

ABSTRACT
Gram-negative bacteria have effective methods of excluding toxic compounds, including a largely impermeable outer membrane (OM) and a range of efflux pumps. Furthermore, when cells become nutrient limited, RpoS enacts a global expression change providing cross-protection against many stresses. Here, we utilized sensitivity to an anionic detergent (sodium dodecyl sulfate [SDS]) to probe changes occurring to the cell's permeability barrier during nutrient limitation. Escherichia coli is resistant to SDS whether cells are actively growing, carbon limited, or nitrogen limited. In actively growing cells, this resistance depends on the AcrAB-TolC efflux pump; however, this pump is not necessary for protection under either carbon-limiting or nitrogen-limiting conditions, suggesting an alternative mechanism(s) of SDS resistance. In carbon-limited cells, RpoS-dependent pathways lessen the permeability of the OM, preventing the necessity for efflux. In nitrogen-limited but not carbon-limited cells, the loss of rpoS can be completely compensated for by the AcrAB-TolC efflux pump. We suggest that this difference simply reflects the fact that nitrogen-limited cells have access to a metabolizable energy (carbon) source that can efficiently power the efflux pump. Using a transposon mutant pool sequencing (Tn-Seq) approach, we identified three genes, sanA, dacA, and yhdP, that are necessary for RpoS-dependent SDS resistance in carbon-limited stationary phase. Using genetic analysis, we determined that these genes are involved in two different envelope-strengthening pathways. These genes have not previously been implicated in stationary-phase stress responses. A third novel RpoS-dependent pathway appears to strengthen the cell's permeability barrier in nitrogen-limited cells. Thus, though cells remain phenotypically SDS resistant, SDS resistance mechanisms differ significantly between growth states.
IMPORTANCE Gram-negative bacteria are intrinsically resistant to detergents and many antibiotics due to synergistic activities of a strong outer membrane (OM) permeability barrier and efflux pumps that capture and expel toxic molecules eluding the barrier. When the bacteria are depleted of an essential nutrient, a program of gene expression providing cross-protection against many stresses is induced. Whether this program alters the OM to further strengthen the barrier is unknown. Here, we identify novel pathways dependent on the master regulator of stationary phase that further strengthen the OM permeability barrier during nutrient limitation, circumventing the need for efflux pumps. Decreased permeability of nutrient-limited cells to toxic compounds has important implications for designing new antibiotics capable of targeting Gram-negative bacteria that may be in a growth-limited state.
KEYWORDS: Escherichia coli, RpoS, efflux pumps, outer membrane, permeability, stationary phase
INTRODUCTION
Antibiotic resistance in Gram-negative bacteria is a matter of increasing importance. In fact, five of seven bacterial groups listed by the WHO as “bacteria of international concern” are Gram negative, and resistance rates for these bacteria to fluoroquinolones and third-generation cephalosporins have exceeded 25% to 50% in countries all over the world (1). Furthermore, the outer membrane (OM) poses a significant challenge to the development of new antibiotics for treatment of Gram-negative bacteria. This barrier consists of an asymmetric bilayer with an inner leaflet consisting of phospholipids and an outer leaflet consisting mainly of lipopolysaccharide (LPS). The tightly packed hydrophilic regions of LPS make the OM a very effective permeability barrier to both large (>700-Da) and hydrophobic molecules (reviewed in reference 2). As most antibiotics must penetrate this barrier as well as the periplasm and inner membrane (IM) to function, designing new antibiotics for Gram-negative bacteria has been problematic (3).
Beyond the strong permeability barrier posed by the OM, antibiotics must also overcome the actions of a wide range of efflux pumps with broad specificity (reviewed in reference 4). Escherichia coli has 29 efflux pumps and putative efflux pumps of which the AcrAB-TolC pump has the greatest effect on the MIC of toxic compounds (4, 5). Many of the efflux pumps (e.g., AcrAB-TolC) consist of tripartite complexes with an IM pump (AcrB), a periplasmic adaptor (AcrA), and an OM channel (TolC), while others are single-component pumps (e.g., EmrD) that pump compounds to the periplasm instead of the extracellular environment (4). Most of the efflux pumps are driven by the proton motive force, although some are ATP driven (4). While many efflux pumps are constitutively expressed (e.g., those encoded by acrAB, emrAB, emrD, and mdfA), the expression of others is controlled by stress responses (e.g., those encoded by mdtABC, mdtD, and acrD) or is dependent on growth phase (e.g., that encoded by mdtEF) (6–8). Thus, the combination of a largely impermeable OM and a wide variety of efflux pumps makes the envelope of Gram-negative bacteria a significant hurdle for antibacterial activity.
One mechanism by which cells survive in the presence of antibiotics that penetrate the envelope is persistence. Persisters are cells without resistance mutations that comprise a small portion of an isogenic bacterial population, remain viable during antibiotic treatment, and can generally resume growth following antibiotic treatment (reviewed in reference 9). Clinically, persisters have been found to be important to the pathogenesis of infections, including persistent tuberculosis, reoccurring uropathogenic E. coli infections, and Pseudomonas aeruginosa infections in cystic fibrosis patients (10–14). The rate of persister formation is much higher in stationary-phase cells than in actively growing cells (10, 15–19). In addition to increasing rates of persister formation, stationary phase can also increase rates of resistance and tolerance to antibiotics (18, 20–23). Although there is an effect of decreased metabolic activity in stationary-phase cells, the resistance to and tolerance for antibiotics observed in stationary-phase cells are largely due to RpoS, the stationary-phase alternative sigma factor, which induces a global gene expression change in nutrient-limited cells that prepares cells to survive under stressful conditions for long time periods (reviewed in reference 24). As it has been estimated that approximately 60% of the world's biomass is made up of quiescent microbes (25), understanding the changes that occur due to regulatory factors such as RpoS in these nongrowing microbes leading to antibiotic resistance and persistence is imperative.
Stationary-phase E. coli cells incur changes to their morphology, metabolism, transcriptional programs, and translational programs, which induce cross-protection from many stresses, including osmotic shock, oxidative stress, heat shock, and acid and base shock (24, 26–28). These changes include alterations to the cell's envelope to make it more stress resistant. For example, the IM becomes more highly ordered with greater proportions of cyclopropyl fatty acid derivatives and cardiolipin, the thickness and cross-linking of peptidoglycan (PG) increase, and trehalose, membrane-derived oligosaccharides, and other stress response factors are secreted into the periplasm (reviewed in references 29 and 24). However, very little is known about changes that may occur to the OM during stationary phase. It has been suggested that during stationary phase, the overall amount of protein in the OM is decreased and that the OM lipoprotein-PG cross-linking increases (30, 31). However, these studies were limited by the techniques available at the time of their publication. Therefore, we set out to elucidate changes that occur in the cell's permeability barrier during stationary phase.
We utilized sodium dodecyl sulfate (SDS) resistance to probe the strength of the cell's permeability barrier since SDS does not rely on the cell's metabolism for its toxicity as many antibiotics do. Previous studies have determined that SDS resistance in E. coli correlates well with antibiotic resistance in actively growing cells with mutations in envelope biosynthesis pathways (32–35). As SDS resistance correlates with antibiotic resistance in these mutants, we focused on SDS resistance to assess the cell's permeability barrier, thus avoiding any effects of altered metabolism between different growth states. Here, we demonstrate that the mechanisms of SDS resistance differ between actively growing cells, carbon-limited stationary-phase cells, and nitrogen-limited stationary-phase cells. Furthermore, we have elucidated genes involved in novel rpoS-dependent pathways that strengthen the OM in carbon-limited cells. Our results highlight the decreased envelope permeability of nongrowing cells, a result with important implications for antibiotic design strategies targeting Gram-negative bacteria.
RESULTS
E. coli is SDS resistant regardless of growth stage.
For many stresses, such as heat shock, osmotic shock, and oxidative stress, the resistance of stationary-phase E. coli cells, whether carbon or nitrogen starved, is greater than that of exponentially growing cells (26, 27); therefore, we hypothesized that stationary-phase cells would be more resistant to SDS than actively growing cells. In order to test this hypothesis, we grew MG1655 cells overnight to stationary phase in minimal medium with limiting concentrations of either glucose (carbon limited) or ammonium sulfate (nitrogen limited), treated them with 2% SDS or a vehicle control, and then assayed the viability over 24 h. For treatment of actively growing cells, we diluted carbon-limited cells into fresh medium with excess carbon and nitrogen and added SDS after 30 min of adaptation to the new conditions. In contrast to other stresses, actively growing cells showed no decrease in viability or growth rate after 6 h of SDS treatment relative to that of untreated cells (Fig. 1A). In 24 h of 2% SDS treatment, carbon-limited cells demonstrated only a minimal decrease in viability (4.9-fold) (Fig. 1B), while nitrogen-limited cells demonstrated no decrease in viability (Fig. 1C). Given the impressive resistance to SDS of the cells in all growth stages, we then set out to determine the mechanism of SDS resistance in these cells.
FIG 1.
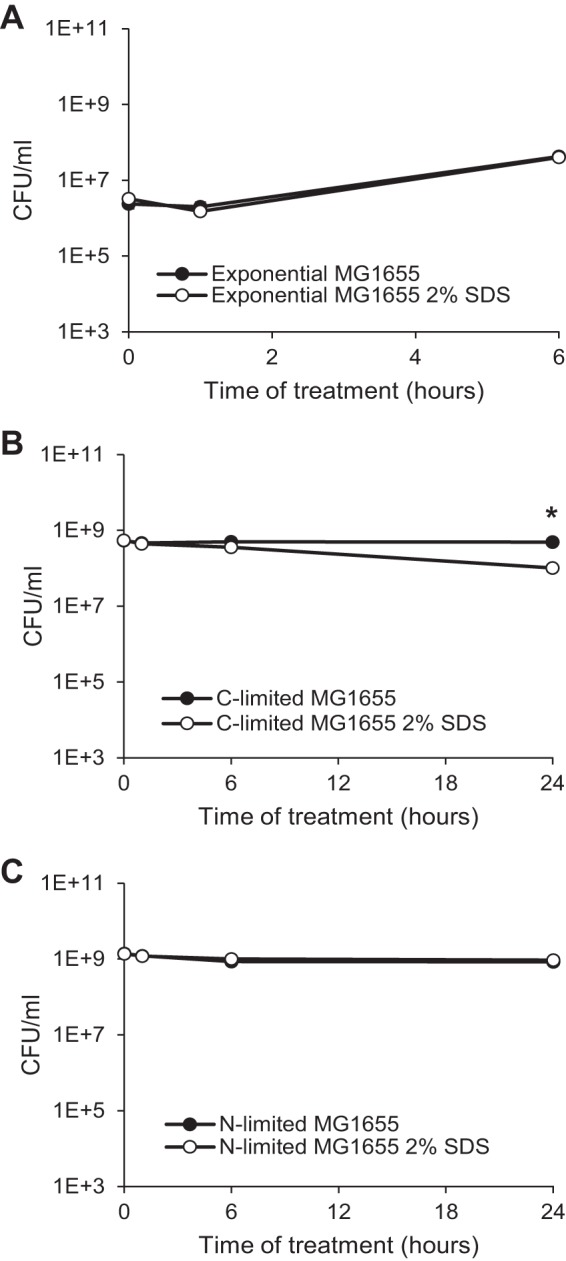
E. coli is resistant to SDS regardless of growth stage. Cells prepared to be actively growing (A), carbon limited (B), or nitrogen limited (C) were treated with 2% SDS, and viability was assayed at the indicated time points. Filled data points indicate untreated samples, while open data points indicate SDS-treated samples. *, P < 0.05, for comparisons of results for treated and untreated samples.
The AcrAB-TolC efflux pump is the primary mediator of SDS resistance in actively growing cells but not in stationary-phase cells.
When the MIC of SDS is examined by growth on plates, the presence of the AcrAB-TolC efflux pump is a major determinant of SDS resistance (5). This pump needs AcrA, AcrB, and TolC present in order to function, although AcrA and TolC can also interact with several other IM pump components (36–38). Thus, we deleted acrA and acrB and examined the effect on SDS resistance of cells in the three growth states. Strains lacking TolC were not used due to the pleiotropic effects of tolC mutation on the OM (39–43) that would confound interpretation of the results. Compared to wild-type cells in which SDS had no effect (Fig. 1A), actively growing cells with deletions in acrA or acrB demonstrated a 120- to 170-fold decrease in viability in 8 h, leading to a 4,000- to 5,000-fold difference in viability, respectively, between treated and nontreated cells (Fig. 2A). In contrast, in carbon-limited cells, the viability of acrA and acrB cells treated with SDS was within 2.5-fold of that of wild-type cells (Fig. 2B). There was, however, a kinetic difference in viability between the wild-type and acrA or acrB strains. Whereas wild-type cells did not demonstrate a significant difference in viability between treated and untreated cells until 24 h posttreatment (Fig. 1B), acrA and acrB cells had a significant decrease in viability starting at 1 h posttreatment (Fig. 2B). Similarly, nitrogen-limited cells demonstrated a small (1.8- to 2.6-fold) but significant decrease in viability starting at 1 h posttreatment (Fig. 2C). Nevertheless, these data illustrate that, unlike actively growing cells, the AcrAB-TolC efflux pump is not the main determinant of SDS resistance in stationary phase, suggesting that stationary-phase cells must employ a different mechanism of resistance, perhaps involving strengthening their envelope permeability barrier.
FIG 2.
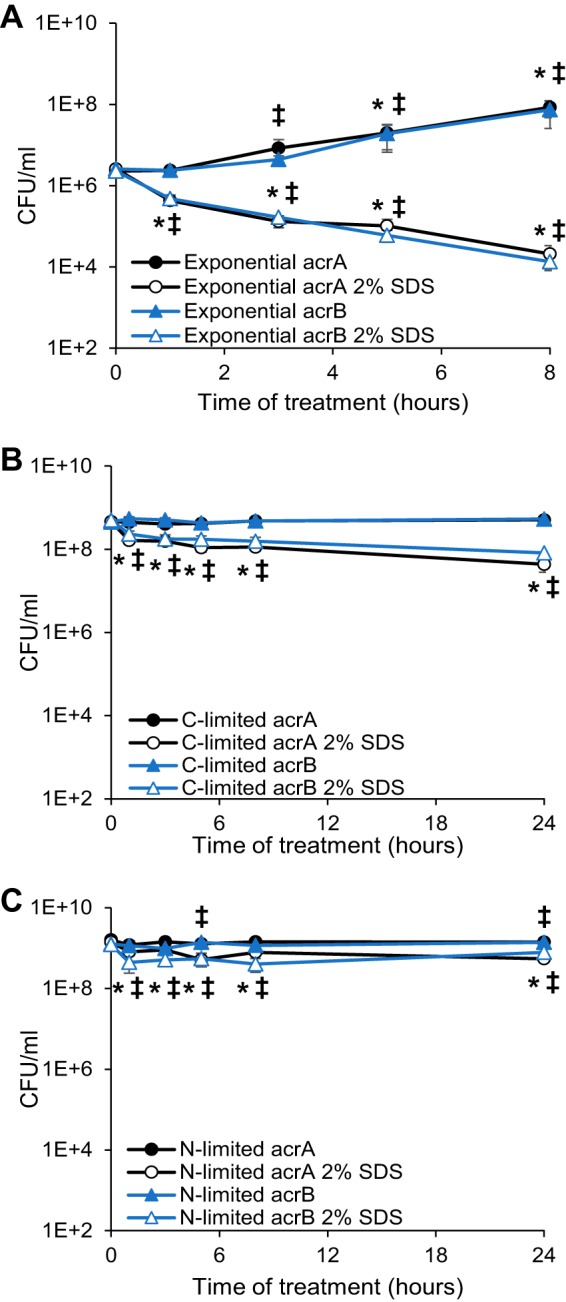
The AcrAB-TolC efflux pump is responsible for SDS resistance in exponential phase. Actively growing (A), carbon-limited (B), or nitrogen-limited (C) cells of the indicated acrA or acrB deletion strains were treated with 2% SDS, and viability was assayed at the indicated time points. Filled data points indicate untreated samples, while open data points indicate SDS-treated samples. *, P < 0.05, for comparisons with initial time point for acrA strain; ‡, P < 0.05, for comparisons with initial time point for acrB strain.
SDS resistance in carbon-limited cells requires an RpoS-dependent mechanism.
There are two possible explanations for SDS resistance in stationary phase. First, the nongrowing state induced by lack of nutrients might be directly responsible for the increase in envelope resistance through the limitation of cell division or metabolic activity. Second, a specific change in gene expression in stationary phase may produce the increase in resistance through activation of protective pathways. RpoS drives a change in gene expression in stationary phase that affects, directly or indirectly, 10% of the genome (24). Therefore, we investigated the SDS resistance of rpoS deletion cells in the three growth phases to determine whether a specific change in gene expression was necessary for stationary-phase SDS resistance. In actively growing cells, deletion of rpoS had no effect on SDS resistance (Fig. 3A); however, in carbon-limited cells, rpoS deletion led to a 940-fold decrease in viability in 24 h in SDS-treated cells, demonstrating that rpoS is required for SDS resistance in carbon-limited cells (Fig. 3B). These data suggest that RpoS-dependent pathways activated in carbon-limited cells are responsible for the SDS resistance of carbon-limited cells. Surprisingly, in nitrogen-limited cells, deletion of rpoS had no effect on the viability of SDS-treated cells (Fig. 3C). These data suggest that RpoS is the main determinant of SDS resistance in carbon-limited cells but not in actively growing cells, which are protected by efflux, or in nitrogen-limited cells. This is interesting considering that the levels of RpoS are much lower in nitrogen-limited cells than in carbon-limited cells and are lower still in exponentially growing cells (44).
FIG 3.
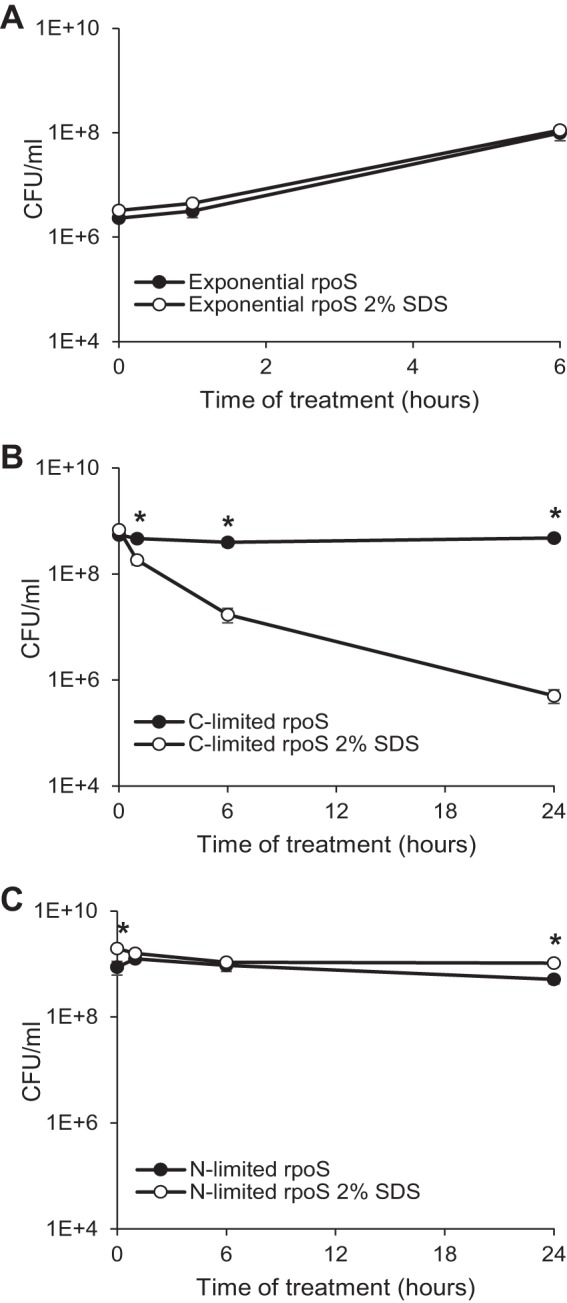
RpoS mediates SDS resistance in carbon-limited cells. Actively growing (A), carbon-limited (B), or nitrogen-limited (C) rpoS deletion cells were treated with 2% SDS, and viability was assayed at the indicated time points. Filled data points indicate untreated samples, while open data points indicate SDS-treated samples. *, P < 0.05, for comparisons of results for treated and untreated samples.
We next investigated whether the lack of a protective effect of RpoS in exponentially growing cells, demonstrated by a decrease in viability with only acrA or acrB deletion, was due to the low levels of RpoS present in these cells or to protective mechanisms activated by RpoS which can operate only in stationary-phase cells. To investigate these possibilities, we utilized sprE deletion strains. SprE is responsible for the ClpXP-dependent degradation of RpoS during exponential phase, and deleting sprE increases RpoS levels in both exponential cells and stationary-phase cells (45). In actively growing cells, deletion of sprE alone did not cause any SDS sensitivity; however, acrA sprE deletion cells were significantly more resistant to SDS than were acrA deletion cells (Fig. 4A). Deletion of sprE was also protective in carbon-limited cells alone or in combination with acrA deletion but was not protective in rpoS deletion cells (Fig. 4B). These data demonstrate that RpoS-dependent mechanisms can be protective in actively growing cells and suggest that wild-type levels of RpoS in these cells are too low to be protective. These data also illustrate that increasing RpoS levels in carbon-limited cells can further increase SDS resistance, suggesting that the effect of RpoS on SDS resistance is dependent on RpoS levels.
FIG 4.
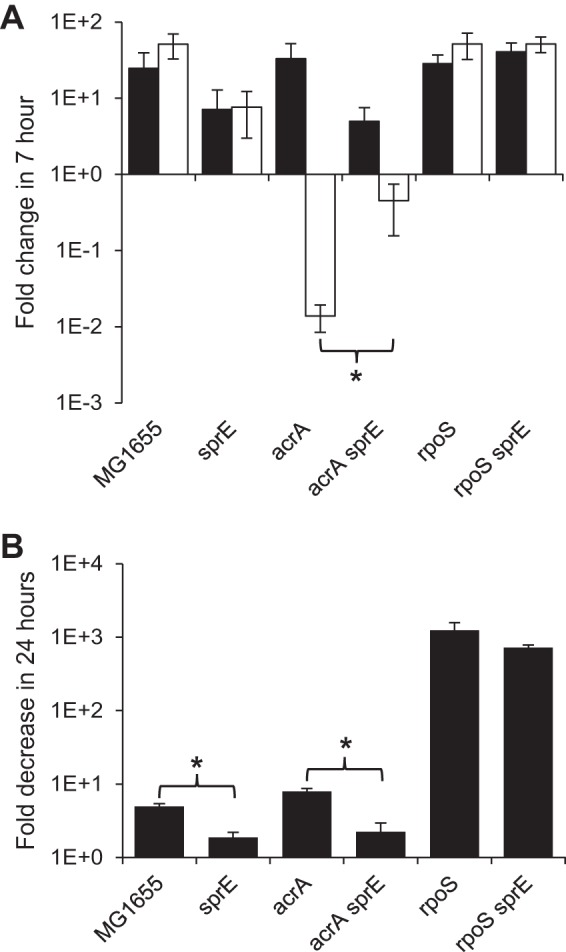
RpoS can be protective in exponential phase. (A) Actively growing cells of the indicated strains were treated with 2% SDS, and viability was assayed at 0 and 7 h. Fold changes between the 0- and 7-h time points (7-h sample/0-h sample) are shown. Filled bars indicate untreated samples, while open bars indicate treated samples. *, P < 0.05, for comparisons both between the treated and untreated samples and between the indicated strains. (B) Carbon-limited cells of the indicated strains were treated with 2% SDS, and viability was assayed after 24 h. The fold decrease in the treated samples versus the untreated samples is shown (untreated sample/treated sample). *, P < 0.05, for comparisons of results for the indicated strains.
In nitrogen-limited cells, the AcrAB-TolC efflux pump can compensate for the loss of rpoS-dependent resistance mechanisms.
As we found AcrAB-TolC to be the main determinant of SDS resistance in actively growing cells and RpoS to be the main determinant of SDS resistance in carbon-limited cells, we then investigated the effect of simultaneously removing these two factors. Therefore, we examined the SDS resistance of acrA rpoS and acrB rpoS double mutants in the three growth states. In actively growing cells, acrA rpoS and acrB rpoS double mutants were even more sensitive to SDS than acrA or acrB single mutants, and viability decreased to near our limit of detection by 3 h of treatment and below our level of detection by 5 h of treatment (Fig. 5A). These results emphasize that although RpoS-dependent mechanisms can be protective in actively growing cells, allowing some remaining viability without the AcrAB-TolC efflux pump, the levels of RpoS are too low to be fully protective. Carbon-limited double mutant cells were also more sensitive to SDS than single mutant cells, with viability decreasing to near or below our limit of detection in 24 h (Fig. 5B). These data demonstrate that AcrAB-TolC can play a protective role in carbon-limited cells in the absence of RpoS but that AcrAB-TolC cannot compensate for the loss of RpoS in these cells. The lack of full protection by the AcrAB-TolC efflux pump under carbon-limiting conditions suggests that the RpoS-dependent mechanisms of SDS resistance must prevent the SDS from penetrating the OM. Under nitrogen-limiting conditions, acrA rpoS and acrB rpoS double mutant cells were SDS sensitive, with 880,000-fold and 4,000-fold decreases in viability, respectively, at 7 h posttreatment, followed by suppressor mutant outgrowth (Fig. 5C). These data combined with the lack of SDS sensitivity in rpoS, acrA, or acrB mutants (Fig. 2C and 3C) demonstrate that under nitrogen-limiting conditions, AcrAB-TolC- and RpoS-dependent protective mechanisms are fully functioning and that each can fully compensate for the loss of the other. Thus, cells in all three growth phases can be protected by both RpoS-dependent mechanisms and by efflux, but the dominance of each mechanism depends on the growth conditions.
FIG 5.
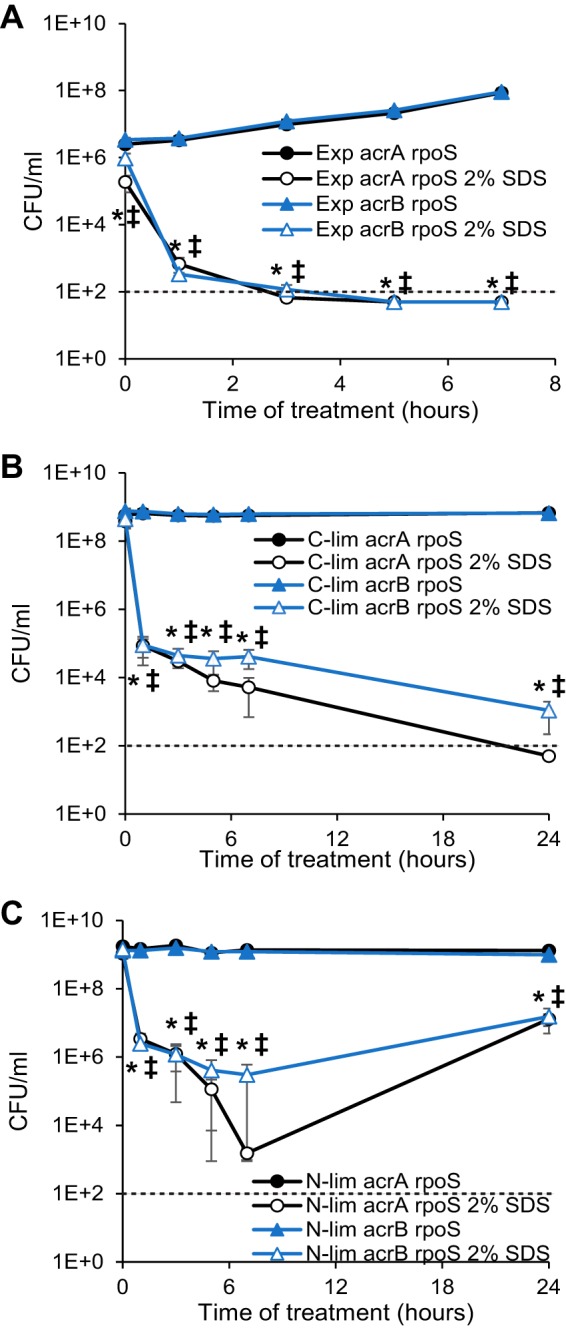
AcrAB-TolC can compensate for the loss of RpoS in nitrogen-limited cells. Actively growing (A) (Exp, exponential), carbon-limited (B), or nitrogen-limited (C) cells of the indicated strains (acrA rpoS or acrB rpoS) were treated with 2% SDS, and viability was assayed at the indicated time points. Filled data points indicate untreated samples, while open data points indicate SDS-treated samples. A dotted line indicates the limit of detection. *, P < 0.05, for comparisons of treated and untreated samples for the acrA rpoS strain; ‡, P < 0.05, for comparisons of treated and untreated samples for the acrB rpoS strain.
Tn-Seq identifies genes causing SDS sensitivity in carbon-limited but not nitrogen-limited conditions.
Given the global changes effected by RpoS in stationary phase (24), we wished to know which RpoS-regulated genes protected cells from SDS in stationary phase. In order to address this question, we utilized a transposon mutant pool sequencing (Tn-Seq) experiment (46). We created a pooled library of approximately 190,000 mutants, each containing an EZ-Tn5 insertion, grew this library overnight to stationary phase under either nitrogen-limiting or carbon-limiting conditions, and collected genomic DNA samples from these cultures before and after 24 h of 2% SDS treatment (Fig. 6A). We isolated the transposon junctions from these mutants and subjected them to deep sequencing to determine the frequency of transposon junctions throughout the genome in the various samples. A sample region of the genome is shown in Fig. 6B. The average and median number of reads per gene across the library were very similar between pre- and posttreatment samples for both carbon- and nitrogen-limiting conditions (see Table S2 in the supplemental material).
FIG 6.

Tn-Seq to identify genes responsible for SDS resistance in C-limited cells. (A) In the experimental strategy for the Tn-Seq experiment, a large library of Tn5 mutants was grown under either carbon or nitrogen limitation and then treated with 2% SDS for 24 h. DNA samples were collected and transposon junctions were sequenced both pre- and posttreatment. (B) Sequenced transposon junctions were mapped to the MG1655 genome, and a histogram for a sample region of the genome is shown. The boxed region indicates a gene, sanA, for which sequencing reads decreased more than 3-fold under carbon-limiting conditions but did not change under nitrogen-limiting conditions. Scale, 0 to 500 reads. (C) Strains with deletions in the indicated genes were grown under carbon limitation and treated with 2% SDS. Viability was assayed at 24 h of treatment. The fold decrease in the treated samples versus the untreated samples is shown (untreated sample/treated sample). Open bars indicate control strains, while filled bars represent strains with deletions in genes identified in the Tn-Seq experiment as causing SDS sensitivity only under carbon-limiting conditions. A dotted line indicates the fold decrease in wild-type MG1655. *, P < 0.05, for comparisons with results for MG1655.
In analyzing the resulting data, we took advantage of the differences in SDS resistance mechanisms between carbon- and nitrogen-limiting conditions to distinguish between mutations that cause nonspecific envelope defects and those that cause defects in the rpoS-dependent mechanism of SDS resistance in stationary phase. Based on our data (Fig. 2C, 3C, and 5C), causing SDS sensitivity through the rpoS-dependent pathway in nitrogen-limited cells requires at least two mutations: inactivation of the AcrAB-TolC efflux pump and inactivation of a component of the rpoS-dependent pathway. In contrast, one mutation that affects envelope biogenesis in a nonspecific manner in an rpoS-independent pathway could cause SDS sensitivity in N-limited cells. Therefore, to enrich for genes important for the rpoS-dependent pathway of SDS sensitivity, we identified genes for which the frequency of sequencing reads decreased in the posttreatment carbon-limited sample compared to the pretreatment carbon-limited sample but did not change between the nitrogen-limited samples, utilizing these samples as a control for genes where disruption causes envelope defects unrelated to RpoS. We chose a minimum of a 3-fold difference between the pre- and posttreatment samples to define a change, as this value defined the edges of the main population of the reads (Table S3 and Fig. S1). An example of a gene that fits these parameters, sanA, is shown in Fig. 6B.
Overall, we identified 12 envelope-related genes (Table 1) and 8 non-enveloped-related genes (Table S4) that had at least a 3-fold decrease in reads under carbon-limiting conditions and less than a 3-fold change in reads under nitrogen-limiting conditions. We then proceeded to examine the effect of the envelope-related genes on SDS resistance under carbon-limiting conditions in a noncompetitive environment. ftsN, which is an essential gene, for which we identified transposon junctions in the nonessential 3′ region, was not examined further. In single deletion settings, we were able to confirm the SDS sensitivity of five of the examined genes: rfaH, sanA, dacA, yhdP, and ydgH (Fig. 6C). Of these genes, rfaH, sanA, and dacA demonstrated the largest effects on SDS resistance, causing 100- to 200-fold decreases in viability in 24 h with SDS treatment. The two other genes, yhdP and ydgH, had smaller but significant effects on viability with SDS treatment, demonstrating 23-fold and 18-fold decreases in viability in 24 h of treatment, respectively. We considered these five genes to be candidates for participants in the rpoS-dependent pathway protecting carbon-limited cells from SDS. Interestingly, wecE deletion had no effect on SDS sensitivity in carbon-limited cells in a noncompetitive environment, despite causing SDS sensitivity in actively growing cells due to the accumulation of lipid II bound to enterobacterial common antigen (ECA) (Fig. S2) (47). These data emphasize the increase in envelope strength that occurs in nutrient-limited cells, as they are able to overcome the effect of mutations that cause envelope permeability in actively growing cells.
TABLE 1.
Envelope-related Tn-Seq hits causing SDS sensitivity only in carbon-limited cells
| Genea | No. of carbon-limited reads/kbpb |
Log2 fold change posttreatment/pretreatmentc |
||
|---|---|---|---|---|
| Pretreatment | Posttreatment | Carbon limited | Nitrogen limited | |
| ydgH | 758 | 60 | −3.7 | 0.7 |
| bssR | 2,000 | 279 | −2.8 | 0.9 |
| tatB | 3,841 | 558 | −2.8 | 0.2 |
| wecA (rfe) | 2,043 | 323 | −2.7 | −0.3 |
| ftsN | 10,134 | 1,618 | −2.6 | −0.5 |
| ompA | 2,108 | 354 | −2.6 | −0.1 |
| dacA | 867 | 162 | −2.4 | −1.2 |
| sanA | 3,261 | 625 | −2.4 | −0.2 |
| ytfK | 7,816 | 2,135 | −1.9 | 0.9 |
| wecE | 1,834 | 531 | −1.8 | 0.9 |
| rfaH | 3,155 | 1,004 | −1.7 | 0.4 |
| yhdP | 2,386 | 760 | −1.7 | 0.3 |
Genes with at least 700 sequence reads decreasing at least 3-fold during treatment under carbon-limiting conditions and changing less than 3-fold under nitrogen-limiting conditions.
The number of sequence reads for each gene was normalized to the length of the gene.
The fold change in read number before and after SDS treatment under either carbon- or nitrogen-limiting conditions.
RpoS acts through sanA, dacA, and yhdP to mediate SDS resistance in stationary phase.
To determine whether the genes from the Tn-Seq that cause SDS sensitivity under carbon-limiting conditions were part of the same pathway as rpoS, we constructed double mutants with deletions of these genes and rpoS or acrA to differentiate between additive and nonadditive effects of these mutations. For three of the genes, yhdP, dacA, and sanA, the effect of their deletion was not additive with rpoS (Fig. 7A, left panel). These data suggest that RpoS acts through sanA, dacA, and yhdP in order to make carbon-limited cells SDS resistant. Combining rpoS and acrA deletions leads to a very strong effect on SDS resistance; in fact, the viability of SDS-treated cultures is below the limit of detection after 24 h of treatment (Fig. 5B). When we combined deletions in the three genes that we identified with deletion of acrA, only the acrA sanA strain recapitulated the strong synthetic phenotype of an acrA rpoS deletion, with viability decreasing to near the limit of deletion in 24 h (Fig. 7A, left panel). These data suggest that sanA plays a dominant role in RpoS-dependent SDS resistance.
FIG 7.

rpoS is epistatic to sanA, dacA, and ydhP. (A) Carbon-limited cells of the indicated strains were treated with 2% SDS, and viability was assayed at 24 h posttreatment. The fold decrease in the treated samples versus the untreated samples is shown (untreated sample/treated sample). Open bars indicate strains in a wild-type background, while black bars indicate strains in an rpoS background and blue bars indicate strains in an acrA background. Control samples (MG1655 and rpoS and acrA strains) are identical between the two panels and include six biological replicates. Strains are separated by those that are not additive with rpoS (left) and those that are additive with rpoS (right). *, P < 0.05, one-tailed test for increase compared to the parent strain with the greater effect. (B) Carbon-limited cells of the indicated strains were treated with 2% SDS, and viability was assayed at 24 h posttreatment. The fold decrease in the treated samples versus the untreated samples is shown (untreated sample/treated sample). The control strain is indicated by a white bar, the single mutants by black bars, double mutants by blue bars, and the triple mutant by an orange bar. *, P < 0.05, one-tailed test for increase compared to the parent strain; n/s, no significant difference between the indicated strains. (C) Nitrogen-limited cells of the indicated strains were treated with 2% SDS, and viability was assayed at 3 h posttreatment. The fold decrease in the treated samples versus the untreated samples is shown (untreated sample/treated sample) on a linear scale. *, P < 0.05, one-tailed test for increase in strain without acrA compared to strain with acrA.
In contrast to effect of the three genes mentioned above, rpoS mutant cells demonstrated additive SDS sensitivity when combined with rfaH and ydgH mutations (Fig. 7A, right panel). As rfaH is required for efficient LPS biosynthesis (48–51), it is likely that rfaH mutants have OM permeability unrelated to the pathway affected by RpoS. YdgH is predicted to be a periplasmic protein, and its levels have been suggested to be downregulated by MicA, an sRNA that increases upon entry to stationary phase but is transcriptionally regulated by sigma E, the envelope stress sigma factor (52–55). Thus, YdgH may play a role in stationary-phase envelope permeability while not being directly regulated by RpoS.
As sanA, dacA, and yhdP appear to work downstream of rpoS to mediate SDS resistance in carbon-limited cells, we then wondered whether they work together on the same pathway or through several different mechanisms to mediate SDS resistance. To address this question, we constructed all possible double and triple deletion mutants of these genes and determined the effect of these mutations on SDS sensitivity under carbon-limiting conditions. A dacA yhdP double mutant did not show an additive effect over that of either a dacA or yhdP single mutant alone, while sanA demonstrated an additive effect with both dacA and yhdP (Fig. 7B). These data suggest that dacA and yhdP work together to mediate SDS resistance under carbon-limiting conditions, while sanA works through a separate rpoS-dependent pathway. These data correlate well with our acrA data, which suggests that sanA plays a more important role in strengthening the OM than dacA and yhdP do. Interestingly, the yhdP dacA sanA triple mutant had a level of SDS sensitivity similar to that of rpoS deletion cells, with a decrease in viability in 24 h of more than 900-fold. In contrast, under nitrogen-limiting conditions, sanA, dacA, and yhdP had no effect on SDS sensitivity even when combined with an acrA mutation (Fig. 7C), suggesting that there may be a third rpoS-dependent mechanism of SDS resistance operating specifically under nitrogen-limiting conditions. Taken together, all of these data demonstrate that the mechanisms of SDS resistance vary greatly depending on growth conditions and involve several novel rpoS-dependent mechanisms for strengthening the envelope permeability barrier.
DISCUSSION
In stationary phase, RpoS enacts a global program of transcriptional regulation that prepares cells for stressful conditions (24). In this work, we have investigated changes occurring to the cell's permeability barrier during times of nutrient limitation, using SDS resistance as a model of the strength of the envelope permeability barrier. Gene deletions causing SDS and SDS-EDTA sensitivity in E. coli have previously been investigated in a high-throughput study (56); however, the study examined colony size on plates and so did not differentiate between different growth states. Several studies examining the SDS resistance of Enterobacteriaceae have touched on the SDS resistance of E. coli (57–61). However, the studies that examined growth phase-dependent effects were conducted in a W3110 background (58, 59), which has mutations in several stress response pathways, including the Rcs pathway, and depending on the source may lack functional RpoS and/or RpoF (62, 63). In addition, no genes involved in stationary-phase SDS resistance were identified.
Therefore, we investigated the SDS resistance of wild-type MG1655. We have determined that although actively growing cells, carbon-limited cells, and nitrogen-limited cells are all resistant to SDS (Fig. 1), the mechanisms of SDS resistance deployed depend on the growth phase of the cells. In actively growing cells, cells are protected from SDS mainly by the AcrAB-TolC efflux pump (Fig. 2). These data suggest that in these cells, SDS penetrates the OM and then AcrAB-TolC pumps it back out through an energy-dependent process (Fig. 8A). Interestingly, it has been previously suggested that SDS could be found in the E. coli periplasm during SDS treatment (64, 65). Although rpoS can play a role in SDS resistance in these cells, it is not the dominant mechanism of resistance employed and is not fully protective (Fig. 4 and 5). With ample carbon and nitrogen available, the cells instead rely on the energy-intensive process of efflux, which in this case is driven by the proton motive force (4) (Fig. 8A).
FIG 8.

Model of interactions between efflux and envelope permeability in SDS resistance. (A) In actively growing cells, SDS is able to penetrate the OM and is then removed by the AcrAB-TolC efflux pump. This energy-intensive process, which relies on the proton motive force, is made efficient by the abundance of nutrients available to the cell. (B) In carbon-limited cells, an RpoS-dependent decrease in OM permeability through pathways that involve SanA, PBP5, and YhdP prevents SDS from entering the cell. Early in treatment, any SDS that does penetrate the OM is removed by the AcrAB-TolC efflux pump; however, lack of energy in carbon-limited cells eventually leads the efflux pump to fail and causes a low level of cell death. When rpoS is removed, SDS penetrates the OM and inefficiency in the efflux pumps due to lack of energy causes the cells to die quickly. (C) In nitrogen-limited cells, an RpoS-dependent mechanism that is different from that employed in carbon-limited cells prevents SDS entry into the cell; however, the loss of rpoS can be compensated for by the activity of the AcrAB-TolC efflux pump, suggesting that although there is a lack of available nitrogen for making new proteins, nitrogen-limited cells have sufficient energy available in the form of the proton motive force for efficient efflux over the time scale of the experiment.
In carbon-limited cells, the resistance to SDS is dependent primarily on rpoS (Fig. 3). Our model for these data suggests that RpoS acts to activate pathways of SDS resistance in carbon-limited cells, which prevent SDS from entering the cell by strengthening the permeability barrier of the OM (Fig. 8B). In the absence of rpoS, the cells can be minimally protected by the AcrAB-TolC efflux pump; however, this pump cannot fully protect the cells in the absence of rpoS likely due to a lack of energy (Fig. 8B). This model explains the kinetic difference in SDS resistance we observed between AcrAB-TolC-deficient cells and wild-type cells under carbon-limiting conditions wherein decreases in viability were observed at earlier time points for acrA and acrB cells than for wild-type cells (compare Fig. 1B and 2B), despite the presence of equal levels of RpoS in these cells (see Fig. S3 in the supplemental material). It is likely that carbon-limited cells can efflux SDS for a brief time, increasing their SDS resistance, but are quickly depleted of protons with which to run efflux, resulting in low levels of cell death. This cell death would occur more quickly in cells in which the efflux pump has been removed. Furthermore, rpoS deletion cells exhibit significant cell death following SDS treatment even when the AcrAB-TolC efflux pump is present. This inability of efflux to protect carbon-limited cells from SDS emphasizes that RpoS-dependent SDS resistance must involve strengthening the OM permeability barrier, preventing the necessity for efflux.
We have identified three genes, sanA, dacA, and yhdP, for which deletion decreases SDS resistance in carbon-limited cells and for which the effect of deletion is not additive with rpoS (Fig. 7). Deletion of these genes has no effect on the levels of RpoS in carbon-limited cells (Fig. S3). These data suggest that RpoS acts through SanA, PBP5 (DacA), and YhdP to provide SDS resistance in carbon-limited cells (Fig. 8B). Given the strong synthetic phenotype observed with sanA and acrA deletions (Fig. 7), our model suggests that RpoS is working primarily through SanA to strengthen the OM in carbon-limiting conditions. It remains possible that PBP5 and YhdP act to decrease SDS sensitivity by altering some other aspect of the cell's envelope (e.g., PG or IM). It is of interest that since RpoS can be protective in exponentially growing cells in the absence of AcrAB-TolC, deletion of sanA, dacA, or yhdP can increase SDS sensitivity in exponentially growing cultures, although this sensitivity cannot be observed for sanA or dacA in an efficiency-of-plating assay on plates with SDS, suggesting that the exponential-phase effect is small (Fig. S4). As these genes have no effect in nitrogen-limiting conditions (Fig. 7), these data suggest that other regulatory factors, in addition to RpoS, may allow these genes to provide a basal level of protection in actively growing cells. Overall, our data suggest that in carbon-limited cells, RpoS acts directly or indirectly to activate pathways that strengthen the OM permeability barrier and involve SanA, PBP5, and YhdP (Fig. 8B).
SanA was first identified as a multicopy suppressor of an unknown mutant with an OM permeability defect, which had a deletion of sanA as well as other mutations leading to OM defects (66). SanA has 239 amino acids and is predicted to have an inner membrane localization with a very small N-terminal cytoplasmic domain (6 amino acids), one transmembrane helix, and the remainder of the protein localized in the periplasm (55, 67). The periplasmic domain of SanA contains a DUF218 domain (68). DUF218 domains contain several charged amino acids, suggesting enzymatic activity (68), and are found in many species throughout the bacterial domain as well as in some archaea, plants, and fungi, mainly in proteins of unknown function (68).
E. coli has three homologs of SanA containing DUF218 domains: YgjQ, YdcF, and ElyC (68, 69). YgjQ has no known function, although it is predicted to have the same topology as SanA (55, 67). YdcF is predicted to be a cytoplasmic protein, which binds to S-adenosyl-l-methionine, and has been suggested to be directly or indirectly regulated by both the Rcs response and FNR (67, 69–71). A deletion mutant of elyC (ycbC), but not sanA, was identified through a high-throughput screen as causing cell lysis at room temperature in LB medium with 1% salt (72). Suppressor and complementation assays suggested that ElyC may be involved in balancing undecaprenyl-phosphate (Und-P) use between PG biosynthesis and the biosynthesis of polysaccharides such as ECA (72). Supporting this role, deletions of genes involved in the biosynthesis of ECA, a glycolipid with trimeric repeats of N-acetyl-d-glucosamine (GlcNAc), N-acetyl-d-mannosaminuronic acid (ManNAcA), and 4-acetamido-4,6-dideoxy-d-galactose (Fuc4NAc), have a suppressive or synthetic phenotype on elyC deletion depending on the Und-P utilization of the mutants. Thus, deletion of wecA acts as a suppressor of lysis with elyC deletion as it prevents Und-P use for ECA synthesis by preventing the formation of Und-P-P-GlcNAc (lipid IECA), while deletion of wecE has a synthetic phenotype with elyC deletion as it causes the buildup of Und-P-P-GlcNAc-ManNAcA (lipid IIECA), preventing the use of Und-P for PG biosynthesis (47, 72, 73). Although these proteins contain DUF218 domains similar to SanA, deletion of ygjQ, ydcF, or elyC had no effect on the SDS sensitivity of carbon-limited cells (Fig. S5). In addition, mutation of wecA or wecE in a sanA deletion strain had no effect on SDS sensitivity (Fig. S5), demonstrating that alteration of Und-P levels available for PG biosynthesis has no effect on the SDS sensitivity of a sanA mutant. These data suggest that SanA plays a different role in strengthening the OM permeability barrier than those played by its paralogs.
Deletion of both sanA and the Salmonella homolog of sanA, sfiX, has been shown to confer vancomycin sensitivity at 43°C but not 42°C, suggesting a temperature-dependent effect of SanA (66, 74). In addition, sanA is annotated to have a heat shock-dependent RpoH promoter (75). This suggests that as the rpoH promoter can be utilized by RpoS and the levels of RpoH increase upon carbon starvation (76, 77), the effect of RpoS on SanA may be indirect through RpoH. The function of SanA in strengthening the OM remains an open question, but our data suggest that SanA is acting independently of PBP5 and YhdP through a mechanism unique from those of its homologs (Fig. 8B). As an acrA sanA strain shows a strong synergistic effect on SDS sensitivity unlike the acrA dacA and acrA yhdP strains (Fig. 7), the RpoS-dependent pathway involving SanA appears to be the dominant pathway strengthening the OM under carbon-limiting conditions. We are currently investigating this pathway.
The second envelope-related gene we identified to be involved with the RpoS-dependent mechanisms for decreasing envelope permeability in stationary phase is PBP5. PBP5 is a d-alanyl–d-alanine carboxypeptidase (DD-CPase) that trims the fifth amino acid from the PG chain after the precursor has been polymerized into the cell wall and is thought to be involved with PG remodeling (78–80). Thus, the PG of exponential-phase cells with a deletion of dacA has a 4-fold increase in pentapeptides compared to the PG of wild-type cells (81). PBP5 is under the transcriptional control of bolA, the stationary-phase morphogene, suggesting that its link to RpoS is likely mediated by BolA (82, 83). E. coli has eight proteins (PBP4, PBP4b, PBP5, PBP6, PBP6b, PBP7, AmpC, and AmpH) capable of removing the terminal d-alanine from PG chains, and removal of more than one of them is necessary to cause visible morphological defects (80, 84–86). In fact, cells remain viable when grown in LB medium after deletion of the genes for at least seven out of the eight proteins responsible for this activity (87). The different proteins with this activity are thought to act under different conditions. For instance, during active growth in neutral conditions, PBP5 is the main protein responsible for DD-CPase activity, while at a pH of 5, PBP6b is mainly responsible for DD-CPase activity (88). In addition, while PBP5 is most active in early log phase, PBP6 and PBP6b are thought to be most active in mid-log and stationary phases (83, 89). It is interesting that although PBP5 is not the DD-CPase that is most active in stationary phase, it is involved in mediating SDS resistance in carbon-limiting conditions. We are currently investigating the possibility that PBP5 activity in carbon-limiting conditions acts as a signal for an envelope-strengthening pathway. As the effects of yhdP and dacA are not additive, we hypothesize that this pathway involves yhdP, the third gene through which RpoS acts to decrease envelope permeability during carbon-limiting conditions.
YhdP, like SanA, is predicted to be an IM protein with a small N-terminal cytoplasmic domain (6 amino acids), one transmembrane helix, and a large periplasmic domain (amino acids 30 through 1266) (55, 67). Homologs of YhdP are common in Gammaproteobacteria and are also found in Betaproteobacteria (90). The periplasmic domain of YhdP contains a DUF3971 domain and an AsmA_2 domain, which is similar to the C-terminal domain of AsmA (68). AsmA has been implicated in the assembly of outer membrane porins (91–93). We are currently investigating whether YhdP may play a similar role. As yhdP does not have an annotated promoter, it is unclear whether RpoS acts directly or indirectly on YhdP. Interestingly, YhdP was suggested through a high-throughput screen to bind to YdgH (94), which also affects SDS resistance under carbon-limiting conditions although not in an RpoS-dependent manner (Fig. 7). A yhdP ydgH double mutant was quite sensitive to SDS under carbon-limiting conditions (Fig. S6), and we are further investigating the possibility of functional interactions between YhdP and YdgH.
In contrast to carbon-limiting conditions, nitrogen-limited cells lacking either rpoS or acrA or acrB alone are resistant to SDS (Fig. 2 and 3); instead, deletions of both rpoS and acrA or acrB are needed in order to cause SDS sensitivity. These data suggest that in wild-type cells, RpoS-dependent mechanisms prevent SDS entry into nitrogen-limited cells but that when rpoS is removed, the AcrAB-TolC efflux pump can compensate for the loss by pumping SDS out of the cell (Fig. 8C). Moreover, the ability of AcrAB-TolC to compensate for the loss of RpoS suggests that the nitrogen-limited cells are not energy limited, likely because of the inhibitory effect of nitrogen limitation on protein synthesis, a highly energy-dependent process (Fig. 8C). Intriguingly, deletion of acrA and sanA, dacA, or yhdP does not cause SDS sensitivity in nitrogen-limited cells (Fig. 7), suggesting that the mechanism of RpoS-dependent SDS resistance present in nitrogen-limited cells is different from that found in carbon-limited cells (Fig. 8C). We are currently investigating this mechanism, which may involve efflux and/or strengthening the resistance of the OM or IM to SDS. Our data for SDS resistance are in contrast to those for other stresses such as heat shock, osmotic stress, and oxidative stress, where RpoS plays the same role in carbon- and nitrogen-limited cells (26, 27). Thus, SDS resistance is an interesting model for investigating the effects of various RpoS levels found with different nutrient limitations (44).
In summary, we have demonstrated that mechanisms of SDS resistance differ between cells in different growth states. Whereas actively growing cells rely on efflux, stationary-phase cells utilize RpoS-dependent mechanisms to strengthen their envelope permeability barrier and may play a role in the persistence, tolerance, and resistance to antibiotics observed during stationary phase. As the vast majority of microbes in the environment are in a quiescent state (25), insights into changes to the cell's permeability barrier that occur in nongrowing cells have important implications for the design of new antibiotics targeting Gram-negative bacteria, for which envelope permeability is a major hurdle. Furthermore, we identified several novel RpoS-dependent pathways through which the cell's envelope permeability barrier can be strengthened. Further investigation of these pathways should lead to insights into the biology of the cell's envelope permeability barrier.
MATERIALS AND METHODS
Strains and growth conditions.
All strains used in this study are listed in Table S1 in the supplemental material. Strains were grown at 37°C in M63 medium (95) supplemented with 1 mM MgSO4 and 100 μg/ml thiamine and with glucose and (NH4)2SO4 concentrations appropriate for the growth state. Carbon-limited cells were grown with 0.05% glucose and 0.2% (NH4)2SO4, while nitrogen-limited cells were grown with 0.4% glucose and 0.05% (NH4)2SO4. Cultures for actively growing cells were grown for carbon limitation overnight and then back diluted 1:200 into M63 medium with 0.2% glucose and 0.2% (NH4)2SO4. Strains were constructed by P1vir transduction (95). Unless otherwise noted, deletion alleles were derived from the Keio collection (96). Where indicated, the kanamycin resistance cassette was removed as has been previously described (97).
Detergent treatment and viability assay.
For evaluation of SDS resistance, strains were grown overnight to either carbon or nitrogen limitation or were grown for 30 min for actively growing cells. Cultures were treated with 10% SDS for a final concentration of 2% SDS or with an equal volume of water for control cultures and incubated at 37°C. Viability was assessed at the indicated time points by plating on LB medium and counting the numbers of CFU. For calculation of the fold decrease in viability, the viability of treated samples was compared to that of untreated samples. Unless otherwise noted, values are averages of the results of at least three independent biological replicates. Error bars represent the standard errors of the mean (SEM). Significance was calculated using the nonparametric Mann-Whitney test. We observed no indication that cell number directly affects SDS sensitivity.
Tn-Seq sample preparation.
A transposon mutant library was constructed from MG1655 by electroporation of the EZ-Tn5<KAN-2>Tnp Transposome (Epicentre) according to the manufacturer's instructions and selected on LB medium with 25 mg/liter kanamycin. Approximately 190,000 individual colonies were pooled for the initial transposon library. Overnight cultures in LB medium were grown from the pooled library and used to inoculate cultures for carbon or nitrogen limitation with approximately 100 copies of the library. The cultures were treated with SDS for 24 h as they were for viability analysis. Samples of 2 × 109 cells were taken before and after SDS treatment, and genomic DNA was isolated using the DNeasy blood and tissue kit (Qiagen) per the manufacturer's instructions. Libraries of transposon junctions were prepared using a method based on the transposon-directed insertion site sequencing (TraDIS) (98), with genomic DNA randomly sheared using a Covaris sonicator. The libraries were pooled and sequenced on the Illumina HiSeq 2500 sequencer in Rapid mode with 67-nt single-end reads in accordance with the standard manufacturer protocol. The sequencing data are available on the Princeton University HTSEQ database (https://htseq.princeton.edu).
Tn-Seq data analysis.
The sequencing reads were trimmed to 25 nt and mapped to the E. coli K-12 genome NC_000913.3 using BWA 1.2.3 (99). The number of reads mapped to each gene was quantified using htseq-count 0.6.0 (http://www-huber.embl.de/users/anders/HTSeq/doc/count.html). Tn-Seq reads across the genome were visualized using Integrative Genomics Viewer (100, 101). The average and median reads per gene were very similar between the different libraries (Table S2). Using the number of reads per gene, the log2 fold change between post- and pretreatment samples was calculated. To ensure the accuracy of fold change data, only genes with at least 700 reads under one or more conditions were analyzed further. Given the standard deviation of the fold values (Table S3), the frequency of transposon insertions in a given gene was considered changed if the change was at least 3-fold.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Silhavy laboratory for productive discussions. We also thank Jessica Wiggins at the Princeton University Genomics Core Facility for preparing libraries and performing sequencing for the Tn-Seq experiment.
This work was funded by National Institute of General Medical Science grants GM065216 and GM118024 and by a National Institute of General Medical Science Fellowship (F32GM116188).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00708-16.
REFERENCES
- 1.World Health Organization. 2014. Antimicrobial resistance: global report on surveillance. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X-Z, Plésiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. 2001. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother 45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baranova N, Nikaido H. 2002. The BaeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol 184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. 2005. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol Microbiol 55:1113–1126. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi A, Hirakawa H, Hirata T, Nishino K, Yamaguchi A. 2006. Growth phase-dependent expression of drug exporters in Escherichia coli and its contribution to drug tolerance. J Bacteriol 188:5693–5703. doi: 10.1128/JB.00217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brauner A, Fridman O, Gefen O, Balaban NQ. 2016. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 10.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 11.Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Putrinš M, Kogermann K, Lukk E, Lippus M, Varik V, Tenson T. 2015. Phenotypic heterogeneity enables uropathogenic Escherichia coli to evade killing by antibiotics and serum complement. Infect Immun 83:1056–1067. doi: 10.1128/IAI.02725-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schilling JD, Hultgren SJ. 2002. Recent advances into the pathogenesis of recurrent urinary tract infections: the bladder as a reservoir for uropathogenic Escherichia coli. Int J Antimicrob Agents 19:457–460. doi: 10.1016/S0924-8579(02)00098-5. [DOI] [PubMed] [Google Scholar]
- 14.Stewart GR, Robertson BD, Young DB. 2003. Tuberculosis: a problem with persistence. Nat Rev Microbiol 1:97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- 15.Knudsen GM, Ng Y, Gram L. 2013. Survival of bactericidal antibiotic treatment by a persister subpopulation of Listeria monocytogenes. Appl Environ Microbiol 79:7390–7397. doi: 10.1128/AEM.02184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuczyńska-Wiśnik D, Stojowska K, Matuszewska E, Leszczyńska D, Algara MM, Augustynowicz M, Laskowska E. 2015. Lack of intracellular trehalose affects formation of Escherichia coli persister cells. Microbiology 161:786–796. doi: 10.1099/mic.0.000012. [DOI] [PubMed] [Google Scholar]
- 17.Luidalepp H, Jõers A, Kaldalu N, Tenson T. 2011. Age of inoculum strongly influences persister frequency and can mask effects of mutations implicated in altered persistence. J Bacteriol 193:3598–3605. doi: 10.1128/JB.00085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol 183:6746–6751. doi: 10.1128/JB.183.23.6746-6751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tkachenko AG, Kashevarova NM, Karavaeva EA, Shumkov MS. 2014. Putrescine controls the formation of Escherichia coli persister cells tolerant to aminoglycoside netilmicin. FEMS Microbiol Lett 361:25–33. doi: 10.1111/1574-6968.12613. [DOI] [PubMed] [Google Scholar]
- 20.Davey P, Barza M, Stuart M. 1988. Tolerance of Pseudomonas aeruginosa to killing by ciprofloxacin, gentamicin and imipenem in vitro and in vivo. J Antimicrob Chemother 21:395–404. doi: 10.1093/jac/21.4.395. [DOI] [PubMed] [Google Scholar]
- 21.McKay SL, Portnoy DA. 2015. Ribosome hibernation facilitates tolerance of stationary-phase bacteria to aminoglycosides. Antimicrob Agents Chemother 59:6992–6999. doi: 10.1128/AAC.01532-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami K, Ono T, Viducic D, Kayama S, Mori M, Hirota K, Nemoto K, Miyake Y. 2005. Role for rpoS gene of Pseudomonas aeruginosa in antibiotic tolerance. FEMS Microbiol Lett 242:161–167. doi: 10.1016/j.femsle.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Rami A, Toutain CM, Jacq A. 2005. An increased level of alternative sigma factor RpoS partially suppresses drug hypersensitivity associated with inactivation of the multidrug resistance pump AcrAB in Escherichia coli. Res Microbiol 156:356–360. doi: 10.1016/j.resmic.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 24.Navarro Llorens JM, Tormo A, Martinez-Garcia E. 2010. Stationary phase in gram-negative bacteria. FEMS Microbiol Rev 34:476–495. doi: 10.1111/j.1574-6976.2010.00213.x. [DOI] [PubMed] [Google Scholar]
- 25.Lewis DL, Gattie DK. 1991. The ecology of quiescent microbes. ASM News 57:27–32. [Google Scholar]
- 26.Jenkins DE, Chaisson SA, Matin A. 1990. Starvation-induced cross protection against osmotic challenge in Escherichia coli. J Bacteriol 172:2779–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins DE, Schultz JE, Matin A. 1988. Starvation-induced cross protection against heat or H2O2 challenge in Escherichia coli. J Bacteriol 170:3910–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Small P, Blankenhorn D, Welty D, Zinser E, Slonczewski JL. 1994. Acid and base resistance in Escherichia coli and Shigella flexneri: role of rpoS and growth pH. J Bacteriol 176:1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huisman GW, Siegele DA, Zambrano MM, Kolter R. 1996. Morphological and physiological changes during stationary phase. ASM Press, Washington, DC. [Google Scholar]
- 30.Allen RJ, Scott GK. 1979. Biosynthesis and turnover of outer-membrane proteins in Escherichia coli ML308-225. Biochem J 182:407–412. doi: 10.1042/bj1820407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wensink J, Gilden N, Witholt B. 1982. Attachment of lipoprotein to the murein of Escherichia coli. Eur J Biochem 122:587–590. [DOI] [PubMed] [Google Scholar]
- 32.Dwyer RS, Ricci DP, Colwell LJ, Silhavy TJ, Wingreen NS. 2013. Predicting functionally informative mutations in Escherichia coli BamA using evolutionary covariance analysis. Genetics 195:443–455. doi: 10.1534/genetics.113.155861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the Gram-negative outer membrane. Proc Natl Acad Sci U S A 106:8009–8014. doi: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricci DP, Hagan CL, Kahne D, Silhavy TJ. 2012. Activation of the Escherichia coli beta-barrel assembly machine (Bam) is required for essential components to interact properly with substrate. Proc Natl Acad Sci U S A 109:3487–3491. doi: 10.1073/pnas.1201362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiz N, Wu T, Kahne D, Silhavy TJ. 2006. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem Biol 1:385–395. doi: 10.1021/cb600128v. [DOI] [PubMed] [Google Scholar]
- 36.Aires JR, Nikaido H. 2005. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J Bacteriol 187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elkins CA, Nikaido H. 2003. Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J Bacteriol 185:5349–5356. doi: 10.1128/JB.185.18.5349-5356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge Q, Yamada Y, Zgurskaya H. 2009. The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J Bacteriol 191:4365–4371. doi: 10.1128/JB.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bleuel C, Große C, Taudte N, Scherer J, Wesenberg D, Krauß GJ, Nies DH, Grass G. 2005. TolC is involved in enterobactin efflux across the outer membrane of Escherichia coli. J Bacteriol 187:6701–6707. doi: 10.1128/JB.187.19.6701-6707.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deininger KNW, Horikawa A, Kitko RD, Tatsumi R, Rosner JL, Wachi M, Slonczewski JL. 2011. A requirement of TolC and MDR efflux pumps for acid adaptation and GadAB induction in Escherichia coli. PLoS One 6:e18960. doi: 10.1371/journal.pone.0018960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misra R, Reeves PR. 1987. Role of micF in the tolC-mediated regulation of OmpF, a major outer membrane protein of Escherichia coli K-12. J Bacteriol 169:4722–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vega DE, Young KD. 2014. Accumulation of periplasmic enterobactin impairs the growth and morphology of Escherichia coli tolC mutants. Mol Microbiol 91:508–521. doi: 10.1111/mmi.12473. [DOI] [PubMed] [Google Scholar]
- 43.Wandersman C, Delepelaire P. 1990. TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc Natl Acad Sci U S A 87:4776–4780. doi: 10.1073/pnas.87.12.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandel MJ, Silhavy TJ. 2005. Starvation for different nutrients in Escherichia coli results in differential modulation of RpoS levels and stability. J Bacteriol 187:434–442. doi: 10.1128/JB.187.2.434-442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pratt LA, Silhavy TJ. 1996. The response regulator SprE controls the stability of RpoS. Proc Natl Acad Sci U S A 93:2488–2492. doi: 10.1073/pnas.93.6.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Opijnen T, Lazinski DW, Camilli A. 2001. Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms. Curr Protoc Mol Biol 106:7.16.1-24. doi: 10.1002/0471142727.mb0716s106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danese PN, Oliver GR, Barr K, Bowman GD, Rick PD, Silhavy TJ. 1998. Accumulation of the enterobacterial common antigen lipid II biosynthetic intermediate stimulates degP transcription in Escherichia coli. J Bacteriol 180:5875–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey MJA, Koronakis V, Schmoll T, Hughes C. 1992. Escherichia coli HIyT protein, a transcriptional activator of haemolysin synthesis and secretion, is encoded by the rfaH (sfrB) locus required for expression of sex factor and lipopolysaccharide genes. Mol Microbiol 6:1003–1012. doi: 10.1111/j.1365-2958.1992.tb02166.x. [DOI] [PubMed] [Google Scholar]
- 49.Creeger ES, Schulte T, Rothfield LI. 1984. Regulation of membrane glycosyltransferases by the sfrB and rfaH genes of Escherichia coli and Salmonella typhimurium. J Biol Chem 259:3064–3069. [PubMed] [Google Scholar]
- 50.Moores A, Chipper-Keating S, Sun L, McVicker G, Wales L, Gashi K, Blomfield IC. 2014. RfaH suppresses small RNA MicA inhibition of fimB expression in Escherichia coli K-12. J Bacteriol 196:148–156. doi: 10.1128/JB.00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pradel E, Schnaitman CA. 1991. Effect of rfaH (sfrB) and temperature on expression of rfa genes of Escherichia coli K-12. J Bacteriol 173:6428–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EGH, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11:941–950. doi: 10.1016/S0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 53.Hammann P, Parmentier D, Cerciat M, Reimegård J, Helfer A-C, Boisset S, Guillier M, Fo. Vandenesch Wagner EGH, Romby P, Fechter P. 2014. A method to map changes in bacterial surface composition induced by regulatory RNAs in Escherichia coli and Staphylococcus aureus. Biochimie 106:175–179. doi: 10.1016/j.biochi.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 54.Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P. 2006. Conserved small non-coding RNAs that belong to the σE regulon: role in down-regulation of outer membrane proteins. J Mol Biol 364:1–8. doi: 10.1016/j.jmb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 55.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 56.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adamowicz M, Kelley PM, Nickerson KW. 1991. Detergent (sodium dodecyl sulfate) shock proteins in Escherichia coli. J Bacteriol 173:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aspedon A, Nickerson KW. 1994. The energy dependence of detergent resistance in Enterobacter cloacae: a likely requirement for ATP rather than a proton gradient or a membrane potential. Can J Microbiol 40:184–191. doi: 10.1139/m94-031. [DOI] [PubMed] [Google Scholar]
- 59.Aspedon A, Nickerson KW. 1993. A two-part energy burden imposed by growth of Enterobacter cloacae and Escherichia coli in sodium dodecyl sulfate. Can J Microbiol 39:555–561. doi: 10.1139/m93-080. [DOI] [PubMed] [Google Scholar]
- 60.Rajagopal S, Eis N, Bhattacharya M, Nickerson KW. 2003. Membrane-derived oligosaccharides (MDOs) are essential for sodium dodecyl sulfate resistance in Escherichia coli. FEMS Microbiol Lett 223:25–31. doi: 10.1016/S0378-1097(03)00323-9. [DOI] [PubMed] [Google Scholar]
- 61.Rajagopal S, Sudarsan N, Nickerson KW. 2002. Sodium dodecyl sulfate hypersensitivity of clpP and clpB mutants of Escherichia coli. Appl Environ Microbiol 68:4117–4121. doi: 10.1128/AEM.68.8.4117-4121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, Horiuchi T. 2006. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol Syst Biol 2:2006.0007. doi: 10.1038/msb4100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jishage M, Ishihama A. 1997. Variation in RNA polymerase sigma subunit composition within different stocks of Escherichia coli W3110. J Bacteriol 179:959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adamowicz M, Conway T, Nickerson KW. 1991. Nutritional complementation of oxidative glucose metabolism in Escherichia coli via pyrroloquinoline quinone-dependent glucose dehydrogenase and the Entner-Doudoroff pathway. Appl Environ Microbiol 57:2012–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nickerson KW, Aspedon A. 1992. Detergent-shock response in enteric bacteria. Mol Microbiol 6:957–961. doi: 10.1111/j.1365-2958.1992.tb02161.x. [DOI] [PubMed] [Google Scholar]
- 66.Rida S, Caillet J, Alix JH. 1996. Amplification of a novel gene, sanA, abolishes a vancomycin-sensitive defect in Escherichia coli. J Bacteriol 178:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes1. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 68.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chao KL, Lim K, Lehmann C, Doseeva V, Howard AJ, Schwarz FP, Herzberg O. 2008. The Escherichia coli YdcF binds S-adenosyl-l-methionine and adopts an α/β-fold characteristic of nucleotide-utilizing enzymes. Proteins 72:506–509. doi: 10.1002/prot.22046. [DOI] [PubMed] [Google Scholar]
- 70.Hagiwara D, Sugiura M, Oshima T, Mori H, Aiba H, Yamashino T, Mizuno T. 2003. Genome-wide analyses revealing a signaling network of the RcsC-YojN-RcsB phosphorelay system in Escherichia coli. J Bacteriol 185:5735–5746. doi: 10.1128/JB.185.19.5735-5746.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salmon K, Hung SP, Mekjian K, Baldi P, Hatfield GW, Gunsalus RP. 2003. Global gene expression profiling in Escherichia coli K12: the effects of oxygen availability and Fnr. J Biol Chem 278:29837–29855. doi: 10.1074/jbc.M213060200. [DOI] [PubMed] [Google Scholar]
- 72.Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. 2014. A genome-wide screen for bacterial envelope biogenesis mutants identifies a novel factor involved in cell wall precursor metabolism. PLoS Genet 10:e1004056. doi: 10.1371/journal.pgen.1004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meier-Dieter U, Barr K, Starman R, Hatch L, Rick PD. 1992. Nucleotide sequence of the Escherichia coli rfe gene involved in the synthesis of enterobacterial common antigen. Molecular cloning of the rfe-rff gene cluster. J Biol Chem 267:746–753. [PubMed] [Google Scholar]
- 74.Mouslim C, Cano AD, Casadesús J. 1998. The sfiX, rfe and metN genes of Salmonella typhimurium and their involvement in the His(c) pleiotropic response. Mol Gen Genet 259:46–53. doi: 10.1007/s004380050787. [DOI] [PubMed] [Google Scholar]
- 75.Huerta AM, Collado-Vides J. 2003. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J Mol Biol 333:261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 76.Janaszak A, Nadratowska-Wesołowska B, Konopa G, Taylor A. 2009. The P1 promoter of the Escherichia coli rpoH gene is utilized by σ70-RNAP or σS-RNAP depending on growth phase. FEMS Microbiol Lett 291:65–72. doi: 10.1111/j.1574-6968.2008.01436.x. [DOI] [PubMed] [Google Scholar]
- 77.Jenkins DE, Auger EA, Matin A. 1991. Role of RpoH, a heat shock regulator protein, in Escherichia coli carbon starvation protein synthesis and survival. J Bacteriol 173:1992–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amanuma H, Strominger JL. 1980. Purification and properties of penicillin-binding proteins 5 and 6 from Escherichia coli membranes. J Biol Chem 255:11173–11180. [PubMed] [Google Scholar]
- 79.Ghosh AS, Chowdhury C, Nelson DE. 2008. Physiological functions of d-alanine carboxypeptidases in Escherichia coli. Trends Microbiol 16:309–317. doi: 10.1016/j.tim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 80.Typas A, Banzhaf M, Gross CA, Vollmer W. 2011. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Potluri L-P, de Pedro MA, Young KD. 2012. Escherichia coli low-molecular-weight penicillin-binding proteins help orient septal FtsZ, and their absence leads to asymmetric cell division and branching. Mol Microbiol 84:203–224. doi: 10.1111/j.1365-2958.2012.08023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen LH, Burgess RR. 1997. Comparative analysis of the interactions of Escherichia coli σS and σ70 RNA polymerase holoenzyme with the stationary-phase-specific bolAp1 promoter. Biochemistry 36:1748–1754. doi: 10.1021/bi961175h. [DOI] [PubMed] [Google Scholar]
- 83.Santos JM, Lobo M, Matos APA, De Pedro MA, Arraiano CM. 2002. The gene bolA regulates dacA (PBP5), dacC (PBP6) and ampC (AmpC), promoting normal morphology in Escherichia coli. Mol Microbiol 45:1729–1740. doi: 10.1046/j.1365-2958.2002.03131.x. [DOI] [PubMed] [Google Scholar]
- 84.Henderson TA, Young KD, Denome SA, Elf PK. 1997. AmpC and AmpH, proteins related to the class C beta-lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli. J Bacteriol 179:6112–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meberg BM, Paulson AL, Priyadarshini R, Young KD. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J Bacteriol 186:8326–8336. doi: 10.1128/JB.186.24.8326-8336.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nelson DE, Young KD. 2000. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol 182:1714–1721. doi: 10.1128/JB.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol 181:3981–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peters K, Kannan S, Rao VA, Biboy J, Vollmer D, Erickson SW, Lewis RJ, Young KD, Vollmer W. 2016. The redundancy of peptidoglycan carboxypeptidases ensures robust cell shape maintenance in Escherichia coli. mBio 7:e00819-16. doi: 10.1128/mBio.00819-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sarkar SK, Dutta M, Chowdhury C, Kumar A, Ghosh AS. 2011. PBP5, PBP6 and DacD play different roles in intrinsic β-lactam resistance of Escherichia coli. Microbiology 157:2702–2707. doi: 10.1099/mic.0.046227-0. [DOI] [PubMed] [Google Scholar]
- 90.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, Von Mering C. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deng M, Misra R. 1996. Examination of AsmA and its effect on the assembly of Escherichia coli outer membrane proteins. Mol Microbiol 21:605–612. doi: 10.1111/j.1365-2958.1996.tb02568.x. [DOI] [PubMed] [Google Scholar]
- 92.Misra R, Miao Y. 1995. Molecular analysis of asmA, a locus identified as the suppressor of OmpF assembly mutants of Escherichia coli K-12. Mol Microbiol 16:779–788. doi: 10.1111/j.1365-2958.1995.tb02439.x. [DOI] [PubMed] [Google Scholar]
- 93.Xiong X, Deeter JN, Misra R. 1996. Assembly-defective OmpC mutants of Escherichia coli K-12. J Bacteriol 178:1213–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu P, Janga SC, Babu M, Díaz-Mejía JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P, Chandran S, Christopoulos C, Nazarians-Armavil A, Nasseri NK, Musso G, Ali M, Nazemof N, Eroukova V, Golshani A, Paccanaro A, Greenblatt JF, Moreno-Hagelsieb G, Emili A. 2009. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol 7:e96. doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 96.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008-2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Langridge GC, Phan M-D, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK. 2009. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.