

Abstract
Continental islands provide opportunities for testing the effects of isolation and migration on genetic variation in plant populations. In characteristic of continental islands is that the geographic connections between these islands, which are currently distinguished by seaways, have experienced fluctuations caused by sea‐level changes due to climate oscillations during the Quaternary. Plant populations on the islands have migrated between these islands via the exposed seafloors or been isolated. Here, we examined the demographic history of a temperate shrub, Rhododendron weyrichii, which is distributed in the southwestern parts of the Japanese archipelago and on an island of South Korea, using statistical phylogeographic approaches based on the DNA sequences of two chloroplast and eight nuclear loci in samples analyzed from 18 populations on eight continental islands, and palaeodistribution modeling. Time estimates for four island populations indicate that the durations of vicariance history are different between these populations, and these events have continued since the last glacial or may have predated the last glacial. The constancy or expansion of population sizes on the Japanese islands, and in contrast a bottleneck in population size on the Korean island Jeju, suggests that these islands may have provided different conditions for sustaining populations. The result of palaeodistribution modeling indicates that the longitudinal range of the species as a whole has not changed greatly since the last glacial maximum. These results indicate that exposed seafloors during the glacial period formed both effective and ineffective migration corridors. These findings may shed light on the effects of seafloor exposure on the migration of plants distributed across continental islands.
Keywords: ecological niche modeling, historical vicariance, island biogeography, isolation with migration model, population demography
1. Introduction
Continental islands, because they represent isolated distributions of terrestrial habitats, provide suitable settings in which to examine the effects of geographic isolation on evolution in plant species (Bittkau & Comes, 2005; MacArthur & Wilson, 1967; Nakamura, Suwa, Denda, & Yokota, 2009). Continental islands are separated from their adjacent mainlands by shallow seas, which prevent plant populations with low capacities for dispersal from migrating among islands and/or the mainlands, thus leading to increased genetic differentiation. During the late Quaternary, glacial–interglacial climate cycles occurred repeatedly on a global scale (Lisiecki & Raymo, 2005; Petit et al., 1999), and these oscillations have produced drastic changes in the connectivity of isolated populations by reconfiguring the spatial arrangements of islands due to exposure of seafloors that connected continental islands. Spatiotemporal changes in the continuity of species distributions during climatic oscillations could therefore have been a key factor affecting the evolutionary dynamics of species on continental islands by influencing isolation and migration history. In addition, it is important for understanding how the flora of the islands has been shaped.
The Japanese Archipelago, continental islands in East Asia, is a good region for investigating effects of spatiotemporal changes in the continuity of species distributions. It has been inferred that in the Japanese Archipelago, the distribution of temperate forests became shrunken and fragmented across small patches along the coastal regions during glacial periods (Gotanda & Yasuda, 2008; Harrison, Yu, Takahara, & Prentice, 2001; Tsukada, 1985). It is increasingly recognized that the current genetic structure of plant populations has been shaped by descent from populations that sheltered in refugia (Aoki, Suzuki, Hsu, & Murakami, 2004; Iwasaki, Aoki, Seo, & Murakami, 2012; Worth, Sakaguchi, Tanaka, Yamasaki, & Isagi, 2013). However, processes of isolation and migration among refugium populations on islands via exposed seafloor are not clear and have received less attention (Burridge et al., 2013; Duncan, Worth, Jordan, Jones, & Vaillancourt, 2016). In addition, although some studies discussed effects of the last glacial maximum (LGM, Clark et al., 2009) to current genetic variation, these were based on fossil records without focal species, not based on time estimations from genetic data (Iwasaki et al., 2012; Worth et al., 2013).
Understanding the complexity of population demography (population expansion, population decline, divergence, admixture, migration, etc.) on the islands presents several difficulties. Firstly, most studies to date have examined only a few DNA sequences (e.g., regions of the chloroplast DNA [cpDNA] and/or nuclear ribosomal DNA), and stochastic variance between gene genealogies in such a limited number of genomic regions has made it difficult to obtain reliable estimates of population demographies. Secondly, if population genetic analyses are not conducted in the framework of an appropriate time scale, it is not possible to make any biogeographically meaningful inferences about the drivers of population divergence. This is particularly problematic in phylogeographic analyses of continental island species, because here population divergence can be triggered by interglacial geographic isolation and/or glacial habitat isolation. In the last decade, demographic modeling of natural populations based on the coalescent approach has become commonplace (Hey & Nielsen, 2004; Nielsen & Beaumont, 2009). This approach can statistically estimate demographic parameters such as migration rate, divergence time, and changes in population size within a given time scale. Multilocus DNA sequences offer the potential for inferring species’ demographic histories in great detail because they provide many genetically informative segregating sites and independent genealogies (Wang et al., 2014). Ecological niche modeling can also provide useful information about historical range shifts by reconstructing palaeodistributions during key periods, for example, the LGM. These approaches enable us to integrate phylogeographic inferences and information about spatial range shifts, and as a result, they can lead to deeper insights into species’ biogeographic histories during the late Quaternary (Phillips, Anderson, & Schapire, 2006; Svenning, Fløjgaard, Marske, Nógues‐Bravo, & Normand, 2011).
Rhododendron weyrichii Maxim. (Ericaceae) is a deciduous shrub species, occurring mainly in temperate regions that experience high rainfall during summer in southwestern parts of the Japanese Archipelago including the Kii Peninsula of Honshu, Shikoku, and Kyushu islands and small islands surrounding them, and in Jeju Island in the southwest of the Korean Peninsula, disjunctly (Figure 1; Chamberlain & Rae, 1990). These regions are thought to have been important glacial refugia for temperate plant species (Gotanda & Yasuda, 2008; Harrison et al., 2001). Rhododendron weyrichii produces numerous small seeds (30–40 mg/100 seeds; Yoichi W., personal observation). Its pollinators are butterflies and bumblebees, and thus, exchange of pollen between island populations is infrequent under present‐day interglacial conditions. However, the dispersal barriers formed by seaways among neighboring islands disappeared during the LGM (ca. 21 kya) due to the shallow depth of the sea (<120 m, Clark et al., 2009; Park, Kimura, & Taira, 1996; Siddall et al., 2003), and this could have opened up effective dispersal corridors for the species. As R. weyrichii is widespread across a number of islands and shows a moderate level of genetic differentiation among populations (Yoichi & Tomaru, 2014), and it provides a suitable study system for analyzing demographic history in the islands.
Figure 1.
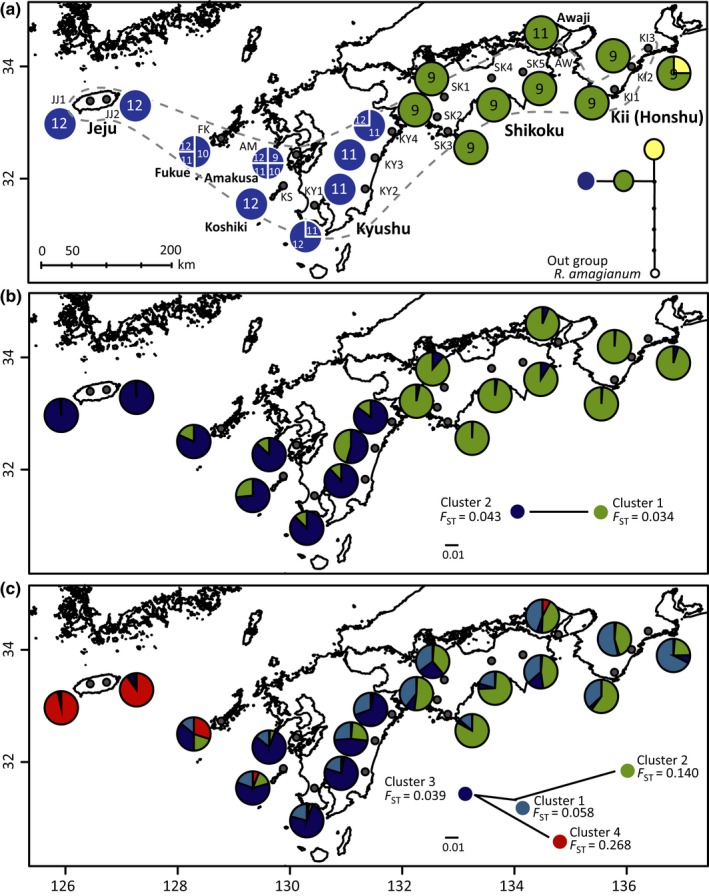
Species range and geographic distribution of (a) three haplotypes, showing the number of tri‐nucleotide repeats (ATT) at chloroplast DNA loci and (b) two and (c) four genetic clusters detected by STRUCTURE analysis based on haplotypes at nuclear DNA loci in Rhododendron weyrichii populations. (a) Small thin letters indicate population codes corresponding to those in Table 1; numbers in pie charts indicate the numbers of tri‐nucleotide repeats; a parsimony network superimposed on the map shows the relationships among the haplotypes using Rhododendron amagianum as an out‐group. (b) and (c) A neighbor‐joining tree superimposed on each map shows the relationships among the clusters based on net nucleotide distances; FST of each cluster indicates the extent of genetic divergence from the ancestral population
The configuration of the landscape in these island regions is believed to have affected the distribution and demographic history of plant populations. In this study, we examined the demographic history of R. weyrichii populations using statistical phylogeographic approaches based on chloroplast and nuclear DNA (nDNA) sequences. We also examined the distribution of the species during the LGM using ecological niche modeling. The specific aims of the study were (1) to estimate population genetic divergence between continental islands and to address the isolation–migration histories of these populations; (2) to evaluate differences in genetic diversity and historical changes in population size among islands; and (3) to make inferences about population survival on each island during the LGM.
2. Materials and Methods
2.1. Plant material
Leaf samples were collected from 142 R. weyrichii individuals from 18 populations on eight continental islands (Figure 1, Table 1). Thirty‐two individuals from four populations of Rhododendron sanctum and one individual of Rhododendron amagianum were used as out‐groups in, respectively, nDNA and cpDNA analyses. Voucher specimens from each population were deposited in the National Museum of Nature and Science, Japan (TNS).
Table 1.
Locality and the numbers of individuals used in chloroplast and nuclear DNA analyses and genetic diversity and neutrality statistics over eight nuclear loci, for 18 populations from eight continental islands
| Continental island | Population code and locality | Coordinate | n c | n n | π | θW | D | |
|---|---|---|---|---|---|---|---|---|
| Jeju Island | JJ1 | Eorimok, Jeju, South Korea | 33.3883N/126.4954E | 4 | 16 | 0.0042 | 0.0032 | 0.656 |
| JJ2 | Saryoni, Jeju, South Korea | 33.4180N/126.6353E | 4 | 16 | 0.0036 | 0.0037 | −0.262 | |
| Fukue Island | FK | Mt. Sasa, Nagasaki, Japan | 32.7216N/128.8099E | 4 | 16 | 0.0042 | 0.0039 | 0.092 |
| Amakusa Island | AM | Mt. Kado, Kumamoto, Japan | 32.3997N/130.0965E | 4 | 16 | 0.0041 | 0.0046 | −0.544 |
| Koshiki Island | KS | Segami, Kagoshima, Japan | 31.8618N/129.8698E | 4 | 16 | 0.0043 | 0.0040 | 0.156 |
| Kyushu Island | KY1 | Mt. Kumagadake, Kagoshima, Japan | 31.4551N/130.4672E | 4 | 16 | 0.0046 | 0.0046 | 0.062 |
| KY2 | Mt. Boroishi, Miyazaki, Japan | 31.8039N/131.3800E | 4 | 16 | 0.0045 | 0.0052 | −0.439 | |
| KY3 | Mt. Kanmuri, Miyazaki, Japan | 32.3849N/131.5385E | 4 | 16 | 0.0042 | 0.0047 | −0.370 | |
| KY4 | Kitaura, Miyazaki, Japan | 32.7336N/131.8325E | 4 | 16 | 0.0054 | 0.0057 | −0.362 | |
| Shikoku Island | SK1 | Kawabe, Ehime, Japan | 33.5020N/132.7661E | 4 | 14 | 0.0035 | 0.0041 | −0.387 |
| SK2 | Mt. Okubo, Kochi, Japan | 33.1855N/132.6120E | 4 | 16 | 0.0042 | 0.0043 | −0.166 | |
| SK3 | Mt. Imano, Kochi, Japan | 32.8590N/132.8537E | 4 | 16 | 0.0024 | 0.0027 | −0.271 | |
| SK4 | Reihoku, Kochi, Japan | 33.7021N/133.6039E | 4 | 16 | 0.0036 | 0.0034 | 0.062 | |
| SK5 | Mt. Syozanji, Tokushima, Japan | 33.9842N/134.3040E | 4 | 16 | 0.0023 | 0.0029 | −0.571 | |
| Awaji Island | AW | Mt. Yuzuriha, Hyogo, Japan | 34.2435N/134.8358E | 4 | 16 | 0.0037 | 0.0037 | −0.076 |
| Kii Peninsula of | KI1 | Mt. Kasane, Wakayama, Japan | 33.5218N/135.8042E | 4 | 16 | 0.0038 | 0.0045 | −0.475 |
| Honshu Island | KI2 | Siro, Mie, Japan | 33.9644N/136.1933E | 4 | 14 | 0.0036 | 0.0044 | −0.321 |
| KI3 | Nisaka, Mie, Japan | 34.2359N/136.3587E | 4 | 16 | 0.0044 | 0.0046 | −0.040 | |
n c and n n, number of alleles used in chloroplast and nuclear DNA analyses, respectively; π, nucleotide diversity; θW, Watterson's θ per site; D, Tajima's D.
Population codes correspond to those in Figure 1.
2.2. DNA extraction and sequencing
Genomic DNA was extracted from leaf samples using the cetyltrimethylammonium bromide method (Murray & Thompson, 1980), after treatment with sorbitol extraction buffer (Wagner et al., 1987). Two cpDNA and eight nDNA regions were sequenced in order to detect DNA polymorphisms. Two cpDNA loci (trnG intron and rpl36‐rps8) were PCR‐amplified from four individuals per population using universal primers (Kress, Wurdack, Zimmer, Weigt, & Janzen, 2005; Shaw et al., 2005). For the eight nDNA loci, primers developed for other Ericaceous species (C16 and C22 [Wei, Fu, & Arora, 2005], EST39, EST65, EST121, and EST136 [De Keyser, De Riek, & Van Bockstaele, 2009], PHYB and PHYE [Ikeda & Setoguchi, 2010]) were used for PCR amplification of seven or eight individuals per population (Table S1). PCR was performed with an initial denaturation for 4 min at 94°C followed by 35 cycles of denaturation for 60 s at 94°C, annealing for 60 s at 55 or 60°C and extension for 60 s at 72°C, and a final extension for 7 min at 72°C, using AmpliTaq Gold Master Mix (Applied Biosystems). After PEG precipitation (Hartley & Bowen, 1996), the PCR products were sequenced directly using the standard method provided with the BigDye Terminator Cycle Sequencing kit v. 3.1 (Applied Biosystems) and separated by electrophoresis on an ABI 3100 Genetic Analyzer (Applied Biosystems).
2.3. Data analysis of chloroplast DNA sequences
The cpDNA sequences acquired were assembled using DNA Baser v. 3 (Heracle BioSoft S.R.L.) and aligned using the Muscle algorithm implemented in MEGA v. 5 (Edgar, 2004; Tamura et al., 2011). Mononucleotide repeats in the sequences were omitted from subsequent analyses to avoid the possibility of homoplasy. The number of tri‐nucleotide repeats ([ATT]n) found in rpl36‐rps8 was counted, but these repeats were excluded from alignments when determining cpDNA haplotypes. Genealogical relationships for all cpDNA haplotypes were constructed as a parsimony network using TCS v. 1.06 (Clement, Posada, & Crandall, 2000).
2.4. Data analysis of nuclear DNA sequences
2.4.1. Estimation of genetic diversity and genetic structure
The nDNA sequences acquired were also assembled using DNA Baser and aligned using Muscle in MEGA; indel polymorphisms in these sequences were omitted from subsequent analyses. The haplotype phases of the aligned sequences were determined using PHASE v. 2.1 (Stephens & Scheet, 2005; Stephens, Smith, & Donnelly, 2001) implemented in DnaSP v. 5 (Librado & Rozas, 2009). Three independent runs employing the general model for recombination rate variation were conducted with a burn‐in period of 1,000 followed by 10,000 iterations with a thinning interval of 100 steps. Those haplotypes with a posterior probability of >0.90 were used in subsequent analyses. Homology search using blastx was conducted against polypeptide sequences from the NCBI and Arabidopsis thaliana (TAIR; http://www.arabidopsis.org) databases to identify a possible function for the product of each locus.
Number of segregating sites (S) and nucleotide diversity (π; Nei, 1987) were calculated for each locus. The minimum number of recombinations (R M) at each locus was estimated based on the four‐gamete test (Hudson & Kaplan, 1985). Neutrality at each locus was tested using Tajima's D (Tajima, 1989) and Fay and Wu's H (Fay & Wu, 2000). Fay and Wu's H was tested with out‐group (R. sanctum) sequences, and significance was determined using 10,000 coalescent simulations. In addition, an HKA test (Hudson, Kreitman, & Aguadé, 1987) was conducted to test for neutral evolution across loci between species (R. weyrichii and R. sanctum). All of these analyses were performed using DnaSP, with the exception of the HKA test which was carried out using the HKA program (http://genfaculty.rutgers.edu/hey/software#HKA). Within‐population genetic diversity was evaluated for each population. Nucleotide diversity, Watterson's θ (Watterson, 1975), and Tajima's D were calculated over all loci within each population using DnaSP.™
Population genetic structure was then estimated as follows. The average number of nucleotide differences (D xy; Nei, 1987) over all loci between populations was calculated. Genealogical relationships among all haplotypes at each locus were estimated by constructing a parsimony network using TCS. Population genetic structure based on genotypes (two haplotypes) at each locus for each individual was estimated by model‐based Bayesian clustering analysis using STRUCTURE v. 2.3.3 (Pritchard, Stephens, & Donnelly, 2000). Admixture, correlated allele frequency and LOCPRIOR models were used (Falush, Stephens, & Pritchard, 2003; Hubisz, Falush, Stephens, & Pritchard, 2009). MCMC simulations were performed with 20 independent runs for each cluster (K = 1–10), with 3.0 × 104 iterations after a burn‐in period of 2.0 × 104. The optimal value of K was estimated by plotting the log likelihood of the data, Ln Pr(X|K) (Pritchard et al., 2000), and the ΔK statistic, which was calculated from the second‐order rates of changes of Ln Pr(X|K) (Evanno, Regnaut, & Goudet, 2005), against K. A neighbor‐joining among‐clusters tree was constructed based on the net nucleotide distances between pairs of clusters, and F ST values between the ancestral population and each cluster, which represent the extent of genetic drift undergone by each cluster after differentiation from the ancestral population, were calculated (Pritchard et al., 2000).
2.4.2. Demographic analysis
Demographic history was firstly inferred using the isolation with migration (IM) model. To estimate the IM history among four major regional population groups (Jeju, Kyushu, Shikoku, and Kii), the population demographic parameters (Figure 3) population split time (t = Tμ), which is a product of the absolute time in years (T) and the mutation rate (μ), population size (θ = 4Nμ, where N is effective population size), migration rate per mutation event (m = M/μ, where M is migration rate), and population migration rate (2NM = 0.5θm) were estimated using IMa2 (Hey & Nielsen, 2004, 2007; see Appendix S1).
Figure 3.
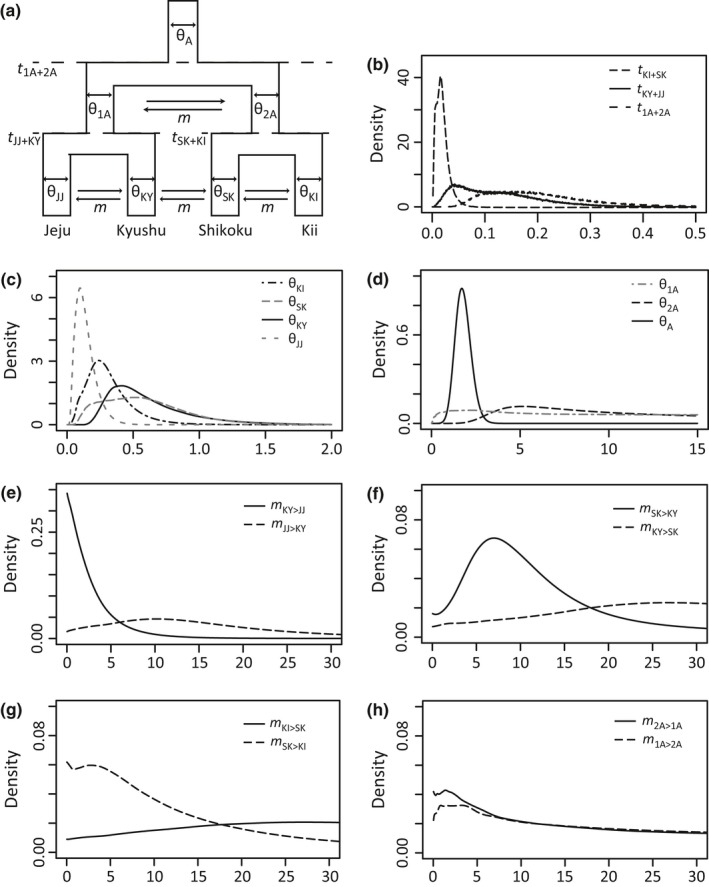
(a) An isolation with migration (IM) model for the four island populations examined in this study (above left), and the marginal distribution of posterior probabilities for (b) the split time (t), (c, d) population size (θ), and (e–h) migration rate between populations (m)
The approximate Bayesian computation (ABC) approach enables us to infer demographic history by estimating the values of parameters in a complex model without calculating its likelihood (Beaumont, 2010; Csillery, Blum, Gaggiotti, & Francois, 2010). In this study, four different demographic models related to changes in population size (Fig. S3), a standard neutral model (Model 1), an exponential growth model (Model 2), an instantaneous size reduction model (Model 3), and an exponential growth after instantaneous size reduction model (Model 2 + Model 3, Model 4) were examined for each of the four island populations using R v. 3.2.1, ms and msABC (Hudson, 2002; Pavlidis, Laurent, & Stephan, 2010; R Core Team, 2015; see Appendix S1).
2.5. Ecological niche modeling
To infer changes in distribution between the LGM and the present, we produced distribution models for R. weyrichii based on modern distribution records and bioclimatic variables using maximum entropy methods (Phillips et al., 2006). Species occurrence data were obtained from the 5th Specific Plant Community Survey (http://www.biodic.go.jp/reports2/5th/vgt_toku/index.html), specimens in two herbaria (the Herbarium of the University of Tokyo and the National Science Museum, Tsukuba), and personal observations. In total, 172 presence records for R. weyrichii were obtained covering the entire range of the species, after removal of duplicate records from within each 2.5‐arc‐minutes cell. Current climate data at a resolution of 2.5 arc‐minutes were obtained from WorldClim (Hijmans, Cameron, Parra, Jones, & Jarvis, 2005). Validation of the distribution model was performed, using default settings with 100 replicates of cross‐validation procedures, with 25% of the data used for model testing, implemented in Maxent v. 3.3.3e (Phillips et al., 2006). Five bioclimatic variables (annual mean temperature, mean temperature in the driest quarter, annual precipitation, precipitation in the wettest quarter, and precipitation in the driest quarter), which showed high values of area under curve (AUC) and are critically important for temperate species that are distributed in humid and warm environments, were chosen for inclusion in the distribution model, as they have been used in ecological niche modeling for other species with similar distribution ranges (Qi, Yuan, Comes, Sakaguchi, & Qiu, 2014; Worth et al., 2013).
The model calibrated with current climate conditions was projected onto the reconstructed climate during the LGM obtained from two climatic simulations, the Community Climate System Model (CCSM; Collins et al., 2006) and the Model for Interdisciplinary Research on Climate (MIROC; Hasumi & Emori, 2004), obtained from the PMIP2 website (http://pmip2.lsce.ipsl.fr/). We prepared the LGM palaeoclimate layers at a resolution of 2.5 arc‐minutes following the methods of Sakaguchi et al. (2012), and the palaeocoastlines were estimated as being −130 m lower than at present on the basis of seafloor topology data (ETOPO1; http://www.ngdc.noaa.gov/mgg/global/).
3. Results
3.1. Genetic diversity and structure of chloroplast DNA
The sequences of two cpDNA loci were obtained from 72 individuals across 18 populations of R. weyrichii (Table 1). The lengths of aligned sequences for the two loci were 557 bp in trnG intron and 455 bp in rpl36‐rps8. There were three haplotypes with three segregating sites across the two loci (Figure 1a). Tri‐nucleotide repeats (ATT) that were identified in a rpl36‐rps8 intergenic spacer showed a high level of variation in Kyushu. The geographic distribution of haplotypes showed a clear division between Shikoku and Kyushu, and a distinct haplotype was recognized in a population from Kii.
3.2. Genetic diversity and neutrality of nuclear DNA
The sequences of eight nDNA loci were obtained from 142 individuals across 18 populations (Table 1). The length of aligned sequences for each locus ranged from 339 bp for PHYE to 470 bp for EST136 (Table 2), and the total length was 3,340 bp. The eight nDNA loci showed a high degree of polymorphism, with the number of segregating sites (S) ranging from 9 to 25 and the nucleotide diversity for all sites (π) ranging from 0.0018 to 0.0060. Recombination events were recognized for seven loci, all except PHYE, and the number of minimum recombination events (R M) ranged from 0 to 9 (Table 2). In the two neutrality tests, which examine intraspecific variation (Tajima's D test) and interspecific (i.e., between R. weyrichii and R. sanctum) variation (Fay and Wu's H test), neutral evolution of every locus was not rejected (Table 2). In addition, the HKA test did not reject neutrality for the divergence between R. weyrichii and R. sanctum (χ2 = 15.043, p = .375). The π, Watterson's θ (θW), and D values for each population ranged from 0.0024 to 0.0054, 0.0027 to 0.0057, and −0.571 to 0.656, respectively (Table 1).
Table 2.
Genetic diversity and neutrality statistics over eight nuclear DNA loci across all the sampled populations
| Locus | Alignment length (bp) | Largest non‐recombining block (bp) | N | S | h | R M | π | D | H |
|---|---|---|---|---|---|---|---|---|---|
| EST39 | 461 | 394 | 278 | 25 | 55 | 9 | 0.0060 | −0.984 | 1.107 |
| EST65 | 387 | 382 | 276 | 18 | 38 | 4 | 0.0027 | −0.415 | 0.289 |
| EST121 | 387 | 380 | 270 | 21 | 28 | 3 | 0.0052 | −1.085 | −2.202 |
| EST136 | 470 | 431 | 272 | 24 | 55 | 5 | 0.0043 | −1.353 | −0.514 |
| C16 | 407 | 389 | 246 | 24 | 30 | 3 | 0.0053 | −1.286 | 1.765 |
| C22 | 425 | 388 | 286 | 9 | 13 | 2 | 0.0037 | 0.202 | −1.573 |
| PHYB | 464 | 464 | 274 | 12 | 18 | 2 | 0.0018 | −1.462 | 0.741 |
| PHYE | 339 | 339 | 286 | 9 | 9 | 0 | 0.0039 | −0.198 | −0.858 |
N, number of alleles sequenced; S, number of segregating sites; h, number of phased haplotypes detected; R M, minimum number of recombination events; π, nucleotide diversity; D, Tajima's D; H, Fay and Wu's H.
Tajima's D and Fay and Wu's H were not significant at any locus.
3.3. Population genetic structure of nuclear DNA
The pairwise genetic divergence (the average number of nucleotide differences, D xy) between populations ranged from 0.0031 to 0.0069 (Table S2). Each nuclear locus had major haplotypes, and variable patterns of haplotype distribution were detected among loci (Figure 2). In the Bayesian clustering analysis, K = 2 was the optimal number of clusters supported by the ΔK statistics, and K = 4 was the optimal number of clusters that showed a plateau in the log‐likelihood value and low variance among runs (Fig. S1). In the case of K = 2, the distribution of clusters showed a division between Kyushu and Shikoku (Figure 1b). In the case of K = 4, clusters 1 and 2 were recognized in the populations of the Japanese Archipelago, cluster 3 was recognized mainly in Kyushu and the adjacent islands (Koshiki, Amakusa, and Fukue), and cluster 4 predominated in Jeju and was also recognized in Fukue (Figure 1c). The F ST values for clusters 2 and 4 showed higher values than those for clusters 1 and 3; in addition, clusters 2 and 4 were distant from clusters 1 and 3. Clear genetic divergence between Jeju–Kyushu and Shikoku–Kii was also supported by the phylogenetic relationships between island populations (Fig. S2).
Figure 2.
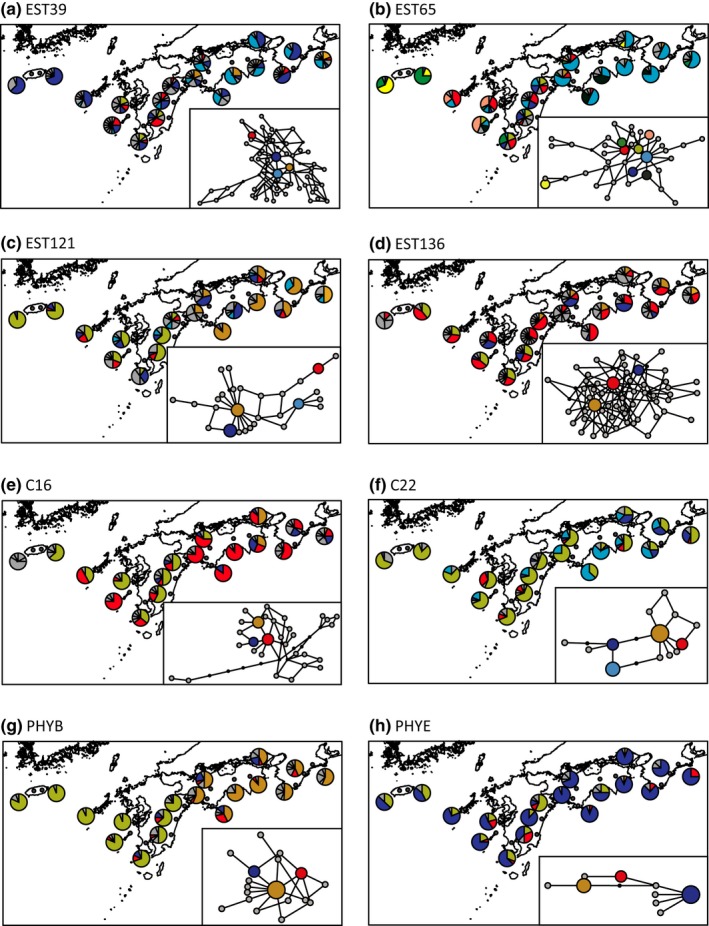
Geographic distributions of haplotypes at nine nuclear loci: (a) EST39, (b) EST65, (c)EST121, (d) EST136, (e) C16, (f) C22, (g) PHYB, and (h) PHYE. A parsimony network superimposed on each map shows the relationships among haplotypes at each locus
3.4. Demographic history of four island populations
The posterior modes and 95% highest posterior density (HPD) intervals for the split time (t) between Jeju and Kyushu (t JJ+KY), between Shikoku and Kii (t SK+KI), and between Jeju–Kyushu and Shikoku–Kii (t 1A+2A) estimated by the IM model were 0.043 (0.008–0.276), 0.006 (0.001–0.041), and 0.151 (0.051–0.384), and the corresponding estimates of the divergence time (T) between these islands were 69 kya (13–443), 9 kya (2–65), and 243 kya (81–616), respectively (Figure 3, Table S3). The nested models, which hypothesized zero migration (i.e., the migration rates [m] and population migrations [2NM] were set on the assumptions of a zero migration hypothesis [m and 2NM = 0]), were not rejected by likelihood ratio tests, with the exceptions of migration from Shikoku to Kyushu and from Kyushu to Jeju in the sense of time forward.
In the ABC analysis of each island population, the standard neutral model (Model 1) was supported for Kii and Kyushu, whereas the exponential growth model (Model 2) was supported for Shikoku and the instantaneous size reduction model (Model 3) was supported in the case of Jeju (Table 3, Figs S3, S4) with good agreement between the posterior predictive simulations for the observed and simulated data sets (Fig. S5). It was estimated that expansion or maintenance of population size in Kii, Shikoku, and Kyushu continued over the last glacial period (Figure 4). The Jeju population exhibited a significantly lower value for population size (mode of θ0 [95% HPD] = 0.136 [0.000–0.673]) than the other populations (Figure 4, Table S4). In addition, a bottleneck event in Jeju was estimated to have happened before the last glacial (467 kya [184–1,026]), and the effective population size has been low since then. When comparing the results of IM and ABC analyses, although the values of θ0 for each island in the ABC analysis showed higher values than those in IM analysis, the relative relationship of θ0 among islands was the same in both analyses, including the finding that the lowest value was that for Jeju.
Table 3.
Posterior probabilities of four demographic models for the populations on four continental islands
| Population | Demographic modela | |||
|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | Model 4 | |
| Jeju | 0.000 | 0.000 | 0.817 | 0.183 |
| Kyushu | 0.547 | 0.158 | 0.145 | 0.150 |
| Shikoku | 0.112 | 0.550 | 0.025 | 0.313 |
| Kii | 0.488 | 0.188 | 0.149 | 0.176 |
The models accepted are shown in boldface.
Model 1, standard neutral model; Model 2, exponential growth model; Model 3, instantaneous size reduction model; Model 4, exponential growth after instantaneous size reduction model.
Figure 4.
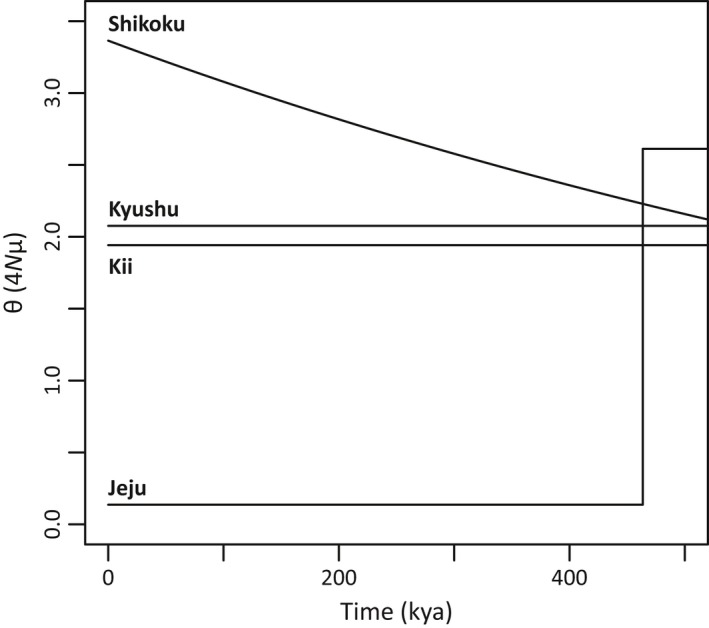
Historical changes in population size (θ) for the populations on four continental islands over time. θ = 4Nμ, where N is effective population size and μ is mutation rate. Time is on a scale of thousands of years ago, kya
3.5. Differences in species distribution between the last glacial maximum and the present time
The Maxent model showed a relatively high accuracy (area under curve, AUC = 0.940 ± 0.009). The predicted distribution based on current climatic conditions was similar to the actual distribution of the species with high probability (>0.50), although some areas along the Pacific Ocean side and on the western edge of Honshu were also predicted to be suitable (Figure 5a). The contribution made by environmental variables to the model prediction was higher for precipitation (annual precipitation = 71.8%, precipitation in the driest month = 12.0%) than for temperature (annual mean temperature = 3.8%, mean temperature in the driest season = 6.0%). Under the two LGM climate simulations (CCSM and MIROC), it was estimated that suitable environments had been maintained on three of the four islands (Figure 5b, c), but the areas on the map with high probabilities (>50%) had shrunk. In addition, the area with a suitable environment on Jeju showed low probabilities and restricted distributions in both models. It is noteworthy that although the exposed seafloor physically connected the major islands during the LGM, the ranges suitable for the species were somewhat scattered between neighboring islands.
Figure 5.
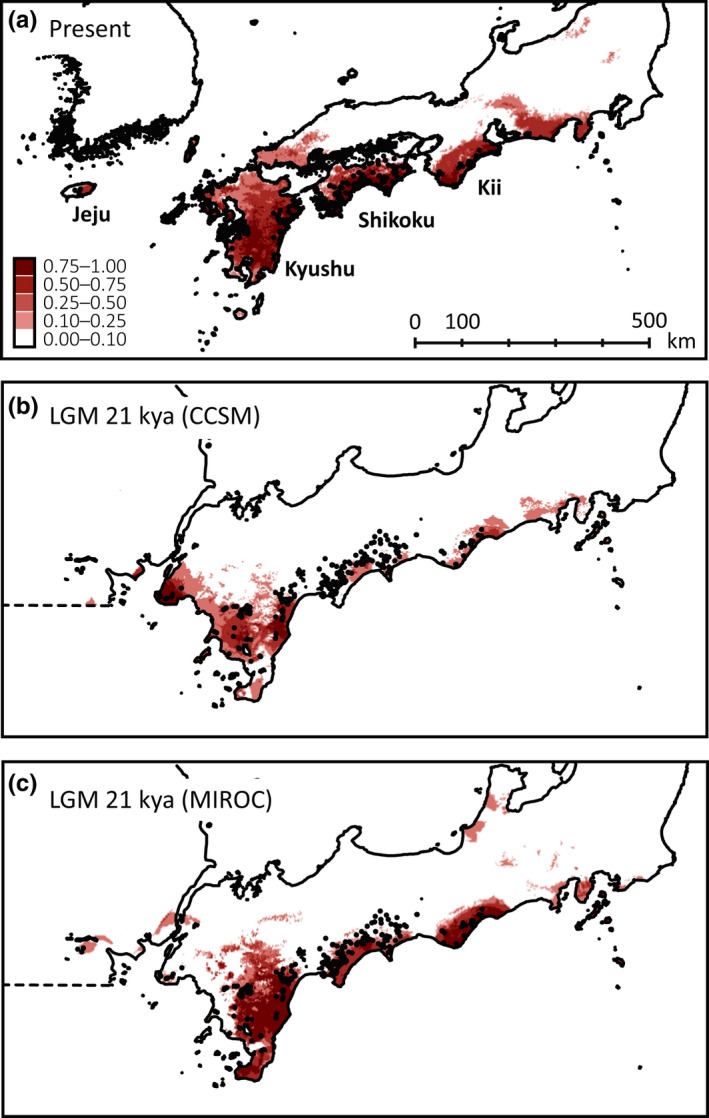
Predicted distribution of Rhododendron weyrichii (a) at present, and at the last glacial maximum (LGM, 21 thousand years ago) under two climatic models (b) the Community Climate System Model, CCSM, and (c) the Model for Interdisciplinary Research on Climate, MIROC. The range of red color intensity indicates the logistic distribution probability, and locations of the present distribution are plotted as black points on the maps
4. Discussion
4.1. Isolation and migration among island populations during the late Quaternary
The estimates of the times of divergence between R. weyrichii populations on these four islands indicate that timing and duration of population divergence is different. In particular, the clear genetic divergence between Jeju–Kyushu and Shikoku–Kii was supported by the genetic structure given by both nuclear and chloroplast DNA sequences, and the time estimate suggests that the vicariance may have continued since the last glacial or have predated the last glacial (Petit et al., 1999). While, the most recent divergence between Shikoku and Kii may indicate a role of exposed seafloor for migration corridor. However, because time estimates are seriously influenced by mutation rate, and by variations in mutation rate among sites depending, for example, on whether substitutions are synonymous or nonsynonymous (Wolf, Li, & Sharp, 1987), careful consideration is necessary when interpreting population history.
In addition, the results of a likelihood ratio test of the IM analysis suggest that a few migration events among island populations happened after their divergence. The distribution of genetic clusters detected by STRUCTURE analysis indicates that Fukue, which is located midway between Jeju and Kyushu, may have been a region of land bridges that were used like stepping stones. These evidences may mean that the exposed seafloors during the last glacial have acted as both effective and ineffective migration corridors between islands (Siddall et al., 2003).
4.2. Differences in population survival among islands
The three island populations in the Japanese Archipelago (those on Kyushu, Shikoku, and Kii) showed large population sizes (θ0) than those on Jeju according to ABC analysis; the pattern was similar for the result of IM analysis. Palynological studies have suggested that warm‐temperate forests may have retreated to southern refugia and been replaced by cold‐tolerant coniferous and broadleaf vegetation throughout most of the Japanese Archipelago during the LGM (Gotanda & Yasuda, 2008). In addition, previous genetic studies on many warm‐temperate species have indicated that southwestern parts of the Japanese Archipelago provided important refugia (Aoki et al., 2004; Zhai, Comes, Nakamura, Yan, & Qiu, 2012). These results suggest that climate oscillations had negligible impacts at the southern tips of the merged “Japanese island” at that time, and the populations may therefore have been maintained in situ in each region throughout at least one cycle of climate change (Clark et al., 2009). Another minor factor that affects genetic diversity and population size of certain species is introgression from related species. A chloroplast haplotype unique among R. weyrichii was recognized in the north of Kii. This haplotype was shared with R. kiyosumense Makino (Yoichi unpublished data), which is distributed on the Pacific Ocean side of Honshu. An intraspecific taxon with a different flower color, which is recognized as R. weyrichii f. purpureum Hara, in the northern part of Kii is considered to have resulted from introgression events from the other species, as has also occurred in other closely related Rhododendron species (Morimoto et al., 2005; Tagane, Hiramatsu, & Okubo, 2008).
In contrast to the populations in the Japanese Archipelago, the estimated value of effective population size on Jeju was low, and this is likely to have been caused not by a recent bottleneck event. In addition, the effective population size on Jeju has not expanded after the bottleneck event. Although the time estimates given by IM and ABC analyses exhibited different values, this difference may be the result of an estimation error due to difference in examined models and model simplification. At least the accepted model and the time estimate suggested that the Jeju population is not likely to have been formed by recent migration from the Japanese island. The Jeju population is currently restricted along rivers in warm temperate forests characterized by trees of Carpinus or Quercus whereas fossil pollen records suggest that the island has been dominated by grassland with patches of cool‐temperate deciduous broadleaved woodland and that conditions were dry during the LGM (Chung, 2007; Lee, Lee, Yoon, & Yoo, 2008). In addition, ecological niche modeling predicted that relatively small areas of the Jeju region were suitable, and the probability values for the distribution were not high. These lines of evidences indicated that Jeju was not suitable for R. weyrichii; however, Bayesian clustering and demographic analyses suggested that R. weyrichii may have survived in restricted and scattered habitats on or around the island during the LGM. The possibility of the invisible refugia is supported by the high genetic diversity of tree species on the island (Chung et al., 2013; Lee, Lee, & Choi, 2013; Lee, Lee, Choi, & Choi, 2014). These regional differences in population size between the islands suggest that environmental sustainability is a key factor in maintaining a population under conditions in which there are few migrations among populations (Frankham, 2005; Morjan & Rieseberg, 2004).
4.3. Survivals of island populations from the last glacial maximum up to the present
When considering isolation and migration among islands, the LGM is an important period in history (Hewitt, 2000, 2004). Under both the CCSM and the MIROC simulation, suitable areas were predicted on each of the four islands, but these areas appeared to lack spatial connections across the exposed seafloor. The existence of at least possible two regions on the islands containing refugia during the LGM, Kii–Shikoku and Kyushu–Jeju, was also supported by IM analysis. Thus, the latitudinal and/or longitudinal range of the species may not have changed greatly since the LGM as is also the case for other forest species on continental islands (Duncan et al., 2016; Qi et al., 2014; Qiu et al., 2009; Worth et al., 2013), and the current distribution may not have been shaped by northward range expansion such as has occurred for widespread species (Fujii et al., 2002; Magri et al., 2006; Sakaguchi, Takeuchi, Yamasaki, Sakurai, & Isagi, 2011).
5. Conclusion
Statistical phylogeographic analyses of R. weyrichii populations using model‐based approaches revealed that history of population isolation between islands is likely to have a considerable influence on population survival on each island, even if the islands were sometimes connected geographically during climatic oscillations (Qi et al., 2014). Especially, differences in survival history due to the suitability of isolated habitats are reflected in the current effective population size on each island. The genetic structure of long‐lived woody species would certainly have been influenced by environments during the last glacial.
Conflict of Interest
None declared.
Supporting information
Acknowledgments
We are grateful to T. Isogimi, K. Hayashi, N. Kawashima, K. Higuchi, and K. Kurokawa for their help in collecting plant materials.
Yoichi, W. , Tamaki, I. , Sakaguchi, S. , Song, J.‐S. , Yamamoto, S.‐i. and Tomaru, N. (2016), Population demographic history of a temperate shrub, Rhododendron weyrichii (Ericaceae), on continental islands of Japan and South Korea. Ecology and Evolution, 6: 8800–8810. doi: 10.1002/ece3.2576
References
- Aoki, K. , Suzuki, T. , Hsu, T.‐W. , & Murakami, N. (2004). Phylogeography of the component species of broad‐leaved evergreen forests in Japan, based on chloroplast DNA variation. Journal of Plant Research, 117, 77–94. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A. (2010). Approximate Bayesian computation in evolution and ecology. Annual Review of Ecology, Evolution and Systematics, 41, 379–406. [Google Scholar]
- Bittkau, C. , & Comes, H. P. (2005). Evolutionary processes in a continental island system: Molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Molecular Ecology, 14, 4065–4083. [DOI] [PubMed] [Google Scholar]
- Burridge, C. P. , Brown, W. E. , Wadley, J. , Nankervis, D. L. , Olivier, L. , Gardner, M. G. , … Austin, J. J. (2013). Did postglacial sea‐level changes initiate the evolutionary divergence of a Tasmanian endemic raptor from its mainland relative? Proceedings of the Royal Society of London B: Biological Sciences, 280, 20132448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, D. F. , & Rae, S. J. (1990). A revision of Rhododendron IV subgenus Tsutsusi . Edinburgh Journal of Botany, 47, 89–200. [Google Scholar]
- Chung, C.‐H. (2007). Vegetation response to climate change on Jeju Island, South Korea, during the last deglaciation based on pollen record. Geosciences Journal, 11, 147–155. [Google Scholar]
- Chung, M. Y. , Moon, M. O. , López‐Pujol, J. , Maki, M. , Yamashiro, T. , Yukawa, T. , … Chung, M. G. (2013). Was Jeju Island a glacial refugium for East Asian warm‐temperate plants? Insights from the homosporous fern Selliguea hastata (Polypodiaceae). American Journal of Botany, 100, 2240–2249. [DOI] [PubMed] [Google Scholar]
- Clark, P. U. , Dyke, A. S. , Shakun, J. D. , Carlson, A. E. , Clark, J. , Wohlfarth, B. , … McCabe, A. M. (2009). The last glacial maximum. Science, 325, 710–714. [DOI] [PubMed] [Google Scholar]
- Clement, M. , Posada, D. , & Crandall, K. A. (2000). TCS: A computer program to estimate gene genealogies. Molecular Ecology, 9, 1657–1659. [DOI] [PubMed] [Google Scholar]
- Collins, W. D. , Bitz, C. M. , Blackmon, M. L. , Bonan, G. B. , Bretherton, C. S. , Carton, J. A. , … Smith, R. D. (2006). The community climate system model version 3 (CCSM3). Journal of Climate, 19, 2122–2143. [Google Scholar]
- Csillery, K. , Blum, M. G. B. , Gaggiotti, O. E. , & Francois, O. (2010). Approximate Bayesian computation in practice. Trends in Ecology & Evolution, 25, 410–418. [DOI] [PubMed] [Google Scholar]
- De Keyser, E. , De Riek, J. , & Van Bockstaele, E. (2009). Discovery of species‐wide EST‐derived markers in Rhododendron by intron‐flanking primer design. Molecular Breeding, 23, 171–178. [Google Scholar]
- Duncan, C. J. , Worth, J. R. P. , Jordan, G. J. , Jones, R. C. , & Vaillancourt, R. E. (2016). Genetic differentiation in spite of high gene flow in the dominant rainforest tree of southeastern Australia, Nothofagus cunninghamii . Heredity, 116, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C. , & Wu, C. I. (2000). Hitchhiking under positive Darwinian selection. Genetics, 155, 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham, R. (2005). Genetics and extinction. Biological Conservation, 126, 131–140. [Google Scholar]
- Fujii, N. , Tomaru, N. , Okuyama, K. , Koike, T. , Mikami, T. , & Ueda, K. (2002). Chloroplast DNA phylogeography of Fagus crenata (Fagaceae) in Japan. Plant Systematics and Evolution, 232, 21–33. [Google Scholar]
- Gotanda, K. , & Yasuda, Y. (2008). Spatial biome changes in southwestern Japan since the Last Glacial Maximum. Quaternary International, 184, 84–93. [Google Scholar]
- Harrison, S. P. , Yu, G. , Takahara, H. , & Prentice, I. C. (2001). Diversity of temperate plants in east Asia. Nature, 413, 129–130. [DOI] [PubMed] [Google Scholar]
- Hasumi, H. , & Emori, S. (2004). K‐1 coupled GCM (MIROC) description. Center for Climate System Research, University of Tokyo, National Institute for Environmental Studies, Frontier Research Center for Global Change, Tokyo, Japan. 34. [Google Scholar]
- Hartley JL, Bowen H (1996) PEG precipitation for selective removal of small DNA fragments. Focus, 18, 27 [Google Scholar]
- Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature, 405, 907–913. [DOI] [PubMed] [Google Scholar]
- Hewitt, G. M. (2004). Genetic consequences of climatic oscillations in the Quaternary. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 359, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J. , & Nielsen, R. (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis . Genetics, 167, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J. , & Nielsen, R. (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences of the United States of America, 104, 2785–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25, 1965–1978. [Google Scholar]
- Hubisz, M. J. , Falush, D. , Stephens, M. , & Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources, 9, 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R. (2002). Generating samples under a Wright‐Fisher neutral model. Bioinformatics, 18, 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R. , & Kaplan, N. L. (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics, 111, 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R. , Kreitman, M. , & Aguadé, M. (1987). A test of neutral molecular evolution based on nucleotide data. Genetics, 116, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, H. , & Setoguchi, H. (2010). Natural selection on PHYE by latitude in the Japanese archipelago: Insight from locus specific phylogeographic structure in Arcterica nana (Ericaceae). Molecular Ecology, 19, 2779–2791. [DOI] [PubMed] [Google Scholar]
- Iwasaki, T. , Aoki, K. , Seo, A. , & Murakami, N. (2012). Comparative phylogeography of four component species of deciduous broad‐leaved forests in Japan based on chloroplast DNA variation. Journal of Plant Research, 125, 207–221. [DOI] [PubMed] [Google Scholar]
- Kress, W. J. , Wurdack, K. J. , Zimmer, E. A. , Weigt, L. A. , & Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences of the United States of America, 102, 8369–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.‐H. , Lee, D.‐H. , & Choi, B.‐H. (2013). Phylogeography and genetic diversity of East Asian Neolitsea sericea (Lauraceae) based on variations in chloroplast DNA sequences. Journal of Plant Research, 126, 193–202. [DOI] [PubMed] [Google Scholar]
- Lee, J.‐H. , Lee, D.‐H. , Choi, I.‐S. , & Choi, B.‐H. (2014). Genetic diversity and historical migration patterns of an endemic evergreen oak, Quercus acuta, across Korea and Japan, inferred from nuclear microsatellites. Plant Systematics and Evolution, 300, 1913–1923. [Google Scholar]
- Lee, S. H. , Lee, Y. I. , Yoon, H. I. , & Yoo, K. C. (2008). East Asian monsoon variation and climate changes in Jeju Island, Korea, during the latest Pleistocene to early Holocene. Quaternary Research, 70, 265–274. [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lisiecki, L. E. , & Raymo, M. E. (2005). A Pliocene‐Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography, 20, PA1003. [Google Scholar]
- MacArthur, R. H. , & Wilson, E. O. (1967). The theory of island biogeography. Princeton, NJ: Princeton University Press. [Google Scholar]
- Magri, D. , Vendramin, G. G. , Comps, B. , Dupanloup, I. , Geburek, T. , Gömöry, D. , … Beaulieu, J‐L . (2006). A new scenario for the quaternary history of European beach populations: Palaeobotanical evidence and genetic consequences. New Phytologist, 171, 199–221. [DOI] [PubMed] [Google Scholar]
- Morimoto, J. , Kamichi, T. , Mizumoto, I. , Hasegawa, S. , Nomura, M. , & Kobayashi, T. (2005). Natural hybridization of Japanese Rhododendron section Brachycaryx in Mount Kintoki in eastern Japan and concerns for genetic diversity in restoring their habitat. Landscape and Ecological Engineering, 1, 149–156. [Google Scholar]
- Morjan, C. L. , & Rieseberg, L. H. (2004). How species evolve collectively: Implications of gene flow and selection for the spread of advantageous alleles. Molecular Ecology, 13, 1341–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M. , & Thompson, W. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research, 8, 4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, K. , Suwa, R. , Denda, T. , & Yokota, M. (2009). Geohistorical and current environmental influences on floristic differentiation in the Ryukyu Archipelago, Japan. Journal of Biogeography, 36, 919–928. [Google Scholar]
- Nei, M. (1987). Molecular evolutionary genetics. New York, NY: Columbia University Press. [Google Scholar]
- Nielsen, R. , & Beaumont, M. A. (2009). Statistical inferences in phylogeography. Molecular Ecology, 18, 1034–1047. [DOI] [PubMed] [Google Scholar]
- Park, J.‐O. , Kimura, M. , & Taira, A. (1996). Late Pleistocene unconformity of the Tsushima and Korea Straits revealed by seismic reflection profiles. Journal of Geography, 105, 297–305. [Google Scholar]
- Pavlidis, P. , Laurent, S. , & Stephan, W. (2010). msABC: A modification of Hudson's ms to facilitate multi‐locus ABC analysis. Molecular Ecology Resources, 10, 723–727. [DOI] [PubMed] [Google Scholar]
- Petit, J. R. , Jouzel, J. , Raynaud, D. , Barkov, N. I. , Barnola, J.‐M. , Basile, I. , … Stievenard, M. (1999). Climate and atmospheric history of the past 420, 000 years from the Vostok ice core, Antarctica. Nature, 399, 429–436. [Google Scholar]
- Phillips, S. J. , Anderson, R. P. , & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190, 231–259. [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, X.‐S. , Yuan, N. , Comes, H. P. , Sakaguchi, S. , & Qiu, Y.‐X. (2014). A strong ‘filter’ effect of the East China Sea land bridge for East Asia's temperate plant species: Inferences from molecular phylogeography and ecological niche modelling of Platycrater arguta (Hydrangeaceae). BMC Evolutionary Biology, 14, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y.‐X. , Sun, Y. , Zhang, X.‐P. , Lee, J. , Fu, C.‐X. , & Comes, H. P. (2009). Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytologist, 183, 480–495. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2015). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Sakaguchi, S. , Takeuchi, Y. , Yamasaki, M. , Sakurai, S. , & Isagi, Y. (2011). Lineage admixture during postglacial range expansion is responsible for the increased gene diversity of Kalopanax septemlobus in a recently colonised territory. Heredity, 107, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi, S. , Qiu, Y.‐X. , Liu, Y.‐H. , Qi, X.‐S. , Kim, S.‐H. , Han, J. , … Isagi, Y. (2012). Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Molecular Ecology, 21, 3823–3838. [DOI] [PubMed] [Google Scholar]
- Shaw, J. , Lickey, E. B. , Beck, J. T. , Farmer, S. B. , Liu, W. , Miller, J. , … Small, R. L. (2005). The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany, 92, 142–166. [DOI] [PubMed] [Google Scholar]
- Siddall, M. , Rohling, E. J. , Almogi‐Labin, A. , Hemleben, C. , Meischner, D. , Schmelzer, I. , & Smeed, D. (2003). Sea‐level fluctuations during the last glacial cycle. Nature, 423, 853–858. [DOI] [PubMed] [Google Scholar]
- Stephens, M. , & Scheet, P. (2005). Accounting for decay of linkage disequilibrium in haplotype inference and missing‐data imputation. American Journal of Human Genetics, 76, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M. , Smith, N. J. , & Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenning, J.‐C. , Fløjgaard, C. , Marske, K. A. , Nógues‐Bravo, D. , & Normand, S. (2011). Applications of species distribution modeling to paleobiology. Quaternary Science Reviews, 30, 2930–2947. [Google Scholar]
- Tagane, S. , Hiramatsu, M. , & Okubo, H. (2008). Hybridization and asymmetric introgression between Rhododendron eriocarpum and R. indicum on Yakushima Island, southwest Japan. Journal of Plant Research, 121, 387–395. [DOI] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. , & Kumar, S. (2011). MEGA5: Molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada, M. (1985). Map of vegetation during the Last Glacial Maximum in Japan. Quaternary Research, 23, 369–381. [Google Scholar]
- Wagner, D. B. , Furnier, G. R. , Saghai‐Maroof, M. A. , Williams, S. M. , Dancik, B. P. , & Allard, R. W. (1987). Chloroplast DNA polymorphisms in lodgepole and jack pines and their hybrids. Proceedings of the National Academy of Sciences of the United States of America, 84, 2097–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Källman, T. , Liu, J. , Guo, Q. , Wu, Y. , Lin, K. , & Lascoux, M. (2014). Speciation of two desert poplar species triggered by Pleistocene climatic oscillations. Heredity, 112, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A. (1975). On the number of segregating sites in genetical models without recombination. Theoretical Population Biology, 7, 256–276. [DOI] [PubMed] [Google Scholar]
- Wei, H. , Fu, Y. , & Arora, R. (2005). Intron‐flanking EST–PCR markers: From genetic marker development to gene structure analysis in Rhododendron . TAG. Theoretical and Applied Genetics., 111, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Wolf, K. H. , Li, W. H. , & Sharp, P. M. (1987). Rates of nucleotide substitutions vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences of the United States of America, 84, 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth, J. R. P. , Sakaguchi, S. , Tanaka, N. , Yamasaki, M. , & Isagi, Y. (2013). Northern richness and southern poverty: Contrasting genetic footprints of glacial refugia in the relictual tree Sciadopitys verticillata (Coniferales: Sciadopityaceae). Biological Journal of the Linnean Society, 108, 263–277. [Google Scholar]
- Yoichi, W. , & Tomaru, N. (2014). Patterns of geographic distribution have a considerable influence on population genetic structure in one common and two rare species of Rhododendron (Ericaceae). Tree Genetics and Genomes, 10, 827–837. [Google Scholar]
- Zhai, S.‐N. , Comes, H. P. , Nakamura, K. , Yan, H.‐F. , & Qiu, Y.‐X. (2012). Late Pleistocene lineage divergence among populations of Neolitsea sericea (Lauraceae) across a deep sea‐barrier in the Ryukyu Islands. Journal of Biogeography, 39, 1347–1360. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials