-4272 has been given to humans at doses similar to allopurinol (38). From the current study, XOR accounts for at least 50% of the total enzymatic activity governing
to NO• in biological systems, because inhibitors of XOR activity, both allopurinol and (–)BOF-4272 (which inhibit the molybdenum site of XOR) (29), profoundly suppress NO• production. Furthermore, the concentration of allopurinol (100 μM) used is therapeutically relevant: The upper limit of the recommended concentration of oxypurinol (the single active metabolite of allopurinol) is 100 μM for a sample collected 6–9 h after an oral allopurinol dose in humans (37). Also, BOF-4272 has been given to humans at doses similar to allopurinol (38). From the current study, XOR accounts for at least 50% of the total enzymatic activity governing 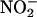 reduction in human as well as rat heart homogenates. The level of expression of XOR in human heart tissue is similar to that in the rat,§,¶ and significant activity of this enzyme in atherosclerotic human coronary arteries has been demonstrated recently (20).
reduction in human as well as rat heart homogenates. The level of expression of XOR in human heart tissue is similar to that in the rat,§,¶ and significant activity of this enzyme in atherosclerotic human coronary arteries has been demonstrated recently (20).
Our data demonstrate up-regulation of XOR activity, in terms of 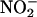 -reduction, at low O2 tension. Although NO• is produced in oxygenated conditions, consistent with recent findings (41), the rate is increased by 3.3- to 6.3-fold in anoxia (at pH 5.5). Although
-reduction, at low O2 tension. Although NO• is produced in oxygenated conditions, consistent with recent findings (41), the rate is increased by 3.3- to 6.3-fold in anoxia (at pH 5.5). Although 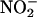 is reduced to NO• at the molybdenum site of XOR, O2, if present, would be reduced at its FAD site, resulting in competition for electrons that otherwise would be available for the reduction of
is reduced to NO• at the molybdenum site of XOR, O2, if present, would be reduced at its FAD site, resulting in competition for electrons that otherwise would be available for the reduction of 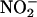 . Moreover, the superoxide
. Moreover, the superoxide 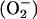 produced from the reduction of O2 will scavenge the NO• produced from the reduction of
produced from the reduction of O2 will scavenge the NO• produced from the reduction of 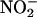 (producing peroxynitrite, ONOO–) (14, 16, 42, 43). Indeed, inhibition of
(producing peroxynitrite, ONOO–) (14, 16, 42, 43). Indeed, inhibition of 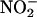 reduction by O2 has been demonstrated recently with purified XOR (41): This reaction was competitive with xanthine as the electron donor (to the molybdenum site), but with NADH as the electron donor, NO• generation rates were maintained at ≈70% of anaerobic levels (NADH occupies the FAD site, preventing O2 binding). In the current study, the aerobic NO• generation rates were only 15–30% of anaerobic levels (at pH 5.5), representing the endogenous mix of substrates present in the tissue homogenates, which will include both hypoxanthine/xanthine and NADH (and also superoxide dismutase).
reduction by O2 has been demonstrated recently with purified XOR (41): This reaction was competitive with xanthine as the electron donor (to the molybdenum site), but with NADH as the electron donor, NO• generation rates were maintained at ≈70% of anaerobic levels (NADH occupies the FAD site, preventing O2 binding). In the current study, the aerobic NO• generation rates were only 15–30% of anaerobic levels (at pH 5.5), representing the endogenous mix of substrates present in the tissue homogenates, which will include both hypoxanthine/xanthine and NADH (and also superoxide dismutase).
The method we developed, collecting NO• gas from the isolated perfused heart and measuring its concentration by using ozone chemiluminescence, confirmed that the ischemic heart could support the conversion of 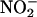 to authentic NO•. In addition, this approach enabled successive measurements of NO• during ischemia, as well as on reperfusion, demonstrating a progressive and concentration-dependent increase in NO• throughout ischemia, with a subsequent drop in NO• levels back to baseline by 30-min reperfusion. No changes in NO• production were detected in the control group, subjected to normal flow throughout without a period of ischemia, despite being infused with
to authentic NO•. In addition, this approach enabled successive measurements of NO• during ischemia, as well as on reperfusion, demonstrating a progressive and concentration-dependent increase in NO• throughout ischemia, with a subsequent drop in NO• levels back to baseline by 30-min reperfusion. No changes in NO• production were detected in the control group, subjected to normal flow throughout without a period of ischemia, despite being infused with 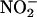 (1,000 μM), indicating that this production of NO• was specific to ischemia. Furthermore, as with the homogenate studies, both allopurinol and (–)BOF-4272 inhibited this NO• production, indicating that the majority of the
(1,000 μM), indicating that this production of NO• was specific to ischemia. Furthermore, as with the homogenate studies, both allopurinol and (–)BOF-4272 inhibited this NO• production, indicating that the majority of the 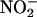 -derived NO• depended on XOR activity. It is likely that the XOR involved in this response is located on the endothelium, because endothelial removal significantly reduced NO• levels. In support of such localization is histochemical evidence locating XOR to the endothelium of blood vessels of the human heart (44).
-derived NO• depended on XOR activity. It is likely that the XOR involved in this response is located on the endothelium, because endothelial removal significantly reduced NO• levels. In support of such localization is histochemical evidence locating XOR to the endothelium of blood vessels of the human heart (44).
In the present study, 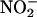 infusion attenuated the deleterious effects of I/R injury, improving cardiac function and reducing necrosis. This finding is a clear demonstration that
infusion attenuated the deleterious effects of I/R injury, improving cardiac function and reducing necrosis. This finding is a clear demonstration that 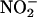 is protective in I/R. Although acidified
is protective in I/R. Although acidified 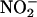 , as a source of authentic NO•, reduces necrosis when applied to the feline heart in vivo during a regional I/R insult (27, 28), the release of NO• from acidified
, as a source of authentic NO•, reduces necrosis when applied to the feline heart in vivo during a regional I/R insult (27, 28), the release of NO• from acidified 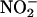 is instantaneous and generalized. In contrast, in the current study application of (unacidified)
is instantaneous and generalized. In contrast, in the current study application of (unacidified) 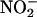 releases NO• specifically during ischemia and does not require the use of a highly acidified medium (pH 2) (27). Use of carboxy-PTIO provided further evidence indicating that it was NO•, rather than
releases NO• specifically during ischemia and does not require the use of a highly acidified medium (pH 2) (27). Use of carboxy-PTIO provided further evidence indicating that it was NO•, rather than 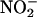 per se, in that mediated the beneficial effects in this study. Furthermore carboxy-PTIO was infused during ischemia only, demonstrating that the benefit from
per se, in that mediated the beneficial effects in this study. Furthermore carboxy-PTIO was infused during ischemia only, demonstrating that the benefit from 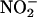 was specifically because of its conversion to NO• during ischemia.
was specifically because of its conversion to NO• during ischemia.
Although the NO• production from 10 μM 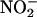 seemed to be much less than that from 100 μM, the degree of protection did not differ. This observation suggests that a maximal level of protection had been reached with the apparently small amounts of NO• produced by
seemed to be much less than that from 100 μM, the degree of protection did not differ. This observation suggests that a maximal level of protection had been reached with the apparently small amounts of NO• produced by 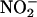 (10 μM). However, these findings also could indicate a limitation of the method of collection, and it is possible that
(10 μM). However, these findings also could indicate a limitation of the method of collection, and it is possible that 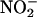 (10 μM) actually produced considerably more NO than we could detect. It is almost certain that at least some of the NO• produced by the heart will have been scavenged by molecules such as myoglobin (45), and, therefore, our measurements may reflect not the actual total NO• generated but, rather, the approximate levels produced.
(10 μM) actually produced considerably more NO than we could detect. It is almost certain that at least some of the NO• produced by the heart will have been scavenged by molecules such as myoglobin (45), and, therefore, our measurements may reflect not the actual total NO• generated but, rather, the approximate levels produced.
Although there is considerable support for the concept that NO• is cardioprotective (21), the exact mechanisms of this effect are unclear. Coronary vasodilatation, and therefore improvement of blood flow, to ischemic areas may be one potential mechanism. However, whether 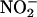 at physiological concentrations vasodilates the arterial circulation is controversial, with evidence supporting (5) and against (2) such an action. Although 100 μM
at physiological concentrations vasodilates the arterial circulation is controversial, with evidence supporting (5) and against (2) such an action. Although 100 μM 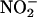 did vasodilate the coronary vasculature before ischemia (≈15% reduction in coronary perfusion pressure compared with baseline, P < 0.001), 10 μM
did vasodilate the coronary vasculature before ischemia (≈15% reduction in coronary perfusion pressure compared with baseline, P < 0.001), 10 μM 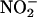 did not, in line with previous studies with aortic rings (5, 46); however, both concentrations resulted in a similar degree of protection. Despite the lack of any vasodilatory activity before ischemia in this study,
did not, in line with previous studies with aortic rings (5, 46); however, both concentrations resulted in a similar degree of protection. Despite the lack of any vasodilatory activity before ischemia in this study, 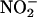 (10 μM) significantly lowered coronary perfusion pressure on reperfusion compared with control (data not shown). This result suggests that functionally relevant conversion of low concentrations of
(10 μM) significantly lowered coronary perfusion pressure on reperfusion compared with control (data not shown). This result suggests that functionally relevant conversion of low concentrations of 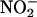 to NO• can occur, but only as a result of a period of ischemia. NO• also may have direct beneficial effects on cardiomyocytes. The protective effects of NO• donors are associated with elevations in cGMP and activation of cGMP-dependent protein kinases; however, the final effector of this signal transduction pathway is uncertain (22). In addition, it has been suggested that NO• may provide protection by modifying mitochondrial respiration by means of interaction with specific complexes of the respiratory chain, resulting in alterations in cellular ATP concentration (47, 48).
to NO• can occur, but only as a result of a period of ischemia. NO• also may have direct beneficial effects on cardiomyocytes. The protective effects of NO• donors are associated with elevations in cGMP and activation of cGMP-dependent protein kinases; however, the final effector of this signal transduction pathway is uncertain (22). In addition, it has been suggested that NO• may provide protection by modifying mitochondrial respiration by means of interaction with specific complexes of the respiratory chain, resulting in alterations in cellular ATP concentration (47, 48).
XOR is generally considered to be damaging in I/R through its generation of reactive oxygen species, including 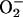 (16, 39, 49). The findings of this study suggest that if XOR is presented with
(16, 39, 49). The findings of this study suggest that if XOR is presented with 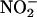 as an alternative substrate, the resultant effects of its activity may be protective, by means of its production of NO•, rather than damaging. In addition, a secondary mechanism for protection might relate to the possibility that
as an alternative substrate, the resultant effects of its activity may be protective, by means of its production of NO•, rather than damaging. In addition, a secondary mechanism for protection might relate to the possibility that 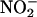 would compete for electrons necessary for the reduction of O (42), thus inhibiting
would compete for electrons necessary for the reduction of O (42), thus inhibiting 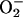 production. Recent evidence demonstrates that, under anaerobic conditions, purified XOR can further reduce NO• (using millimolar concentrations of NO• donor) to the nitroxyl anion (NO– or HNO) and that this inactivates XOR (40). However it is unlikely that the nitroxyl anion accounts for the activity of
production. Recent evidence demonstrates that, under anaerobic conditions, purified XOR can further reduce NO• (using millimolar concentrations of NO• donor) to the nitroxyl anion (NO– or HNO) and that this inactivates XOR (40). However it is unlikely that the nitroxyl anion accounts for the activity of 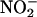 in the current study because, at least in the heart, nitroxyl increases the deleterious effects of myocardial I/R injury (24).
in the current study because, at least in the heart, nitroxyl increases the deleterious effects of myocardial I/R injury (24).
Thus, in conclusion, we have shown that during myocardial ischemia, the hypoxic and acidic environment generated by this insult catalyzes, by means of XOR, the reduction of 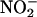 to cardioprotective NO•. Moreover, this conversion of
to cardioprotective NO•. Moreover, this conversion of 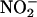 to NO• occurs selectively within ischemic regions and, as such, may provide a previously uncharacterized means of administering NO• at sites of ischemia while avoiding the systemic effects inherent with NO• and NO• donors.
to NO• occurs selectively within ischemic regions and, as such, may provide a previously uncharacterized means of administering NO• at sites of ischemia while avoiding the systemic effects inherent with NO• and NO• donors.
Acknowledgments
We thank the Otsuka Pharmaceutical Factory for the generous gift of (–)BOF-4272. A.W. was supported by The Research Advisory Board of St. Bartholomew's, The Royal London Charitable Foundation, and Servier.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: (–)BOF-4272, levorotatory stereoisomer of sodium 8-(3-methoxy-4-phenylsulfinylphenyl)pyrazolo[1,5-α]-1,3,5-triazine-4-olate monohydrate; carboxy-PTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide; I/R, ischemia–reperfusion; L-NAME, Nω-nitro-l-arginine methyl ester; NOS, NO• synthase; XOR, xanthine oxidoreductase.
Footnotes
Abadeh, S., Case, P. C. & Harrison, R. (1992) Biochem. Soc. Trans. 20, 346S (abstr.).
Abadeh, S., Case, P. C. & Harrison, R. (1993) Biochem. Soc. Trans. 21, 99S (abstr.).
References
- 1.Bredt, D. S., Hwang, P. M., Glatt, C. E., Lowenstein, C., Reed, R. R. & Snyder, S. H. (1991) Nature 351, 714–718. [DOI] [PubMed] [Google Scholar]
- 2.Lauer, T., Preik, M., Rassaf, T., Strauer, B. E., Deussen, A., Feelisch, M. & Kelm, M. (2001) Proc. Natl. Acad. Sci. USA 98, 12814–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benjamin, N., O'Driscoll, F., Dougall, H., Duncan, C., Smith, L., Golden, M. & McKenzie, H. (1994) Nature 368, 502 (lett.). [DOI] [PubMed] [Google Scholar]
- 4.Bjorne, H. H., Petersson, J., Phillipson, M., Weitzberg, E., Holm, L. & Lundberg, J. O. (2004) J. Clin. Invest. 113, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., Yang, B. K., Waclawiw, M. A., Zalos, G., Xu, X. et al. (2003) Nat. Med. 9, 1498–1505. [DOI] [PubMed] [Google Scholar]
- 6.Zweier, J. L., Wang, P., Samouilov, A. & Kuppusamy, P. (1995) Nat. Med. 1, 804–809. [DOI] [PubMed] [Google Scholar]
- 7.Lundberg, J. O., Weitzberg, E., Lundberg, J. M. & Alving, K. (1994) Gut 35, 1543–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gladwin, M. T., Shelhamer, J. H., Schechter, A. N., Pease-Fye, M. E., Waclawiw, M. A., Panza, J. A., Ognibene, F. P. & Cannon, R. O., III (2000) Proc. Natl. Acad. Sci. USA 97, 11482–11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez, J., Maloney, R. E., Rassaf, T., Bryan, N. S. & Feelisch, M. (2003) Proc. Natl. Acad. Sci. USA 100, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari, R., Cargnoni, A., Bernocchi, P., Pasini, E., Curello, S., Ceconi, C. & Ruigrok, T. J. (1996) Circulation 94, 2587–2596. [DOI] [PubMed] [Google Scholar]
- 11.Gabel, S. A., Cross, H. R., London, R. E., Steenbergen, C. & Murphy, E. (1997) Am. J. Physiol. 273, H2257–H2262. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, Z., Naughton, D., Winyard, P. G., Benjamin, N., Blake, D. R. & Symons, M. C. (1998) Biochem. Biophys. Res. Commun. 249, 767–772. [DOI] [PubMed] [Google Scholar]
- 13.Millar, T. M., Stevens, C. R., Benjamin, N., Eisenthal, R., Harrison, R. & Blake, D. R. (1998) FEBS Lett. 427, 225–228. [DOI] [PubMed] [Google Scholar]
- 14.Godber, B. L., Doel, J. J., Sapkota, G. P., Blake, D. R., Stevens, C. R., Eisenthal, R. & Harrison, R. (2000) J. Biol. Chem. 275, 7757–7763. [DOI] [PubMed] [Google Scholar]
- 15.Li, H., Samouilov, A., Liu, X. & Zweier, J. L. (2001) J. Biol. Chem. 276, 24482–24489. [DOI] [PubMed] [Google Scholar]
- 16.Harrison, R. (2002) Free Radical Biol. Med. 33, 774–797. [DOI] [PubMed] [Google Scholar]
- 17.Hassoun, P. M., Yu, F. S. & Shedd, A. L. (1994) Am. J. Physiol. 266, L163–L171. [DOI] [PubMed] [Google Scholar]
- 18.Kayyali, U. S., Donaldson, C., Huang, H., Abdelnour, R. & Hassoun, P. M. (2001) J. Biol. Chem. 276, 14359–14365. [DOI] [PubMed] [Google Scholar]
- 19.Poss, W. B., Huecksteadt, T. P., Panus, P. C., Freeman, B. A. & Hoidal, J. R. (1996) Am. J. Physiol. 270, L941–L946. [DOI] [PubMed] [Google Scholar]
- 20.Spiekermann, S., Landmesser, U., Dikalov, S., Bredt, M., Gamez, G., Tatge, H., Reepschlager, N., Hornig, B., Drexler, H. & Harrison, D. G. (2003) Circulation 107, 1383–1389. [DOI] [PubMed] [Google Scholar]
- 21.Bolli, R. (2001) J. Mol. Cell. Cardiol. 33, 1897–1918. [DOI] [PubMed] [Google Scholar]
- 22.Du Toit, E. F., Meiring, J. & Opie, L. H. (2001) J. Cardiovasc. Pharmacol. 38, 529–538. [DOI] [PubMed] [Google Scholar]
- 23.Brunner, F., Leonhard, B., Kukovetz, W. R. & Mayer, B. (1997) Cardiovasc. Res. 36, 60–66. [DOI] [PubMed] [Google Scholar]
- 24.Ma, X. L., Gao, F., Liu, G. L., Lopez, B. L., Christopher, T. A., Fukuto, J. M., Wink, D. A. & Feelisch, M. (1999) Proc. Natl. Acad. Sci. USA 96, 14617–14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, S. P., Girod, W. G., Palazzo, A. J., Granger, D. N., Grisham, M. B., Jourd'Heuil, D., Huang, P. L. & Lefer, D. J. (1999) Am. J. Physiol. 276, H1567–H1573. [DOI] [PubMed] [Google Scholar]
- 26.Sumeray, M. S., Rees, D. D. & Yellon, D. M. (2000) J. Mol. Cell. Cardiol. 32, 35–42. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, G., III, Tsao, P. S. & Lefer, A. M. (1991) Crit. Care Med. 19, 244–252. [DOI] [PubMed] [Google Scholar]
- 28.Johnson, G., III, Tsao, P. S., Mulloy, D. & Lefer, A. M. (1990) J. Pharmacol. Exp. Ther. 252, 35–41. [PubMed] [Google Scholar]
- 29.Okamoto, K. & Nishino, T. (1995) J. Biol. Chem. 270, 7816–7821. [DOI] [PubMed] [Google Scholar]
- 30.McLean, P. G., Perretti, M. & Ahluwalia, A. (1999) Br. J. Pharmacol. 128, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rees, D. D., Palmer, R. M., Schulz, R., Hodson, H. F. & Moncada, S. (1990) Br. J. Pharmacol. 101, 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chauhan, S., Rahman, A., Nilsson, H., Clapp, L., MacAllister, R. & Ahluwalia, A. (2003) Cardiovasc. Res. 57, 207–216. [DOI] [PubMed] [Google Scholar]
- 33.Nachlas, M. M. & Shnitka, T. K. (1963) Am. J. Pathol. 42, 379–405. [PMC free article] [PubMed] [Google Scholar]
- 34.Zacharowski, K., Olbrich, A., Otto, M., Hafner, G. & Thiemermann, C. (1999) Br. J. Pharmacol. 126, 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giraldez, R. R., Panda, A., Xia, Y., Sanders, S. P. & Zweier, J. L. (1997) J. Biol. Chem. 272, 21420–21426. [DOI] [PubMed] [Google Scholar]
- 36.Alikulov, Z. A., L'vov, N. P. & Kretovich, V. L. (1980) Biokhimiya 45, 1714–1718. [PubMed] [Google Scholar]
- 37.Saji, M. (1996) Nippon Jinzo Gakkai Shi 38, 640–650. [PubMed] [Google Scholar]
- 38.Uematsu, T. & Nakashima, M. (1994) J. Pharmacol. Exp. Ther. 270, 453–459. [PubMed] [Google Scholar]
- 39.Granger, D. N., Hollwarth, M. E. & Parks, D. A. (1986) Acta Physiol. Scand. Suppl. 548, 47–63. [PubMed] [Google Scholar]
- 40.Saleem, M. & Ohshima, H. (2004) Biochem. Biophys. Res. Commun. 315, 455–462. [DOI] [PubMed] [Google Scholar]
- 41.Li, H., Samouilov, A., Liu, X. & Zweier, J. L. (2004) J. Biol. Chem. 279, 16939–16946. [DOI] [PubMed] [Google Scholar]
- 42.Godber, B. L., Doel, J. J., Durgan, J., Eisenthal, R. & Harrison, R. (2000) FEBS Lett. 475, 93–96. [DOI] [PubMed] [Google Scholar]
- 43.Millar, T. M. (2004) FEBS Lett. 562, 129–133. [DOI] [PubMed] [Google Scholar]
- 44.Moriwaki, Y., Yamamoto, T., Suda, M., Nasako, Y., Takahashi, S., Agbedana, O. E., Hada, T. & Higashino, K. (1993) Biochim. Biophys. Acta 1164, 327–330. [DOI] [PubMed] [Google Scholar]
- 45.Brunori, M. (2001) Trends Biochem. Sci. 26, 209–210. [DOI] [PubMed] [Google Scholar]
- 46.Modin, A., Bjorne, H., Herulf, M., Alving, K., Weitzberg, E. & Lundberg, J. O. (2001) Acta Physiol. Scand. 171, 9–16. [DOI] [PubMed] [Google Scholar]
- 47.Beltran, B., Mathur, A., Duchen, M. R., Erusalimsky, J. D. & Moncada, S. (2000) Proc. Natl. Acad. Sci. USA 97, 14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moncada, S. & Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell. Biol. 3, 214–220. [DOI] [PubMed] [Google Scholar]
- 49.Berry, C. E. & Hare, J. M. (2004) J. Physiol. 555, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]