

Summary
Drug resistance in protein targets is an increasingly common phenomenon that reduces the efficacy of both existing and new antibiotics. However, knowledge of future resistance mutations during pre-clinical phases of drug development would enable the design of novel antibiotics that are robust against not only known resistant mutants, but also against those that have not yet been clinically observed. Computational structure-based protein design (CSPD) is a transformative field that enables the prediction of protein sequences with desired biochemical properties such as binding affinity and specificity to a target. The use of CSPD to predict previously unseen resistance mutations represents one of the frontiers of computational protein design. In a recent study (1), we used our OSPREY (Open Source Protein REdesign for You) suite of CSPD algorithms to prospectively predict resistance mutations that arise in the active site of the dihydrofolate reductase enzyme from methicillin-resistant Staphylococcus aureus (SaDHFR) in response to selective pressure from an experimental competitive inhibitor. We demonstrated that our top predicted candidates are indeed viable resistant mutants.
Since that study, we have significantly enhanced the capabilities of OSPREY with not only improved modeling of backbone flexibility, but also efficient multi-state design, fast sparse approximations, partitioned rotamers for more accurate energy bounds, and a computationally efficient representation of molecular-mechanics and quantum-mechanical energy functions. Here, using SaDHFR as an example, we present a protocol for resistance prediction using the latest version of OSPREY. Specifically, we show how to use a combination of positive and negative design to predict active site escape mutations that maintain the enzyme’s catalytic function but selectively ablate binding of an inhibitor.
Keywords: OSPREY, Computational protein design, Positive and negative design, Antibiotic resistance prediction
1 Introduction
Antibiotic resistance is an unfortunate consequence of evolutionary pressures on drug targets. In particular, selective pressures from competitive inhibitors that target enzymes elicit single nucleotide polymorphisms that give rise to amino acid changes that preserve catalytic function in the target but disrupt inhibitor binding. Dihydrofolate reductase (DHFR) in Staphylococcus aureus is a clinically important example of this mode of resistance. A single amino acid polymorphism in DHFR confers resistance to trimethoprim, a commonly prescribed antibiotic (2). This and other drug-resistant strains – collectively referred to as methicillin-resistant Staphylococcus aureus (MRSA) – cause pneumonia as well as skin, bloodstream, and surgical site infections. Additional mutations in MRSA DHFR (SaDHFR) result in even higher levels of drug resistance.
Successfully predicting resistance-conferring SaDHFR mutations before they emerge can enable the development of more robust inhibitors. However, because 20 amino acids can occur at every residue position, the combinatorially large number of candidate sequences that must be evaluated for resistance far exceeds the capabilities of current experimental methods. Fortunately, computational structure-based protein design (CSPD) is a practical alternative strategy to predict drug resistance over a large set of mutations.
OSPREY (Open Source Protein REdesign for You) (3, 4, 5, 6, 7, 8, 9, 10) is a state-of-the-art, free, and open-source suite of computational protein design algorithms. To date, a number of research groups have successfully used OSPREY to perform biomedically important protein designs. For example, we previously used OSPREY to predict escape mutations in SaDHFR that confer resistance to a lead inhibitor (11). More recently, we used OSPREY to predict escape mutations that grant SaDHFR resistance to a different experimental inhibitor, compound 1; we showed that two novel, predicted mutants (V31L and V31G) were selected in resistance selection experiments along with an additional compensating mutation (F98Y) (1). Additionally, we used OSPREY to alter the specificity of Gramicidin S Synthetase A (12, 13), to design epitope-specific HIV antibody probes (14), to design peptides to inhibit the interaction between the protein CAL and cystic fibrosis transmembrane conductance regulator (CFTR) (15), and to screen inhibitors of a leukemia-associated protein-protein interaction (16). Furthermore, the Vaccine Research Center (VRC) used OSPREY to design HIV antibodies that are easier to induce (17). In (18), we collaborated with the VRC to use OSPREY to design broader and more potent anti-HIV antibodies. Finally, Bailey-Kellogg and colleagues used OSPREY to optimize stability and immunogenicity of therapeutic proteins (19, 20, 21).
OSPREY is based on the following principles:
Accurate modeling of flexibility in the protein (backbone and side-chains) and ligand captures conformational changes induced by amino acid mutations. Other CSPD algorithms typically represent amino acid side-chain rotational isomers (rotamers) as discrete points in χ-angle space, resulting in sub-optimal design predictions (5, 7). OSPREY overcomes the limitations imposed by discrete rotamers by implementing continuous rotamers: continuous regions of χ-angle space that more accurately reflect empirically-observed side-chain placements (3, 5, 15). In contrast to protein designs using discrete rotamers, those using continuous rotamers find lower energy conformations and different sequences, leading to more accurate biological predictions (5, 7).
Ensemble-based design enables more accurate predictions of binding free energy. Traditional protein design methods focus on locating the global minimum energy conformation (GMEC). However, a protein in solution exists not as a single low-energy structure but as a thermodynamic ensemble of conformations. Since a thermodynamic ensemble of low-energy conformations governs protein-ligand binding (22), models that only consider the GMEC may incorrectly predict binding (15). OSPREY improves upon GMEC-based protein design by using the K * algorithm (3, 4), which efficiently approximates the association constant, Ka, of a protein-ligand complex using structural ensembles. In particular, K * only considers the most probable low-energy conformations and discards the high energy conformations that are rarely populated by either the protein or the ligand.
Mathematical guarantees of accuracy. Because CSPD algorithms must search vast sequence and conformation spaces, computational complexity remains a limiting factor in protein design. Accordingly, CSPD programs must rely on a simplified input model, which defines a computationally tractable simplification of the protein design space. Briefly, the input model consists of the initial protein structure(s), the permitted set of mutations to the wild type structure, the allowed protein flexibility, and an energy function to rank the generated conformations. Nevertheless, protein design remains NP-hard (23). Because of this complexity, heuristic search methods based on stochastic optimization, such as Monte Carlo (24, 25), are often used. However, these methods cannot guarantee to find the lowest energy conformations nor sequences. In contrast, OSPREY uses provable algorithms to determine the lowest energy conformations satisfying the input model. As a consequence, OSPREY determines protein sequences that satisfy the design objective with mathematical guarantees of accuracy (up to the accuracy of the input model). Crucially, this means that discrepancies between experimental results and predictions by OSPREY are attributable solely to errors in the input model; when using OSPREY any such discrepancies are substantially easier to resolve by making corrections to the input model. On the other hand, the causes of erroneous design predictions are much more difficult to ascertain when using heuristic methods.
Below, we describe the specific application of OSPREY to predict novel, viable resistance mutations that arise in SaDHFR in response to our novel propargyl-linked antifolate inhibitor, compound 1 (Fig. 1A) (1, 26). The combination of positive and negative design (to maintain native substrate binding and to abrogate inhibitor binding, respectively) in OSPREY is sufficient to predict novel escape mutations in this system (Fig. 1B–C). We use this specific example to illustrate the more general problem of predicting resistance in drug targets in other systems. These extensions may require the modeling of backbone flexibility (6), multi-state specificity (8), faster energy functions (10), or efficient sparse approximations (9), all of which are available in OSPREY.
Figure 1.
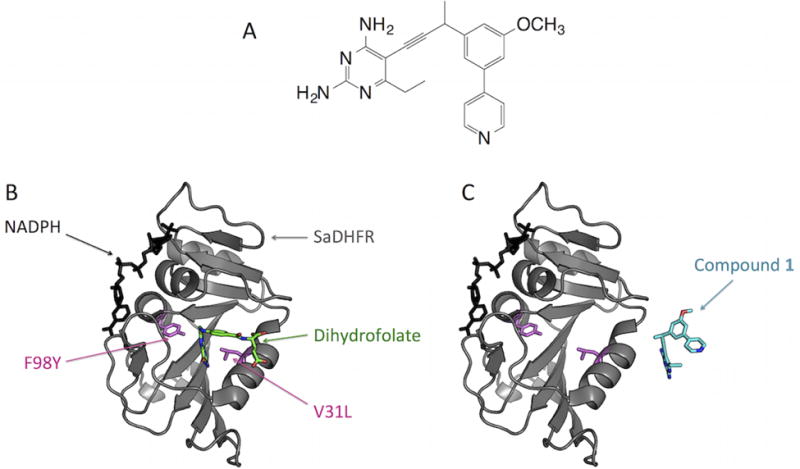
Positive design to maintain SaDHFR:dihydrofolate binding and negative design to destabilize SaDHFR:compound 1 binding using OSPREY. (A) Compound 1, an experimental SaDHFR inhibitor. (B) OSPREY positive design objective. OSPREY predicts mutations (pink) of SaDHFR (gray) that maintain binding of dihydrofolate (green) in the SaDHFR active site. These mutations allow SaDHFR to preserve its catalytic activity. The co-factor NADPH is shown in black. (C) OSPREY negative design objective. OSPREY predicts mutations that destabilize the binding of an inhibitor (compound 1) to SaDHFR. OSPREY predicts SaDHFR candidate escape mutations that bind dihydrofolate but selectively disrupt binding of compound 1.
We begin with a detailed description of the input model for OSPREY’s positive and negative design steps. The input model consists of 3D structures (determined by nuclear magnetic resonance, X-ray crystallography, or homology modeling), the allowable set of mutations, protein and ligand flexibility parameters, and an energy function (Fig. 2A–B). To predict candidate resistance mutations (i.e., those that bind SaDHFR’s natural substrate but not compound 1 (Fig. 1A)), we perform positive (Fig. 2A) and negative (Fig. 2B) designs using structures of SaDHFR:dihydrofolate:NADPH and SaDHFR:compound 1:NADPH, respectively. Since crystal structures of these complexes were unavailable, we created the respective homology models from (2) and PDB ID 3FQC (27). Having constructed these models, we considered a sequence space consisting of the most prevalent modes of mutational resistance: single nucleotide polymorphisms to active site residues (28). These residues are also subject to OSPREY’s flexibility model, which specifies the empirically determined set of energetically favorable protein side-chain and ligand rotational isomers in a rotamer library (29). For improved prediction accuracy, OSPREY’s continuous rotamer model extends this rigid definition of a rotamer to a bounded, yet continuously flexible region of side-chain conformation space (5). Ligands (dihydrofolate and compound 1), which are also modeled using continuous rotamers, are further allowed rigid body rotational and translational degrees of freedom within the active site. Together, the 3D structures, allowable mutations, and protein and ligand flexibility parameters define the conformation space for all candidate resistant mutants. The fourth component of the input model, a computationally-efficient all-atom residue-pairwise energy function, is used to evaluate structures in this conformation space. Several energy functions are available in OSPREY (7), but usually, and for this example, the energy function consists of the Amber96 (30) energy function for van der Waals, electrostatic, and dihedral energies and the EEF1 solvation model (31).
Figure 2.
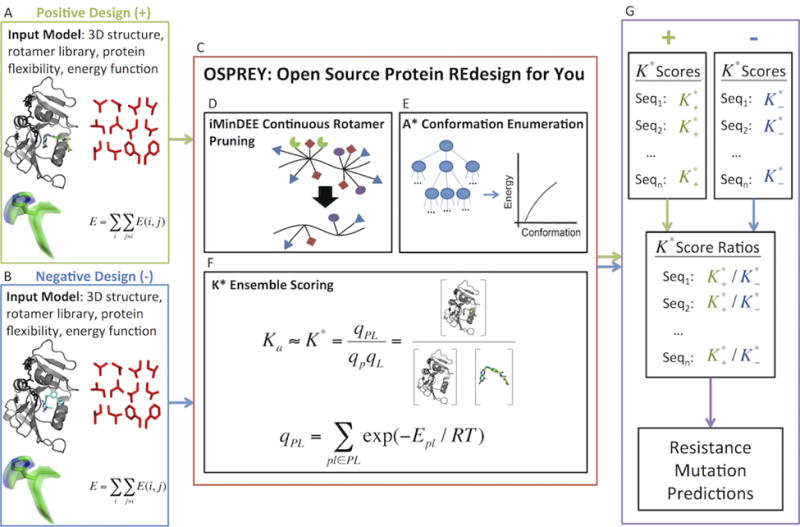
Processing of positive and negative design input models in OSPREY. (A) Input model for positive design. The 3D structure is a model of SaDHFR bound to dihydrofolate and NADPH. (B) Input model for negative design. The 3D structure is a model of SaDHFR bound to compound 1 (Fig. 1A) and NADPH. (C) Pruning, search, and ensemble scoring algorithms in OSPREY. (D) iMinDEE continuous rotamer pruning removes rotamers that provably do not belong to the ensemble of lowest energy conformations. (E) A * conformation enumeration generates an ensemble of conformations in a gap-free, energetically increasing order. (F) K * ensemble scoring approximates Boltzmann-weighted partition functions for the bound and unbound states and subsequently approximates the association constant, Ka, with mathematical guarantees of accuracy relative to the input model. (G) Prediction of resistance mutations in OSPREY. (Top Left) Positive design K * scores, , generated by OSPREY for each sequence. (Top Right) Negative design K * scores, , generated by OSPREY for each sequence. (Middle) A ratio of the positive design score to the negative design score, , for each sequence. (Bottom) Sequences are sorted in decreasing order of K * score ratios. The top predicted mutants, which have the highest ratio of scores, are evaluated experimentally.
Having presented the components of the input model, we now describe the use of OSPREY to predict novel SaDHFR escape mutations. For each mutation defined in the input model, OSPREY performs a positive design step to predict the mutant’s binding affinity for SaDHFR’s natural substrate (dihydrofolate) and a negative design step to predict its affinity for compound 1. Mutants with both tight binding affinity for dihydrofolate and poor binding affinity for compound 1 are selected as the best candidate mutants. We discuss OSPREY’s procedure to predict binding affinity below.
Positive design and negative design are performed and scored separately for each candidate mutant using the iMinDEE (5), A * (3, 32), and K * (3, 4, 15) algorithms in OSPREY (Fig. 2C). In a pre-processing step, the iMinDEE algorithm (Fig. 2D) efficiently prunes rotamers that are provably incompatible with the ensemble of lowest energy conformations. Importantly, iMinDEE extends the provable guarantees of the original dead-end elimination algorithms (33, 34) to OSPREY’s continuous rotamer model, allowing both biophysically accurate protein modeling and an exponential reduction in the size of the conformation space. Subsequently, the A * algorithm (Fig. 2E) enumerates the remaining conformations in gap-free energetically-increasing order, starting from the global minimum energy conformation (GMEC). The K * module (Fig. 2F) of OSPREY approximates a Boltzmann-weighted partition function, q, from this energetically-ordered ensemble, S, of conformations:
where Es is the energy of conformation s ∈ S, T is the temperature in Kelvin, and R is the gas constant. To efficiently approximate the full partition function q defined over all conformations in S, K * halts A * conformation enumeration when the partial partition function q*, computed from the ensemble of lowest energy conformations in S, is provably within a factor ε of q. The user specifies ε ahead of time as part of the input model. In practice, K * achieves a provably accurate ε-approximation to q using only a small fraction of the lowest energy conformations in S. Subsequently K * approximates the association constant, Ka, for a protein-ligand complex as the ratio of ε-approximated partition functions for the bound and unbound states:
where PL, P, and L represent the protein-ligand complex, the unbound protein, and unbound ligand, respectively. For each candidate mutant, separate positive and negative design K * scores are computed (Fig. 2G, Top). Since a higher K * score denotes tighter predicted binding affinity, a resistant mutant would have a high positive design score (for dihydrofolate) and a low negative design score (for compound 1). Therefore, mutants were ranked by their ratio of positive to negative design scores. Mutants with both a higher rank than the wild type and a good positive design score relative to the wild type were considered candidate resistant mutants. Among this set of mutants, a higher ratio of scores indicates a greater degree of predicted resistance to compound 1 (Fig. 2G, Middle). On the other hand, mutants such as L20F, which have high positive to negative design score ratios but low positive design scores, are not considered viable, due to low predicted affinity for dihydrofolate. The top-ranked predicted resistant mutants according to our protocol were recommended for creation and experimental testing.
In summary, we combined positive and negative protein design with the state-of-the-art algorithms in OSPREY to predict viable mutations in SaDHFR that confer resistance to our potent competitive inhibitors (1, 26). Table 1 shows predictions and experimental characterizations for wild type SaDHFR (Sa(WT)DHFR) and OSPREY’s four top-ranked resistance mutations. Each of these mutants (V31L, V31I, L5I, and L5V) had not only higher positive to negative design K * score ratios than Sa(WT)DHFR, but also a comparable or tighter predicted binding affinity for dihydrofolate than Sa(WT)DHFR. To test our top resistance predictions, we created these SaDHFR SNP mutants using site-directed mutagenesis. An evaluation of Michaelis–Menten kinetics confirmed that our top four predicted mutant enzymes are catalytically competent, exhibiting small losses in k cat/KM. Furthermore, the resistance of our top four mutants, as measured by fold loss in Ki relative to the wild type, correlates perfectly with our predicted K * ratio rank (see (1) for details).
Table 1.
K * resistance prediction (columns 1–5) and experimental characterization (columns 6–7) of wild type and mutant SaDHFR enzymes from (1).
| Enzyme | K* ratio rank | K* positive-to-negative design ratio | K* positive design (dihydrofolate) score | K* negative design (compound 1) score | kcat/KM | Fold loss (Ki mut/Ki wt) compound 1 |
|---|---|---|---|---|---|---|
| Sa(WT)DHFR | 18 | 1.96 E + 06 | 7.16 E + 42 | 3.66 E + 36 | 6.1 ± 0.3 | n/a |
| Sa(V31L)DHFR | 1 | 7.11 E + 21 | 2.16 E + 41 | 3.04 E + 19 | 1.60 ± 0.06 | 58 |
| Sa(V31l)DHFR | 2 | 5.95 E + 21 | 4.87 E + 36 | 8.18 E+ 14 | 1.74 ± 0.07 | 36 |
| Sa(L5l)DHFR | 3 | 1.71 E + 15 | 6.06 E + 39 | 3.54 E+ 24 | 2.24 ± 0.1 | 4.4 |
| Sa(L5V)DHFR | 4 | 1.16 E + 14 | 4.01 E + 44 | 3.44 E + 30 | 1.8 ± 0.1 | 1.9 |
Since these predictions were made in (1), we have substantially improved OSPREY’s capabilities with the following algorithmic enhancements: improved backbone flexibility (6), multi-state specificity (8), fast sparse approximations (9), partitioned rotamers for improved energy bounds (35), and a computationally efficient representation of molecular-mechanics and quantum-mechanical energy functions (10). In the following Materials and Methods sections, with this system as an example, we present a protocol to predict the same SaDHFR escape mutations using the most recent release of OSPREY. The Methods section describes how to install and set up OSPREY (Section 3.1), how to perform positive and negative design in OSPREY (Section 3.2), how to predict resistant mutants using OSPREY’s positive and negative design scores (Section 3.3.1), and how to visualize the PDB files that represent OSPREY’s structural ensemble predictions (Section 3.3.2). Importantly, the paradigm described here is applicable to the prediction of novel escape mutations to any antibacterial, antiviral, or antineoplastic drug. In all these cases, the combination of positive and negative design in OSPREY can be used to model selective pressure by inhibitors on other protein targets.
2 Materials
2.1 Operating System Environment
An operating system that supports the Java programming language.
Java Runtime Environment (JRE) 7.0 or later.
Python version 2.7 (required for post-processing scripts).
2.2 Input Files
The input files can be downloaded at: http://www.cs.duke.edu/donaldlab/Supplementary/mimb2015/OSPREY-V2.2B-MIMB2015.zip and consist of the following:
Homology model for positive design: structure of SaDHFR in complex with dihydrofolate, SaDHFR:DHF:NADPH (see Notes 1, 2, 3, and 4).
Homology model for negative design: structure of SaDHFR in complex with compound 1, SaDHFR:compound 1:NADPH (see Notes 1, 2, 3, and 4).
-
Two expanded amino acid rotamer libraries:
LovellRotamer-wt-pos.dat and LovellRotamer-wt-neg.dat
-
Two generic rotamer libraries for non-amino acids:
GenericRotamers-fol.dat and GenericRotamers-pye.dat
Shell scripts necessary to run software.
A Python script to analyze the output.
Other default data files also found in the OSPREY software package.
2.3 OSPREY Suite of Algorithms
OSPREY 2.2 software package, available at http://www.cs.duke.edu/donaldlab/osprey.php
2.4 Other Software
PyMOL 1.6 or later, available at http://www.pymol.org/
AmberTools (see Note 3), available at http://ambermd.org/AmberTools14-get.html
3 Methods
3.1 OSPREY Installation
Download the OSPREY version 2.2 suite of protein design algorithms (Section 2.3.1).
- After downloading the OSPREY software package from the above source, unzip the file to a desired location using the following command:
# tar -xvfz OSPREY.tar.gz
- Next, add the third-party libraries provided with OSPREY to your classpath:
# libpath=/whatever/OSPREY/lib # export CLASSPATH=$CLASSPATH:$libpath/architecture-rules-3.0.0-M1.jar:$libpath/commons-logging-1.1.1.jar:$libpath/colt-1.2.0.jar:$libpath/commons-math3-3.0.jar:$libpath/commons-beanutils-1.6.jar: $libpath/jdepend-2.9.1.jar:$libpath/commons-collections-2.1.jar:$libpath/joptimizer.jar:$libpath/commons-digester-1.6.jar:$libpath/junit-3.8.1.jar: $libpath/commons-io-1.4.jar:$libpath/log4j-1.2.14. jar:$libpath/commons-lang-2.5.jar:$libpath/xml-apis-1.0.b2.jar
Now, change directories to the OSPREY directory and create a new directory, bin.
- Finally, change directories to the src directory and run the following command:
# javac -d ../bin *.java
3.2 Design
In this section, we describe how to run positive and negative design in OSPREY. Nine active site residues were chosen to be continuously-flexible within 9° of the rotamers in the Penultimate rotamer library (29) and mutable up to one nucleotide substitution: L5{L/V/I/R/Q}, V6{V/A/L/I/F/D/G}, L20{L/V/I/F/S}, L28{L/V/M/W/F/S}, V31{V/A/I/F/L/D/G}, T46{T/A/R/I/K/S}, I50{I/V/L/M/F/N/S/T}, L54{L/R/Q/V}, and F92{F/V/L/I/Y/S/C}. We also apply this flexibility model to rotamers of the ligands (i.e. dihydrofolate and compound 1), whose motions also include rigid body translations and rotations in the active site. To empirically determine a ligand rotamer library for compound 1, we began by modeling roughly 10,000 of its binding conformations to SaDHFR. Next, we used OSPREY’s MinDEE/A* algorithm (3) to determine the lowest energy binding conformations beneath a steric threshold. This process yielded 1,660 binding poses for compound 1 (see GenericRotamers-pye.dat in the OSPREY negative design directory in Section 3.2.1). The collection of mutable and flexible residues, including the ligands, resulted in a total of 47 sequences. This set of sequences is used in the following positive and negative designs.
3.2.1 Obtaining Input Files for Design
Download the required files for this section, described in Section 2.2.
- Extract the file to create the project directory:
# unzip OSPREY-V2.2B-MIMB2015.zip
The base directory created is OSPREY-V2.2B-MIMB2015. Its sub-directory, OSPREY-INPUT, is the parent directory for the positive design directory, OSPREY-INPUT/pos-design, and the negative design directory, OSPREY-INPUT/neg-design.
3.2.2 Running Positive Design in OSPREY
This section describes how to run the provided scripts (Section 2.2.5) to complete the positive design. The PDB file pos-design.pdb (Section 2.2.1) consists of all amino acids within an 8 Å radius of dihydrofolate, DHF (see Note 5).
- Change to the directory where the files for positive design are located:
OSPREY-INPUT/pos-design
- Run the provided shell script for positive design.
# ./runPositiveDesign.sh
3.2.3 Running Negative Design in OSPREY
This section describes how to run the provided scripts (Section 2.2.5) to complete the negative design. The PDB file neg-design.pdb (Section 2.2.2) consists of all amino acids within an 8 Å radius of compound 1, PYE (see Note 5).
- Change to the directory where the files for positive design are located:
OSPREY-INPUT/negative-design
- Run the provided shell script for negative design.
# ./runNegativeDesign.sh
3.3 OSPREY Output
3.3.1 Predicting Resistance from the Ratio of OSPREY Positive to Negative Design Scores
This section describes how to rank sequences by their predicted resistance to compound 1. A python script is provided to complete this process (Section 2.2.5).
Move to the OSPREY-INPUT directory.
- Run the provided Python script:
# python summarizeResults.py
Each row of output is formatted as follows: mutation, positive design score (log scale), negative design score (log scale), and ratio of design scores (log scale). The mutations are ordered by increasing order of score ratios. So, the mutation in the last line of the output has the highest positive to negative design ratio. (See Note 6 for the interpretation of a positive or negative design K * score of 0.) From this list, the top candidate resistant mutants are those with both a high positive design score (i.e. high predicted binding affinity for dihydrofolate relative to the wild type) and a high positive to negative design score ratio (see Note 7).
3.3.2 Structural Analysis of OSPREY Output
The script in Methods Section 3.3.1 ranks sequences by increasing order of positive to negative design score ratios. Candidate resistant mutants, which have high positive design scores and high score ratios, can be identified visually in this list. Below, we describe how to view the lowest energy structures from each sequence.
After completing positive and negative design (Methods Sections 3.2.2 & 3.2.3) OSPREY outputs the PDB files for the top ten conformations for each sequence. This section describes these PDB files and how to view them. Each PDB file name takes on one of the following formats:
n_aaaaaaaaa_0_m.pdb n_X_1_m.pdb. n_aaaaaaaaaX_2_m.pdb
where n is an index assigned to each sequence and m is a three digit number ranking one sequence’s set of ten conformations from lowest to highest energy. Each string of a’s corresponds to an amino acid sequence (e.g., LVLLVTILF). X represents the non-amino acid ligand (i.e., dihydrofolate or compound 1). The first format corresponds to SaDHFR unbound to the ligand (either dihydrofolate for the positive design or compound 1 for the negative design). The second format corresponds to the ligand unbound to SaDHFR. Finally, the third format corresponds to SaDHFR in complex with the ligand.
Change directories into OSPREY/pos-design/ksConfs This directory contains all of the PDB files output for the positive design (Methods Section 3.2.2).
Open and view the PDB files using PyMOL (Materials Section 2.4.1). Several files can be opened and viewed simultaneously.
Change directories into OSPREY/neg-design/ksConfs This directory contains all of the PDB files output for the positive design (Methods Section 3.2.3).
Open and view the PDB files using PyMOL (Materials Section 2.4.1). Several files can be opened and viewed simultaneously.
Acknowledgments
The authors would like to thank Mark Hallen and Dr. Kyle Roberts for thoughtful suggestions and technical assistance. This work was supported by NIH grant R01 GM-78031 to B.R.D., R01 AI-111957 to A.C.A., and A.O. was supported in part by NSF Graduate Research Fellowships Program Award 1106401.
Footnotes
In this example, we modeled the inputs for both the positive and negative design steps from structures of related ligands bound to SaDHFR. Other 3D protein structures (i.e., determined by NMR and X-ray crystallography) are also viable input structures for OSPREY.
A structure of dihydrofolate (DHF) or compound 1 bound to SaDHFR was not available when the original predictions were made. As a result, the bound complex of SaDHFR:DHF:NADPH (positive design) was modeled on the coordinates of a single mutant Sa(F98Y)DHFR bound to folate and NADPH (2). (The structure upon which the model is based was not deposited in the Protein Data Bank.) The structure for SaDHFR:compound 1:NADPH (negative design) was modeled using the bound structure of a related SaDHFR inhibitor (PDB ID 3FQC, (27)).
It is often necessary to alleviate steric clashes in the input structures prior to running OSPREY. This is achieved by performing an energy minimization step using AmberTools (See Materials Section 2.4.2). This process is detailed in the Antechamber tutorial: http://ambermd.org/tutorials/basic/tutorial4b/
# antechamber -i x.pdb -fi pdb -o x.prepi -fo prepi -c bcc -s 2and append the contents of output file x.prepi (where x is the base name of the .pdb file containing only the coordinates of the non-protein compound), starting from This is a remark line, to the file all_nuc94_and_gr.in, which is part of OSPREY’s input model. Next, create a file named
GenericRotamers.datto store rotamers for the compound. To determine rotamers for the compound, open the structure in Pymol and use the Wizard > Measurement tool in PyMOL (see Materials Section 2.4.1). Add rotamers in the format specified in the OSPREY manual (see Note 8). Reference this file in System.cfg using the grotFilei keyword.
To create an 8 Å shell of a protein for your own designs, use PyMOL (see Materials Section 2.4.1).
Resistance (i.e. positive to negative design ratio) rankings in which either the positive or negative design K * score is 0 are handled specially. Mutations for which only the negative design score is 0 receive a score ratio of infinity. Mutations for which either only the positive design score is 0 or both positive and negative design scores are 0 receive a design ratio of 0.
A candidate resistant mutant has both a high positive design score (indicating of high predicted binding affinity for dihydrofolate) and a low negative design score (denoting low predicted binding affinity for compound 1). Mutants with a high positive to negative design score ratio but a low positive design score (such as L20F) relative to the wild type are not considered viable, as they are predicted to bind dihydrofolate poorly.
To perform your own protein designs using OSPREY, please refer to the user manual found in the OSPREY software download from Sections 2.3 & 3.1.
The results presented in (1) were performed using OSPREY 1.1a. This can lead to slightly different results than those in the newer version of OSPREY. To reproduce the results in (1) exactly, please download the code from: http://www.cs.duke.edu/donaldlab/Supplementary/mimb2015/OSPREY-V2010-MIMB2015.zip
References
- 1.Reeve Stephanie M, Gainza Pablo, Frey Kathleen M, Georgiev Ivelin, Donald Bruce R, Anderson Amy C. Protein design algorithms predict viable resistance to an experimental antifolate. Proc Natl Acad Sci U S A. 2015 Jan;112(3):749–54. doi: 10.1073/pnas.1411548112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dale GE, Broger C, D’Arcy A, Hartman PG, DeHoogt R, Jolidon S, Kompis I, Labhardt AM, Langen H, Locher H, Page MG, Stüber D, Then RL, Wipf B, Oefner C. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997 Feb;266(1):23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- 3.Georgiev Ivelin, Lilien Ryan H, Donald Bruce R. The minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. J Comput Chem. 2008 Jul;29(10):1527–42. doi: 10.1002/jcc.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donald Bruce R. Algorithms in Structural Molecular Biology. MIT Press; Cambridge, MA: 2011. [Google Scholar]
- 5.Gainza Pablo, Roberts Kyle E, Donald Bruce R. Protein design using continuous rotamers. PLoS Comput Biol. 2012 Jan;8(1):e1002335. doi: 10.1371/journal.pcbi.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallen Mark A, Keedy Daniel A, Donald Bruce R. Dead-end elimination with perturbations (DEEPer): a provable protein design algorithm with continuous sidechain and backbone flexibility. Proteins. 2013 Jan;81(1):18–39. doi: 10.1002/prot.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gainza Pablo, Roberts Kyle E, Georgiev Ivelin, Lilien Ryan H, Keedy Daniel A, Chen Cheng-Yu, Reza Faisal, Anderson Amy C, Richardson David C, Richardson Jane S, Donald Bruce R. OSPREY: protein design with ensembles, flexibility, and provable algorithms. Methods Enzymol. 2013;523:87–107. doi: 10.1016/B978-0-12-394292-0.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallen Mark A, Donald Bruce R. COMETS (constrained optimization of multistate energies by tree search): A provable and efficient algorithm to optimize binding affinity and specificity with respect to sequence. Research in Computational Molecular Biology (RECOMB) 2015 Apr;9029:122–135. doi: 10.1089/cmb.2015.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jou Jonathan, Jain Swati, Georgiev Ivelin, Donald Bruce R. BWM*: A novel, provable, ensemble-based dynamic programming algorithm for sparse approximations of computational protein design. Research in Computational Molecular Biology (RECOMB) 2015;9029:154–166. doi: 10.1089/cmb.2015.0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallen Mark A, Gainza Pablo, Donald Bruce R. Compact representation of continuous energy surfaces for more efficient protein design. Journal of Chemical Theory and Computation. 2015 Mar;11(5):2292–2306. doi: 10.1021/ct501031m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey Kathleen M, Georgiev Ivelin, Donald Bruce R, Anderson Amy C. Predicting resistance mutations using protein design algorithms. Proc Natl Acad Sci U S A. 2010 Aug;107(31):13707–12. doi: 10.1073/pnas.1002162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens Brian W, Lilien Ryan H, Georgiev Ivelin, Donald Bruce R, Anderson Amy C. Redesigning the PheA domain of gramicidin synthetase leads to a new understanding of the enzyme’s mechanism and selectivity. Biochemistry. 2006 Dec;45(51):15495–504. doi: 10.1021/bi061788m. [DOI] [PubMed] [Google Scholar]
- 13.Chen Cheng-Yu, Georgiev Ivelin, Anderson Amy C, Donald Bruce R. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci U S A. 2009 Mar;106(10):3764–9. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgiev I, Schmidt S, Li Y, Wycuff D, Ofek G, Doria-Rose N, Luongo T, Yang Y, Zhou T, Donald BR, Mascola J, Kwong P. Design of epitope-specific probes for sera analysis and antibody isolation. Retrovirology. 2012;9 [Google Scholar]
- 15.Roberts Kyle E, Cushing Patrick R, Boisguerin Prisca, Madden Dean R, Donald Bruce R. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput Biol. 2012;8(4):e1002477. doi: 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorczynski Michael J, Grembecka Jolanta, Zhou Yunpeng, Kong Yali, Roudaia Liya, Douvas Michael G, Newman Miki, Bielnicka Izabela, Baber Gwen, Corpora Takeshi, Shi Jianxia, Sridharan Mohini, Lilien Ryan, Donald Bruce R, Speck Nancy A, Brown Milton L, Bushweller John H. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol. 2007 Oct;14(10):1186–97. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Georgiev Ivelin S, Rudicell Rebecca S, Saunders Kevin O, Shi Wei, Kirys Tatsiana, McKee Krisha, O’Dell Sijy, Chuang Gwo-Yu, Yang Zhi-Yong, Ofek Gilad, Connors Mark, Mascola John R, Nabel Gary J, Kwong Peter D. Antibodies VRC01 and 10E8 neutralize HIV-1 with high breadth and potency even with IG-framework regions substantially reverted to germline. J Immunol. 2014 Feb;192(3):1100–6. doi: 10.4049/jimmunol.1302515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudicell Rebecca S, Kwon Young Do, Ko Sung-Youl, Pegu Amarendra, Louder Mark K, Georgiev Ivelin S, Wu Xueling, Zhu Jiang, Boyington Jeffrey C, Chen Xuejun, Shi Wei, Yang Zhi-Yong, Doria-Rose Nicole A, McKee Krisha, O’Dell Sijy, Schmidt Stephen D, Chuang Gwo-Yu, Druz Aliaksandr, Soto Cinque, Yang Yongping, Zhang Baoshan, Zhou Tongqing, Todd John-Paul, Lloyd Krissey E, Eudailey Joshua, Roberts Kyle E, Donald Bruce R, Bailer Robert T, Ledgerwood Julie, NISC Comparative Sequencing Program. Mullikin James C, Shapiro Lawrence, Koup Richard A, Graham Barney S, Nason Martha C, Connors Mark, Haynes Barton F, Rao Srinivas S, Roederer Mario, Kwong Peter D, Mascola John R, Nabel Gary J. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J Virol. 2014 Nov;88(21):12669–82. doi: 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker Andrew S, Choi Yoonjoo, Griswold Karl E, Bailey-Kellogg Chris. Structure-guided deimmunization of therapeutic proteins. J Comput Biol. 2013 Feb;20(2):152–65. doi: 10.1089/cmb.2012.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salvat Regina S, Choi Yoonjoo, Bishop Alexandra, Bailey-Kellogg Chris, Griswold Karl E. Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads. Biotechnol Bioeng. 2015 Feb; doi: 10.1002/bit.25554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Hongliang, Verma Deeptak, Li Wen, Choi Yoonjoo, Ndong Christian, Fiering Steven N, Bailey-Kellogg Chris, Griswold Karl E. Depletion of T cell epitopes in lysostaphin mitigates anti-drug antibody response and enhances antibacterial efficacy in vivo. Chem Biol. 2015 May;22(5):629–39. doi: 10.1016/j.chembiol.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J. 1997 Mar;72(3):1047–69. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierce Niles A, Winfree Erik. Protein design is np-hard. Protein Eng. 2002 Oct;15(10):779–82. doi: 10.1093/protein/15.10.779. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Farid H, Pistor E, Farid RS. A new approach to the design of uniquely folded thermally stable proteins. Protein Sci. 2000 Feb;9(2):403–16. doi: 10.1110/ps.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000 Sep;97(19):10383–8. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frey Kathleen M, Lombardo Michael N, Wright Dennis L, Anderson Amy C. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase aureus dihydrofolate reductase. J Struct Biol. 2010 Apr;170(1):93–7. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frey Kathleen M, Liu Jieying, Lombardo Michael N, Bolstad David B, Wright Dennis L, Anderson Amy C. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternate conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009 Apr;387(5):1298–308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frey Kathleen M, Viswanathan Kishore, Wright Dennis L, Anderson Amy C. Prospective screening of novel antibacterial inhibitors of dihydrofolate reductase for mutational resistance. Antimicrob Agents Chemother. 2012 Jul;56(7):3556–62. doi: 10.1128/AAC.06263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000 Aug;40(3):389–408. [PubMed] [Google Scholar]
- 30.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, Ferguson D, Seibel G, Kollman P. Amber: a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91(42):1–41. [Google Scholar]
- 31.Lazaridis T, Karplus M. Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J Mol Biol. 1999 May;288(3):477–87. doi: 10.1006/jmbi.1999.2685. [DOI] [PubMed] [Google Scholar]
- 32.Hart PE, Nilsson NJ, Raphael B. A formal basis for the heuristic determination of minimum cost paths. IEEE Trans on SSC. 1968;4:100–114. [Google Scholar]
- 33.Desmet J, De Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992 Apr;356(6369):539–42. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein RF. Efficient rotamer elimination applied to protein side-chains and related spin glasses. Biophys J. 1994 May;66(5):1335–40. doi: 10.1016/S0006-3495(94)80923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts Kyle E, Donald Bruce R. Improved energy bound accuracy enhances the efficiency of continuous protein design. Proteins. 2015 Jun;83(6):1151–64. doi: 10.1002/prot.24808. [DOI] [PMC free article] [PubMed] [Google Scholar]