

Abstract
We report a general method for syntheses of [18F]CHF2–arenes from [18F]fluoride for radiopharmaceutical discovery. The method is practical, operationally simple, tolerates a wide scope of functional groups, enables labeling of a variety of arenes and heteroarenes with radiochemical yields (RCYs, not decay corrected) from 10% to 60%. The 18F-fluorination precursors are readily prepared from aryl chlorides, bromides, iodides and triflates, respectively. Seven 18F-difluoromethylarene drug analogs and radiopharmaceuticals including claritin, fluoxetine (prozac), and [18F]DAA1106 were synthesized to show the potential of the method for applications in PET radiopharmaceutical design.
Keywords: 18F-difluoromethylarene, aryl (pseudo) halides, fluorine-18, positron emission tomography, radiopharmaceuticals
Fluorine-18 (18F) is the most commonly used radionuclide for molecular imaging by positron emission tomography (PET).1 Over the past five years, several modern fluorination reactions have been developed with 18F, many of which are achieved with high specific activity (SA) [18F]fluoride.2 Development of modern 18F-multi-fluoromethylation reactions, for example to incorporate 18F into trifluoromethyl groups, are now also addressed, due to the prospects of having more labeled chemotypes available for imaging.3 A challenge in the preparation of 18F-labeled compounds that contain more than one fluorine atom, especially when located on the same carbon atom, is the probability of isotope exchange that can substantially lower the specific activity. Here we describe a general method for difluoromethyl [18F]CHF2–arenes from [18F]fluoride. The method is practical and general over a wide variety of arenes and heteroarenes. On a fundamental level, the crucial but unusual C-F bond formation occurs by nucleophilic substitution on a fully substituted carbon.
The most extensive work to date on multi-fluorinated 18F-labeled molecules has been directed at the synthesis of 18F-trifluoromethyl groups:3 Gouverneur and co-workers have established a practical method based on [18F]CuCF3 species, with large substrate scope.3f A year later, three other groups independently reported different methods to prepare [18F]CuCF3, which was also explored for [18F]trifluoromethylation of aryl boronic acids.3g-3i Groves and co-workers have reported a direct C–H to C–18F transformation based on Mn–salen-catalyzed benzylic fluorination with [18F]fluoride,2j which was also extended to 18F-trifluoromethylarenes.3j [18F]CHF2 arenes such as [18F]4-(difluoromethyl)-1,1'-biphenyl, can be synthesized through decarboxylative 18F-fluorination with [18F]Selectfluor generated from [18F]F2 or AgI-mediated halogen-exchange with [18F]fluoride ion.3e,k Gouverneur's group improved the substrate scope of AgI-mediated halogen-exchange; but the CHFCl–arene starting materials still require several synthetic steps for preparation.3m To date, no method has been reported for the 18F-labeling of difluoromethylarenes with high specific activity. Here, we describe progress toward the goal of high-specific-activity difluoromethylation in a practical and general reaction.
To overcome the challenge of C–F bond formation to achieve 18F-difluoromethylation from [18F]fluoride, we have designed an 18F-difluoromethylation reaction that features a benzoyl auxiliary, which controls the chemoselectivity of C–H functionalization; [18F]fluoride introduction is observed only at the desired benzylic position. The auxiliary can be installed from aryl (pseudo) halides, and quickly cleaved in situ after radiofluorination to generate 18F-difluoromethylarenes (Scheme 1).
Scheme 1.

Auxiliary Facilitated [18F]Difluoromethylarene Formation
The labeling procedure entails (1) C–H bromination to α-Br-α-F-acetophenone; (2) in situ halogen-exchange with [18F]fluoride; and (3) benzophenone cleavage.4 Treatment of aryl acetophenones 2 with N-bromophthalimide and [18F]fluoride in the presence of tetraethylammonium bicarbonate (TEAB) in chlorobenzene and acetonitrile at 100 °C,5 followed by addition of an aqueous KOH solution after 5 min, gave [18F]CHF2–arenes 3 (Table 1). The procedure was successfully applied to the 18F-difluoromethylation of a wide range of substrates, including both electron-deficient and electron-rich aromatics. The radiochemical yields (RCYs, non-decay corrected; relative to starting [18F]fluoride) range from 10% to 60%.6 Halide, alcohol, ether, aldehyde, ketone, ester, amide, carbamate, cyanide, nitrate, cyclopropane and alkyne functional groups are tolerated. The method is also compatible with heterocycles (e.g. 3m–p). Moisture is tolerated; water-saturated chlorobenzene gave the same radiochemical yield within error ([18F]3m, 47% vs. 49% RCY). For aniline derivative 2g, no expected product was generated under the standard condition: We surmised that the basic nitrogen was oxidized by N-bromophthalimide instead of basic nitrogen was oxidized by N-bromophthalimide instead of the α-C–H bond of acetophenone. Given that bromo-arylaminium can be reduced to aniline, after 18F-labeling of 2g, addition of sodium thiosulfate to the reaction mixture afforded [18F]CHF2-aniline ([18F]3g) in 16% RCY.
Table 1.
18F-Difluoromethylarene Formation of Aryl-acetophenones.[a]

|
To demonstrate the potential utility of the 18F-labeling procedure, we examined a variety of well-known biologically active molecule analogues, including fenofibrate (cholesterol scavenger), fluoxetine (selective serotonin reuptake inhibitor), DAA-1106 (translocator protein 18 kDa radiotracer), SC-58125 (COX2 inhibitor) and analepticon (respiratory stimulant). The labeling method was selective on a claritin derivative, which possesses five benzylic C–H bonds and four allylic C–H bonds. To achieve 18F-labeling of tertiary amines, a stepwise method was developed, with bromination carried out separately, prior to fluorination (Scheme 2). Specifically, the enolate of precursor 2, generated by deprotonation with LiHMDS at low temperature, was trapped by NBS. Without separation, the crude brominated precursor was subjected to radiofluorination, which afforded the corresponding 18F-labeled amines, including [18F]CHF2–hydroquinidine ester in 26% RCY.
Scheme 2.

Step-wise 18F-Difluoromethylarene Formation of Tertiary Amines. Reaction conditions: Arene 2 (10 μmol), LiHMDS (1.0 M in THF, 1.3 equiv), THF (0.25 mL), −78 °C, 45 min. then NBS (1.0 equiv), THF (0.1 mL), −78 °C, 45 min. (b) TEAB (3.5 mg), chlorobenzene (0.2 mL), MeCN (20 μL), 100 °C, 5 min, then KOH aqueous solution (45 w%, 40 μL), 100 °C, 15 min.
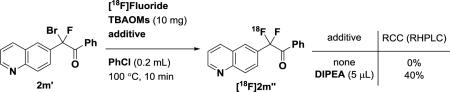
Obtaining high specific activity (SA) is an ongoing challenge for 18F-labeling of multi-fluoromethyl-compounds due to undesired isotope exchange reactions,3e–3l with recent progress reported for 18F-trifluoromethylation.3g The reactions conditions provided in Table 1 are useful to readily access 18F-labeled difluoromethyl arenes, but only with low specific activity (e.g. 0.49 mCi/μmol for [18F]3m). To achieve high specific activity, we attempted to identify the undesired pathway that led to the formation of non-radioactive compounds 3. Control experiments showed that 19F/18F isotope exchange did not occur for 2r, 2r”, and 3r in the presence of [18F]fluoride/TEAB at 100 °C (Scheme 3a). In the absence of additional fluoride, treatment of α-phenyl-α-Br-α-F-acetophenone (2r’), with TEAB gave 2r” in 9% yield, determined by NMR spectroscopy. The results demonstrated that decomposition of 2r’ by TEAB released 19F-fluoride ion, which fluorinated 2r’ to form the non-radioactive byproduct 2r”. However, use of a weaker base, tetrabutylammonium methanesulfonate (TBAOMs), did not decompose α-Br-α-F-acetophenone 2m’, but 18F-fluorination was not observed when TEAB was replaced by TBAOMs. To increase the nucleophilicity of [18F], organic bases such as N,N-diisopropylethylamine (DIPEA) were explored to promote 18F-fluorination.7 With 0.5 μmol of precursor 2m’ and a reaction temperature of 80 °C, we determined a specific activity for [18F]3m at 81 mCi/μmol (3.0 GBq/μmol), with only 39 mCi of starting [18F]fluoride after bombardment (Scheme 3b) for a reaction yielding over 3 mCi of product, which could be sufficient for preclinical PET imaging studies. Given the anticipated increase of specific activity with larger amounts of activity in the mCi range (Scheme 3b), we propose that, with appropriate automation, specific activity higher than 81 mCi/μmol (3.0 GBq/μmol) will be achievable.
Scheme 3.

a. Control Experiments. b. Improvement of Specific Activity.
Starting material preparation from aryl halides and pseudo halides is robust and practical: Prior to our work, palladium-catalyzed cross-coupling of aryl halide with α-fluoroacetophenone was not a general reaction, and yields were generally low (e.g. 17%).8 We extended the transformation to be general, from aryl chlorides, bromides, iodides and triflates, including sterically hindered arenes (full table of Pd-catalyzed fluoroacetophenonation of arenes is shown in the supporting information). To highlight the convenience of precursor preparation, we show six examples in Table 2.
Table 2.
Selected Examples of Pd-Catalyzed Fluoro-acetophenonation of Arenes.[a]

|
In conclusion, we have reported a general method for 18F-labeling of difluoromethylarenes vial aryl (pseudo) halides with broad functional group compatibility. The strategy outlined herein will augment the pool of radiochemical methodologies and enable 18F-labeled difluoromethylarenes to be considered in PET radiotracer design.
Supplementary Material
Acknowledgements
We thank the NIH for funding (GM088237), Erica M. D'Amato, Constanze N. Neumann, Anthony R. Mazzotti, and Ziyang Zhang for helpful discussions. S.H.L is a recipient of an NIH career development award (NIH/NIDA K01DA038000).
Footnotes
Supporting information for this article is given via a link at the end of the document
Contributor Information
Hang Shi, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA, 02138, United States; Division of Nuclear Medicine and Molecular Imaging & Gordon, Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, MA, 02114, United States.
Augustin Braun, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA, 02138, United States.
Lu Wang, Division of Nuclear Medicine and Molecular Imaging & Gordon, Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, MA, 02114, United States.
Prof. Dr. Steven H. Liang, Division of Nuclear Medicine and Molecular Imaging & Gordon, Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, MA, 02114, United States; Department of Radiology, Harvard Medical School, 55 Fruit Street, Boston, MA, 02114, United States.
Prof. Dr. Neil Vasdev, Division of Nuclear Medicine and Molecular Imaging & Gordon, Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, MA, 02114, United States; Department of Radiology, Harvard Medical School, 55 Fruit Street, Boston, MA, 02114, United States.
Prof. Dr. Tobias Ritter, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA, 02138, United States; Division of Nuclear Medicine and Molecular Imaging & Gordon, Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, MA, 02114, United States; Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany.
References
- 1.a Phelps ME. Proc. Natl. Acad. Sci. U.S.A. 2000;97:9226. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matthews PM, Rahiner EA, Passchier J, Gunn RN. Br. J. Clin. Pharmacol. 2012;73:175. doi: 10.1111/j.1365-2125.2011.04085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Miller PW, Long NJ, Vilar R, Gee AD. Angew. Chem. Int. Ed. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120:9136. [Google Scholar]; d Ametamey SM, Honer M, Schubiger PA. Chem. Rev. 2008;108:1501. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]; e Fowler JS, Wolf AP. Acc. Chem. Res. 1997;30:181. [Google Scholar]; f Rohren EM, Turkington TG, Coleman RE. Radiology. 2004;231:305. doi: 10.1148/radiol.2312021185. [DOI] [PubMed] [Google Scholar]; g Cai L, Lu S, Pike VW. Eur. J. Org. Chem. 2008:2853. [Google Scholar]; h Preshlock S, Tredwell M, Gouverneur V. Chem. Rev. 2016;116:719. doi: 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see: Tredwell M, Gouverneur V. Angew. Chem. Int. Ed. 2012;51:11426. doi: 10.1002/anie.201204687.Angew. Chem. 2012;124:11590.Brooks AF, Topczewski JJ, Ichiishi N, Sanford MS, Scott PJH. Chem. Sci. 2014;5:4545. doi: 10.1039/C4SC02099E.Campbell MG, Ritter T. Chem. Rev. 2015;115:612. doi: 10.1021/cr500366b.Le Bars D. J. Fluorine Chem. 2006;127:1488. For selective examples, see: Hollingworth C, Hazari A, Hopkinson MN, Tredwell M, Benedetto E, Huiban M, Gee AD, Brown JM, Gouverneur V. Angew. Chem. Int. Ed. 2011;50:2613. doi: 10.1002/anie.201007307.Angew. Chem. 2011;123:2661.Gao Z, Lim YH, Tredwell M, Li L, Verhoog S, Hopkinson M, Kaluza W, Collier TL, Passchier J, Huiban M, Gouverneur V. Angew. Chem. Int. Ed. 2012;51:6733. doi: 10.1002/anie.201201502.Angew. Chem. 2012;124:6837.Lee E, Hooker JM, Ritter T. J. Am. Chem. Soc. 2012;134:17456. doi: 10.1021/ja3084797.Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639. doi: 10.1126/science.1212625.Graham TJA, Lambert RF, Ploessl K, Kung HF, Doyle AG. J. Am. Chem. Soc. 2014;136:5291. doi: 10.1021/ja5025645.Huang X, Liu W, Ren H, Neelamegam R, Hooker JM, Groves JT. J. Am. Chem. Soc. 2014;136:6842. doi: 10.1021/ja5039819.Ichiishi N, Brooks AF, Topczewski JJ, Rodnick ME, Sanford MS, Scott PJH. Org. Lett. 2014;16:3224. doi: 10.1021/ol501243g.Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nat. Commun. 2014;5:4365. doi: 10.1038/ncomms5365.Buckingham F, Kirjavainen AK, Forsback S, Krzyczmonik A, Keller T, Newington IM, Glaser M, Luthra SK, Solin O, Gouverneur V. Angew. Chem. Int. Ed. 2015;54:13366. doi: 10.1002/anie.201506035.Angew. Chem. 2015;127:13564.Mossine AV, Brooks AF, Makaravage KJ, Miller JM, Ichiishi N, Sanford MS, Scott PJH. Org. Lett. 2015;17:5780. doi: 10.1021/acs.orglett.5b02875.Huang X, Liu W, Hooker JM, Groves JT. Angew. Chem. Int. Ed. 2015;54:5241. doi: 10.1002/anie.201500399.Angew. Chem. 2015;127:5330.Rotstein BH, Wang L, Liu RY, Patteson J, Kwan EE, Vasdev N, Liang SH. Chem. Sci. 2016 doi: 10.1039/c6sc00197a. DOI: 10.1039/C1036SC00197A.
- 3.a Kilbourn MR, Pavia MR, Gregor VE. Appl. Radiat. Isot. 1990;41:823. doi: 10.1016/0883-2889(90)90059-p. [DOI] [PubMed] [Google Scholar]; b Prabhakaran J, Underwood MD, Parsey RV, Arango V, Majo VJ, Simpson NR, Heertum RV, Mann JJ, Kumar JSD. Bioorg. Med. Chem. 2007;15:1802. doi: 10.1016/j.bmc.2006.11.033. [DOI] [PubMed] [Google Scholar]; c Angelini G, Speranza M, Shiue C-Y, Wolf AP. J. Chem. Soc. Chem. Commun. 1986:924. [Google Scholar]; d Angelini G, Speranza M, Wolf AP, Shiue C-Y, Label J. Compd. Radiopharm. 1990;28:1441. [Google Scholar]; e Mizuta S, Stenhagen ISR, O'Duill M, Wolstenhulme J, Kirjavainen A, Forsback S, Tredwell M, Sandford G, Moore PR, Huiban M, Luthra S, Passchier J, Solin O, Gouverneur V. Org. Lett. 2013;15:2648. doi: 10.1021/ol4009377. [DOI] [PubMed] [Google Scholar]; f Huiban M, Tredwell M, Mizuta S, Wan Z, Zhang X, Collier TL, Gouverneur V, Passchier J. Nat. Chem. 2013;5:941. doi: 10.1038/nchem.1756. [DOI] [PubMed] [Google Scholar]; g van der Born D, Sewing C, Herscheid JDM, Windhorst AD, Orru RVA, Vugts DJ. Angew. Chem. Int. Ed. 2014;53:11046. doi: 10.1002/anie.201406221. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126:11226. [Google Scholar]; h Ruhl T, Rafique W, Lien VT, Riss PJ. Chem. Commun. 2014;50:6056. doi: 10.1039/c4cc01641f. [DOI] [PubMed] [Google Scholar]; i Ivashkin P, Lemonnier G, Cousin J, Grégoire V, Labar D, Jubault P, Pannecoucke X. Chem.–Eur. J. 2014;20:9514. doi: 10.1002/chem.201403630. [DOI] [PubMed] [Google Scholar]; j Carroll L, Evans HL, Spivey AC, Aboagy EO. Chem. Commun. 2015;51:8439. doi: 10.1039/c4cc05762g. [DOI] [PubMed] [Google Scholar]; k Khotavivattana T, Verhoog S, Tredwell M, Pfeifer L, Calderwood S, Wheelhouse K, Collier TE, Gouverneur V. Angew. Chem., Int. Ed. 2015;54:9991. doi: 10.1002/anie.201504665. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015;127:10129. [Google Scholar]; l Zheng J, Wang L, Lin J-H, Xiao J-C, Liang SH. Angew. Chem., Int. Ed. 2015;54:13236. doi: 10.1002/anie.201505446. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015;127:13434. [Google Scholar]; m Verhoog S, Pfeifer L, Khotavivattana T, Calderwood S, Collier TL, Wheelhouse K, Trewell M, Gouverneur V. Synlett. 2016;27:25. [Google Scholar]
- 4.Ge S, Chaładaj W, Hartwig J. J. Am. Chem. Soc. 2014;136:4149. doi: 10.1021/ja501117v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For optimization of labeling conditions, see supporting information Table S2..
- 6.RCY (SPE): reaction mixture was purif ied by solid-phase extrac-tion (SPE) to separate 18F-labeled product from polar by -products and unreacted [18F]fluoride. The radiochemical yield was measured based on the filtrate, and is reported relative to starting [18F]fluoride activity. The filtrate was also analyzed by radio-HPLC in conjunction with radio-TLC to determine radiochemical purity. RCY (HPLC): reaction mixture was purif ied by semi-preparative HPLC to separate 18F-labeled product. The radiochemical yield was measured based on the fraction corresponding to radiolabeled product, and is reported relativ e to starting [18F]fluoride activity . The filtrate was also analyzed by radio-HPLC. RCY (SPE) and RCY (HPLC) for [18F]difluoromethy l-SC-58125, [18F]6-difluoromethylquinoline, and [18F]difluoromethy l-feno fibrate were consistent (see supporting information Table S3).
-
7.
- 8.Guo Y, Twamley B, Shreeve J, M. Org. Biomol. Chem. 2009;7:1716. doi: 10.1039/b900311h. [DOI] [PubMed] [Google Scholar]; For palladium-cataly zed cross-coupling of aryl bromide with α-alky l-α-fluoroacetophenone, see: Guo C, Wang R-W, Guo Y, Qing F-L. J. Fluorine Chem. 2012;133:86.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.