

Abstract
The Ewing sarcoma breakpoint region 1 (EWSR1) gene is known to fuse with various partner genes to promote the development of the Ewing sarcoma family of tumors and other sarcomas. In contrast, the association of EWSR1 chimeric fusion genes with leukemia has rarely been reported. We identified a novel EWSR1‐associated chimeric fusion gene in a patient with acute myeloid leukemia harboring 46, XY, t (11; 22) (p13; q12) karyotype abnormality. The patient was refractory to intensified chemotherapy including hematopoietic stem cell transplantation. Total RNA paired‐end sequencing identified a novel chimeric fusion gene as EWSR1/ELF5, a member of the E26 transformation‐specific transcription factor family. Transduction of EWSR1/ELF5 to NIH3T3 cells induced transformation by attenuating with the p53/p21‐dependent pathway. The injection of EWSR1/ELF5‐transduced NIH3T3 cells into NSG‐SCID mice systematically induced the development of tumors in vivo. These results revealed the oncogenic potency of EWSR1/ELF5.
Keywords: Acute myelogenous leukemia, ELF5, EWSR1, senescence, TP53
Ewing sarcoma breakpoint region 1 (EWSR1) belongs to a small family of RNA binding proteins, including FUS, EWSR1 and TAFII68. They are involved in multiple cellular processes, including gene expression, cell signaling and RNA processing and transport.1, 2 The encoding genes are known to fuse with several partner genes encoding transcription factors which are associated with sarcoma and leukemia. The phenotype of the tumor is defined by the fusion partner. For example, the Ewing sarcoma family of tumors (EFT) carries EWSR1 fused to a member of the E26 transformation‐specific (ETS) transcription factor family, such as FLI1, ERG, ETV1, ETV4 or FEV.3, 4, 5, 6, 7 In other types of tumors, EWSR1 fuses with WT1 in desmoplastic small round cell sarcoma, with CHN in myxoid chondrosarcoma, with ATF1 in clear cell sarcoma, and with CHOP in myxoid liposarcoma.8, 9, 10, 11 Although EWSR1 is mainly associated with sarcoma, one report showed the fusion of EWSR1 with ZNF384 (CIZ/NMP4) in patients diagnosed with acute undifferentiated leukemia and acute lymphoblastic leukemia (ALL).12
The molecular mechanism of oncogenic transformation induced by a chimeric gene is relatively well characterized for EWSR1/FLI1. In vitro studies suggest that a disruption of senescence or DNA damage responses, both associated with the tumor barrier, may play a critical role in the tumorigenic effects of EWSR1/ETS.13, 14 In addition, the tumor phenotype is determined by the cell type expressing EWSR1/ETS. Expression of EWSR1/FLI1 in NIH3T3 cells and primary murine bone marrow derived mesenchymal stem cells (MSC) induced oncogenesis, but not in Rat‐1 cells, primary mouse embryonic fibroblasts (MEF) and hTERT‐immortalized human primary fibroblasts. Intriguingly, EWSR1/FLI1 can direct partial neuroectodermal differentiation of primary mesenchymal stem cells.15 However, EWSR1/ETS is rarely associated with leukemia,16, 17 thus preventing hematopoietic lineage analysis in clinical specimens. However, conditional EWSR1/ETS transgenic mice exhibit a leukemia phenotype, suggesting that the expression of EWSR1/ETS in the hematopoietic lineage has leukemogenic potential.18, 19
We identified a 2‐year‐old boy who developed acute myeloid leukemia (AML) and carried a novel EWSR1/ETS chimeric fusion gene, EWSR1/ELF5. Chromosomal and functional assays demonstrate that this fusion gene promotes oncogenesis by interfering with the p53/p21‐dependent pathway.
Materials and Methods
Cytogenetic analysis
FISH analysis was performed following the standard method. The probe used for EWSR1 was the LSI EWSR1 dual‐color break‐apart probe (Abbott Molecular/Vysis, Des Plaines, IL, USA).
Establishment of an Epstein–Barr virus‐transformed lymphoblastoid cell line
An Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL) was established using peripheral lymphocytes from a patient when they had first achieved remission. The Epstein–Barr virus from the B95‐8 strain was used to infect the lymphocytes, and the cells were cultured with RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 20% FBS and cyclosporin 200 ng/mL, as described previously.20 EB‐LCL were maintained in RPMI 1640 with 15% FBS at approximately 3–5 × 105 cells/mL at 37°C in 5% CO2.
Total RNA paired‐end sequencing
The RNA paired‐end sequencing (RNA‐seq) experiments were performed as previously described.21 All samples collected from the patient were obtained after obtaining written informed consent from the parents. The research protocol was approved by the Institutional Review Board of the Tokyo Medical and Dental University (No. 103). Total RNA was extracted from the cells of AML patients, and the patient's Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL) using Sepagene (Eidia, Tokyo, Japan). The cDNA was generated using the SmartPCR cDNA kit (Clontech Laboratories, Mountain View, CA, USA) and fragmented using the Covaris instrument (Covaris, Woburn, MA, USA). The cDNA fragments were used to prepare an Illumina library with the NEBNext reagents (New England Biolabs, Ipswich, MA, USA). The libraries were then submitted for Illumina HiSeq2000 sequencing, according to the standard protocols. Paired‐end 100 nucleotide reads were generated and verified for data quality using the FASTQC software (Babraham Institute, Cambridge, UK) and mapped using the reference human genome (Homo sapiens hg19 sequence). Fusion transcript discovery was performed using the CLC genomics Workbench software 6.0.2 (CLC‐bio, Aarhus, Denmark), which identifies the fusion transcripts by clustering discordantly the aligning paired‐end reads spanning a fusion breakpoint.
RT‐PCR and direct sequencing
The RT‐PCR experiments were performed using standard protocols. The mRNA from the patient's AML cells were reverse‐transcribed into cDNA using SuperScript III (Thermo Fisher Scientific). The EWSR1/ELF5 fusion transcript was confirmed by RT‐PCR using patient cDNA and specific primers for EWSR1 (5′‐CAGCCACTGCACCTACAAGA) and ELF5 (5′‐AATGAGCTTGATGCCTGGAG). The cDNA PCR‐amplicon was detected after electrophoresis on a 1% agarose gel and was then purified and sequenced using a BigDye Terminator kit (version 3.1, Applied Biosystems, Foster City, CA, USA).
Plasmid constructs
FLAG‐tagged EWSR1/ELF5 was generated by PCR amplification of the cDNA of the patient's AML cells using Phusion high‐fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA) and specific primers for EcoRI‐FLAG‐tagged EWSR1 (5′‐ATGCGAATTCGCCACCATGGATTACAAGGATGACGACGATAAGGCGTCCACGGATTACA) and XhoI‐tagged ELF5 (5′‐AGACTCGAGTCATAGCTTGTCTTCCTGCCA). The PCR product was cloned into the pCR2.1‐TOPO TA vector (Invitrogen, Carlsbad, CA, USA) and verified by sequence analysis. Then, the insert was transferred into the EcoRI‐SalI site of the pBABE‐Puro retroviral vector in the correct orientation downstream to the 5′ long terminal repeat.22
Cell lines and transduction of DNA
NIH3T3 cells, H1299 and U2OS cells were purchased from ATCC (Manassas, VA, USA) and grown in DMEM, supplemented with 10% FBS and penicillin–streptomycin (100 units/mL). The patient's EB‐LCL was grown in RPMI medium supplemented with 10% FBS and penicillin–streptomycin (100 units/mL). All cell lines were maintained at 37°C in an atmosphere of 5% CO2. The pBABE‐Puro vectors containing FLAG‐EWSR1/ELF5 or empty vectors (MOCK) were transfected using a polyethyleneimine into PlatE cells, an ecotropic packaging cell line.23 Supernatants containing high titers of retrovirus were collected at 48 and 72 h and used to infect the NIH3T3 cell line. NIH3T3 cells were seeded at a density of 2 × 105 cells/well in a 6‐well plate during 24 h before adding viral supernatant containing 4‐μg/mL protamine. Although infection efficacy was >90%, the infected cells were selected with 3‐μg/mL puromycin during the 48 h after the infection. The U2OS and the H1299 cells were directly transfected using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer's protocol.
Transformation and tumorigenesis assays in vitro
For the focus formation assay, FLAG‐EWSR1/ELF5‐transduced or MOCK‐transduced NIH3T3 cells were seeded at a density of 106 cells/10‐cm culture dish over 3 weeks. The culture medium was replaced twice a week. Soft agar assays were performed as previously described.24 FLAG‐EWSR1/ELF5‐transduced or MOCK‐transduced NIH3T3 cells were plated at a density of 1 × 104 cells/3.5‐cm dish in 0.3% agar. The number of colonies measuring >150 μm in diameter was scored on day 14.
Apoptosis analysis and senescence associated‐β‐gal staining
Apoptotic cells were identified by flow cytometry using Annexin V‐PE (MBL, Nagoya, Japan). Flow cytometry analysis was conducted on a FACSCalibur instrument (Becton Dickinson, Franklin Lakes, NJ, USA) using the CellQuest software. The detection protocol for senescence associated (SA)‐β‐gal activity was as previously described.25
In vivo mouse model
Six‐week‐old female NSG‐SCID mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Experimental and animal care protocols were approved by the Tokyo Medical and Dental University Animal Care and Use Committee (protocol numbers 0150358A and 0150004A). NIH3T3 cells (1 × 106) transduced with FLAG‐EWSR1/ELF5 or MOCK vectors were inoculated subcutaneously into NSG‐SCID mice, and their tumorigenicity was monitored over 6 weeks.
Gene expression profiling and Gene mutation analysis
Gene expression levels were determined according to the entire coverage of a gene, which was defined as the sum of the coverage of each non‐redundant exonic nucleotide normalized by all mapped nucleotides. The CLC genomics Workbench software 6.0.2 (Qiagen, Rewood City, CA, USA) was used to calculate the expression level of the UniProtKB genes annotated in reads per kilobase of transcript per million mapped reads (RPKM) and to identify gene mutations.
Western blotting
Cell line lysates were prepared in TGN buffer (50‐mM Tris‐HCl pH 7.5, 150‐mM NaCl, 1% Tween 20 and 0.5% NP40) containing the protease inhibitor PMSA. The lysates were electrophoresed on SDS‐polyacrylamide gel and then transferred to a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% nonfat milk and then incubated with primary antibodies against the following proteins: FLAG M2 (F3165, Sigma‐Aldrich, St. Louis, MO, USA), β‐Actin (AC‐15, Santa Cruz Biotechnology, Dallas, TX, USA), p53 (Ab‐6, Santa Cruz Biotechnology) and p21 (c‐19, Santa Cruz Biotechnology). The primary antibodies were detected with HRP‐conjugated anti‐rabbit or anti‐mouse secondary antibodies and visualized using an enhanced chemiluminescence (ECL) kit (GE Healthcare, Little Chalfont, UK).
Luciferase assay
Luciferase assay was performed as described previously.26 Briefly, p53 null H1299 cells were cultured in 24‐well plates and transfected with pCMV wild‐type p53 vector, pG13 p21 promoter‐luciferase reporter plasmid or TM861‐2 BAX, pRL‐renilla‐luciferase expression vector, and pBABE‐Puro FLAG‐EWSR1/ELF5 or pBABE‐Puro MOCK vector. Then, 24 h after transfection, firefly and renilla luciferase activities were measured using the Dual Luciferase Assay Kit (Promega, Madison, WI, USA). The firefly luciferase values of each sample were normalized by renilla luciferase activity.
Results
Case presentation
A 2‐year‐old boy was admitted to our hospital with skin lesions presenting extramedullary infiltrations. He was diagnosed with low blast‐count AML immunologically positive for CD7, CD33, CD34, CD45dull, CD61, CD99 and CD117 (c‐kit) but negative for the sarcoma markers desmin, MyoD1, myogenin, neuron‐specific enolase (NSE) and paired box gene 5 (PAX5). The leukemia karyotype was identified as 46, XY, t (11; 22) (p13; q12), and FISH analysis detected EWSR1 split signal (Fig. 1a,b). The patient was given conventional induction therapy, including daunorubicin, cytosine arabinoside, mitoxantrone and etoposide but failed to achieve complete remission. Therefore, he received a hematopoietic stem cell transplantation (HSCT) from his mother, who expressed identical human leukocyte antigen (HLA). The conditioning regimen using melphalan and total body irradiation (total dose: 12 Gy) was employed. However, the AML relapsed 1 year after HSCT. A second HSCT was performed, but remission was not achieved and the patient died.
Figure 1.
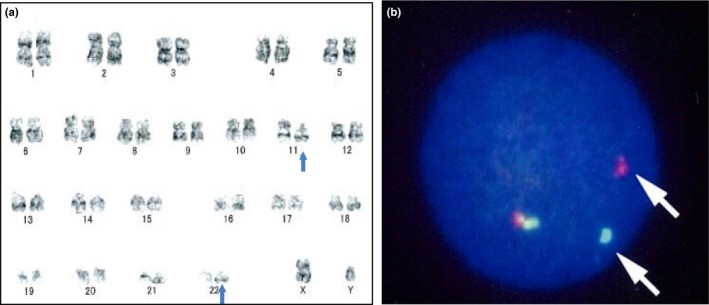
Chromosome analysis providing evidence of EWSR1 rearrangement. (a) Complete G‐banding chromosomal analysis identified a translocation between 11p13 and 22q12 (blue arrows). (b) FISH analysis revealed EWSR1 split signals (green or red) on the acute myeloid leukemia (AML) cells, whereas intact EWSR1 genes appear as fused yellow signals. The EWSR1 rearrangement‐positive cells show one yellow, one red and one green signal pattern.
Total RNA paired‐end sequencing identifies the new EWSR1/ELF5 chimeric fusion transcript
Chromosomal G‐banding analysis using leukemic cell revealed translocation between 22q12 and 11p13 in leukemia cells (Fig. 1a). FISH analysis provided evidence of gene translocation involving EWSR1, the gene located at 22q12 (Fig. 1b). Chromosomal G‐banding analysis suggests that the partner gene is located at 11p (Fig. 1a). The only gene which has been known to fuse with EWSR1 located at 11p was WT1. However, RT‐PCR analysis failed to detect EWSR1/WT1 fusion mRNA (the partner gene(s) fused with EWSR1. The RNA samples were obtained from primary cells data not shown). Therefore, total RNA paired‐end sequencing (RNA‐seq) was performed to identify the patient's AML cells, and EB‐LCL cells were derived from the patient for the internal control. A total of 28.9 million paired‐end AML‐derived RNA fragments and 29.0 million paired‐end EB‐LCL‐derived RNA fragments were sequenced. Among them, the fusion of ELF5 and EWSR1 (19 reads) was detected only in the AML samples (Fig. 2a). The presence of EWSR1/ELF5 fusion mRNA was confirmed by RT‐PCR and Sanger sequencing (Fig. 2b,c). The exon 7 of EWSR1 (NM_013986.3, c.1139) was fused to the exon 3 of ELF5 (NM_001422.3, c250) (Fig. 2c,d). EWSR1 has a Ser–Tyr–Gln–Gly‐rich (SYQG) transactivation domain at the NH2 terminus, followed by an RNA‐binding domain composed of three Arg–Gly–Gly‐rich (RGG) regions and an RNA recognition motif (RRM) at the COOH terminus. ELF5 contains an ETS DNA‐binding domain at the COOH terminus. In the EWSR1/ELF5 fusion transcript, the RNA‐binding domain of EWSR1 is replaced by the ETS DNA‐binding domain of ELF5, as reported for other EWSR1/ETS gene fusions.3, 4, 5, 6, 7
Figure 2.
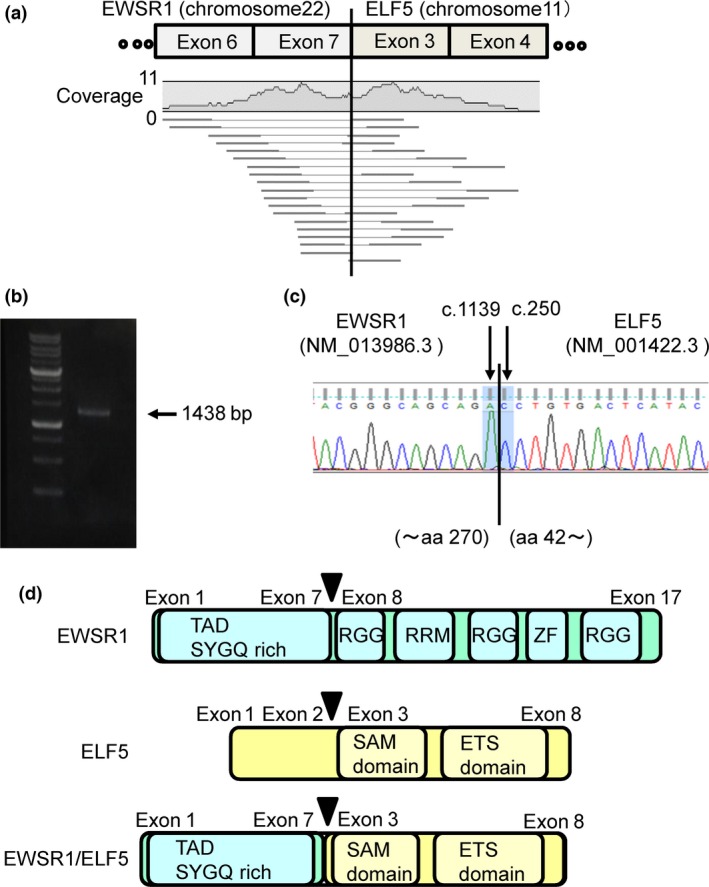
Total RNA paired‐end sequencing (RNA‐seq) identified the EWSR1/ELF5 transcript fusion. (a) Fragments of EWSR1/ELF5 fusion mRNA observed by RNA‐seq. Alignment of 17 mate‐pairs and two single sequences on either side of the breakpoint (pairing EWSR1 and ELF5). The histogram shows the absolute coverage of the sequence reads, and the mean numbers of reads. (b) RT‐PCR with a EWSR1 sense primer and an ELF5 antisense primer showing the EWSR1/ELF5 fusion mRNA product. (c) Direct sequencing of the RT‐PCR product confirmed the identity of the EWSR1/ELF5 fusion. aa, amino acid. (d) Schematic representation of the EWSR1 wild‐type protein (top panel), the ELF5 wild‐type protein (middle panel) and the EWSR1/ELF5 fusion protein (bottom panel). Arrows indicate the breakpoint. SAM, sterile alpha motif; ZF, zinc finger.
Mutation or altered expressed genes in leukemic cells
By comparing with EB‐LCL, tumor‐specific mutations and the altered expression of genes were investigated using RNA‐seq data. In the leukemic cells, 223 mutations were identified, although mutation of the well‐characterized tumor suppressor gene TP53 was not detected. Instead, mutation of CASP9, a central player in common apoptosis pathways, was identified (Data S1). Analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) revealed several patterns of tumor‐specific gene alterations. Among them, gene ontology classification revealed that genes involved in proteolysis were highly mutated (Data S2). Pathway analysis extracted two pathways: protein processing in the endoplasmic reticulum and control of gene expression by the vitamin D receptor (Data S3). Various non‐expressed genes were also identified. This gene silencing likely occurs due to gene deletion or gene silencing by epigenetic alteration (Data S1 and S4). Among them, changes in expression of CDKN1A, a cell cycle inhibitor, and NPM1, known to be mutated in AML, were both observed (Data S1). Intriguingly, pathway analysis revealed that several genes associated with DNA replication were also silenced (Data S5).
The EWSR1/ELF5 chimeric protein possesses transformation properties and tumorigenic potential in vitro and in vivo
The oncogenic potential of EWSR1/ELF5 was investigated by transformation assay using NIH3T3 cells, the cell line traditionally used to test the oncogenicity of other EWSR1/ETS fusion transcripts.27, 28 FLAG‐tagged EWSR1/ELF5 or empty vectors (MOCK) were retrovirally transduced into NIH3T3 cells (Fig. 3a). The EWSR1/ELF5 fusion transcript consistently induced the transformation of NIH3T3 cells, as evidenced by a loss of contact inhibition with more proliferative activity than the MOCK‐transduced cells (Fig. 3b,c). The tumorigenic potential of EWSR1/ELF5 was also analyzed by the substrate‐independent growth assay using EWSR1/ELF5 and MOCK transduced NIH3T3 cells in soft agar. The EWSR1/ELF5‐transduced cells formed over six times more colonies than the MOCK‐transduced cells (Fig. 3d).
Figure 3.
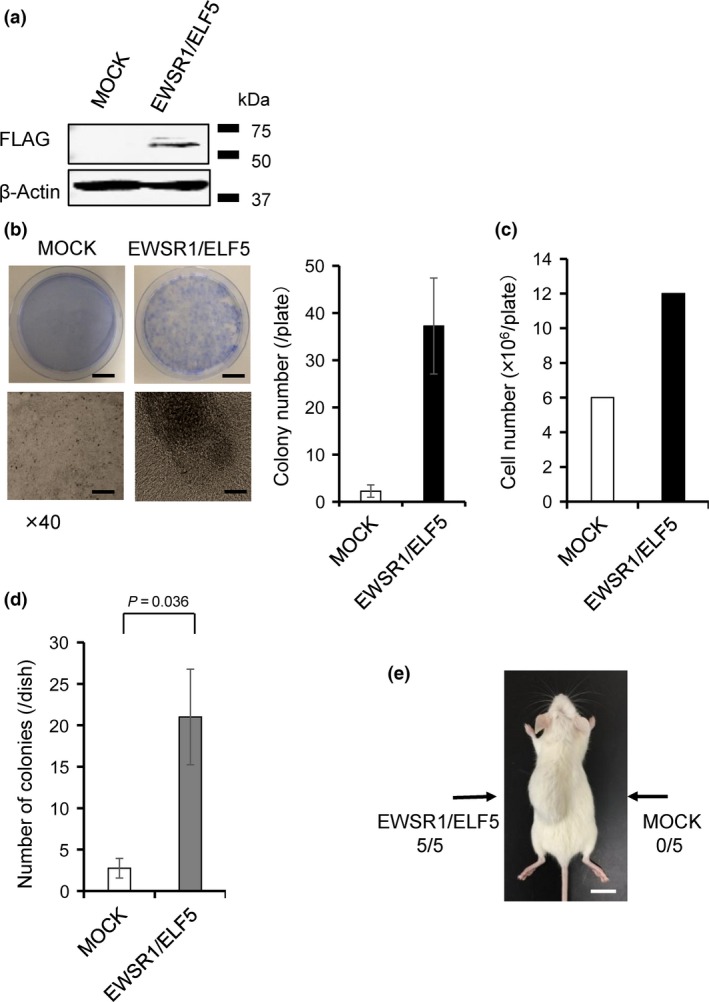
Transforming and tumorigenic properties of the EWSR1/ELF5 fusion protein. (a) Expression of FLAG‐tagged EWSR1/ELF5 in NIH3T3 cells confirmed by immunoblot analysis. (b) Macroscopic images of the focus formation assay taken with crystal violet staining after 3 weeks culture (top panels). Microscopic images (bottom panels) taken at indicated magnifications. Scale bars, top panels 20 mm; bottom panels 1000 μm. Average colony number from four independent experiments are shown as a bar graph. (c) Comparison of the number of NIH3T3 cells observed in (b). (d) Colony assay in soft agar showing that EWSR1/ELF5 promotes the anchorage‐independent growth of NIH3T3 cells. The number of colonies was scored on day 14. The data are presented as the mean ± SE (n = 3). The Student t‐test was used to compare differences. (e) In vivo tumorigenicity assay using NSG‐SCID mice (n = 5). The EWSR1/ELF5‐transduced and MOCK‐transduced cells were injected on opposite sides of the same mice (arrows). Scale bar, 10 mm.
The in vivo oncogenic effect was tested by injecting EWSR1/ELF5‐transduced or MOCK‐transduced NIH3T3 cells into the left and right sides of five NSG‐SCID mice, respectively. After 6 weeks, all the mice presented a large tumor on the left side only (Fig. 3e). The in vitro leukemogenic potential of EWSR1/ELF5 was first investigated using the Ba/F3 hematopoietic cell line. In this murine pro‐B‐cell line, oncogenic transformation was reported to induce growth factor‐independent cell growth or enhanced resistance to apoptosis.29 However, EWSR1/ELF5 transduction did not elicit these transformations (data not shown). Next, the EWSR1/ELF5 fusion gene was transduced into murine bone marrow cells to conduct colony assays, as previously described.30 EWSR1/ELF5 transduction did not stimulate significant colony formation (Fig. S1). In addition, HSCT of EWSR1/ELF5‐transduced murine bone marrow cells into lethally‐irradiated mice did not induce leukemia over a period of 12 months, based on the normal blood counts of low‐level reporter genes in the peripheral blood (data not shown). These experiments suggest that despite the structural differences between the partner genes, EWSR1/ELF5 has transformation potential as the other EWSR1 fusions in NIH3T3 cells but not in hematopoietic cell lineages.
Aberrant EWSR1/ELF5 expression had toxicity in NIH3T3 cells
The oncogenic potential of EWSR1/ELF5 was supported by transductions conducted in NIH3T3 cells but not in the Ba/F3 hematopoietic lineage cell line. We hypothesized that the expression of EWSR1/ELF5 is toxic in primary cells cultured in vitro. Therefore, we investigated whether the transduction of EWSR1/ELF5 induces apoptosis or cellular senescence in NIH3T3 cells. Apoptosis was increased twofold in EWSR1/ELF5‐transduced cells, compared to MOCK‐transduced cells (Fig. 4a,b). In addition, SA‐β‐gal staining was enhanced in EWSR1/ELF5 transduced NIH3T3 cells (Fig. 4c). Together, these data suggest that EWSR1/ELF5 induces senescence and apoptosis, as reported for EWSR1/FLI1, yet is associated with transformation activity.14
Figure 4.
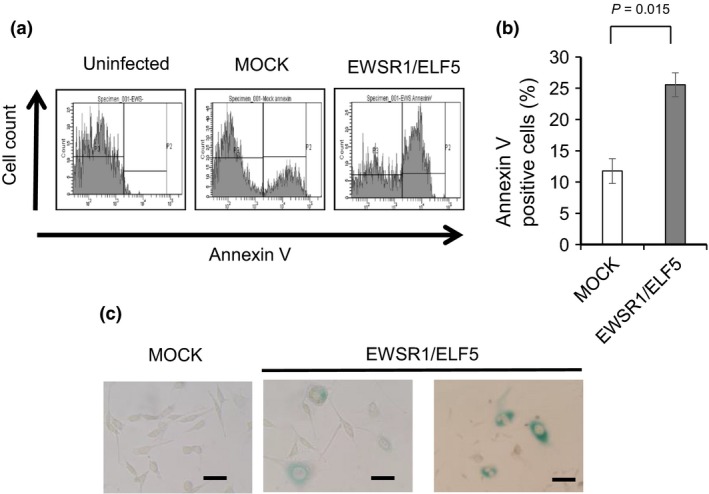
The expression of EWSR1/ELF5 induces toxicity in NIH3T3 cells. The cells were either uninfected, transduced with the empty vector (MOCK) or transduced with EWSR1/ELF5. (a) Flow cytometry analysis of apoptotic cells detected using Annexin V. (b) Comparison of the percentages of apoptotic cells. The data are presented as the mean ± SE (n = 3). The Student t‐test was used to compare differences. (c) Senescence associated (SA)‐β‐gal staining of MOCK‐transduced and EWSR1/ELF5‐transduced cells. Senescent cells were stained in blue. Scale bar, 50 μm.
EWSR1/ELF5 drives an expression signature of transformation
RNA‐seq analysis not only detects fusion mRNA but also provides genome‐wide mapping of quantitative mRNA expressions. A comparison of the expression profiles of the patient's AML cells and the patient's EB‐LCL revealed the alignment of 17 797 annotated genes. DAVID analysis of the most significantly upregulated and downregulated 200 genes in the patient's AML cells in comparison with the patient's EB‐LCL identified several informative Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, including those expressed by the hematopoietic cell lineage pathway, the cytokine–cytokine receptor interaction pathway, and the p53 signaling pathway (Fig. 5a,b; gene lists in Tables S1 and S2).31
Figure 5.
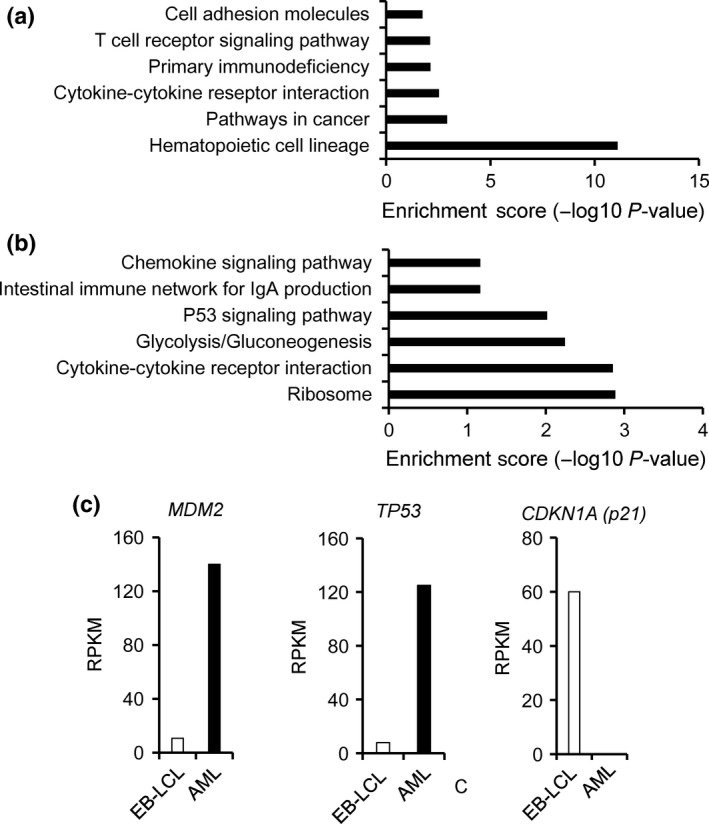
Gene expression profile of acute myeloid leukemia (AML) cells. DAVID analysis of the most (a) upregulated and (b) downregulated 200 genes in the patient's AML cells, compared to the patient's Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL). (c) mRNA expression level of MDM2,TP53 and CDKN1A in the patient's AML cells, compared to the patient's EB‐LCL control cells. RPKM, reads per kilobase of transcript per million mapped reads.
The expression of EWSR1/ELF5 activates p53 but inhibits p21
Given the senescence and apoptotic phenotypes induced by EWSR1/ELF5 (Fig. 4a–c) and the identification of the p53 signaling pathway involved in apoptosis by DAVID analysis (Fig. 5b), we focused on major molecules implicated in the p53 signaling pathway: TP53, which encodes p53; MDM2 and p21 (CDKN1A), which operates downstream of p53. In the patient's AML cells, TP53 and MDM2 are upregulated, whereas p21 (CDKN1A) is downregulated, compared to the patient's control EB‐LCL cells (Fig. 5c). The EWSR1/FLI1 fusion transcript was reported to inhibit p21 activity.32, 33, 34 To determine whether EWSR1/ELF5 also suppresses p21 expression, EWSR1/ELF5 was transduced into p53‐competent U2OS cells. Intriguingly, EWSR1/ELF5 stimulated the baseline expression of p53 and p21, compared to MOCK‐transduced U2OS cells (Fig. 6a,b). In contrast, p21 induction was inhibited by EWSR1/ELF5 after 5‐Gy irradiation, suggesting that the fusion transcript functions as a suppressor of p21 transcription after DNA damage. The effect of EWSR1/ELF5 to p53 downstream gene promoters (p21 and BAX promoter) were investigated using a luciferase reporter assay. Wild‐type p53 expression vectors were co‐transfected with MOCK or EWSR1/ELF5 expressing vector into H1299 cells. Interestingly, basal p21 reporter promoter activity was upregulated by EWSR1/ELF5 expression (Fig. 6c). p21 reporter promoter activity induced by p53 transfection was attenuated by EWSR1/ELF5 expression. In contrast, BAX reporter promoter activity was not suppressed by EWSR1/ELF5 expression (Fig. 6d). These data indicate that the novel EWSR1/ELF5 fusion transcript promotes oncogenesis through a dysregulation of the p53/p21‐dependent pathway (Fig. 7), similar to other EFT.
Figure 6.
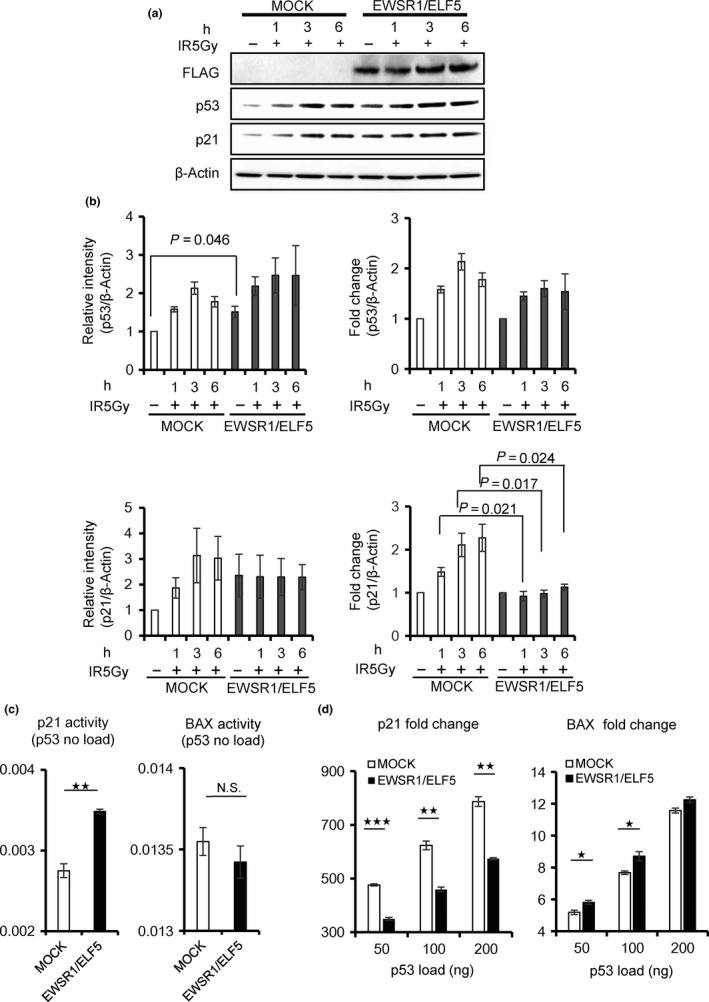
Impact of the EWSR1/ELF5 fusion transcript on the p53/p21 pathway promoting apoptosis. (a) Western blot analysis of U2OS cells transduced with EWSR1/ELF5 or the empty vector (MOCK). Cells were irradiated with 5 Gy, then harvested after 1, 3 or 6 h. (b) Left panel, quantitative analysis of the Western blot from three independent experiments. The intensities of p53 and p21 expression were normalized to β‐Actin expression. Right panel, fold increase in p53 and p21 expression. The values obtained from unirradiated samples are set as 1. The data are presented as the mean ± SE. (c) p53 reporter (p21 or BAX) promoter activity was measured using a luciferase reporter system. Luciferase activity was shown 24 h after the transfection of MOCK (white column) or EWSR1/ELF5 (black column). (d) Fold increase of luciferase activity was shown 24 h after the transfection of MOCK (white column) or EWSR1/ELF5 (black column) with various amounts of p53 expression vectors in H1299 cells. The values obtained from no load of p53 samples are set as 1. The Student t‐test was used to compare differences. *p < 0.05; **p < 0.005; ***p < 0.0005.
Figure 7.
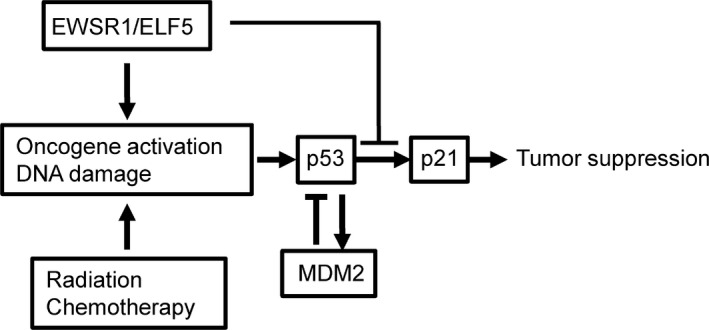
Proposed schematic model of tumorigenicity by EWSR1/ELF5.
Discussion
In the present study, we identified EWSR1/ELF5, a new EWSR1/ETS chimeric fusion gene that is associated with AML. EWSR1/ELF5 was identified from the chromosomal translocation t(11;22)(p13;q12). Cytogenetically, the same translocation t(11;22)(p13;q12) also creates the EWSR1/WT1 fusion in the desmoplastic small round cell tumor. Interestingly, both genes are closely located, with only 2.1 Mb separating them at the genomic level. This region contains various genes involved in oncogenesis, such as LMO2, WT1 and PAX6. In addition, this region is known to be altered in various types of tumors, as well as the inherited cancer‐prone WAGR syndrome. We hypothesized that this region may, therefore, be susceptible to gene rearrangement.
The EWSR1/ELF5 fusion protein exhibits oncogenic potential in vitro and in vivo. The molecular mechanism of oncogenesis involves an aberrant regulation of the p53/p21‐dependent pathway by EWSR1/ELF5. However, the expression of EWSR1/ELF5 in murine hematopoietic cells does not confer leukemia, suggesting that some additional mechanisms or yet identified prerequisite genetic background is needed for full transformation in the hematopoietic system.
The molecular mechanism of the transformation induced by the family of EWSR1/ETS gene fusions is not fully understood. Among them, EWSR1/FLI1 is relatively well characterized owing to the strong association between this chimeric gene fusion and human EFT. The oncogenic potency of EWSR1/FLI1 was demonstrated by its ability to transform NIH3T3 cells, to grow in semisolid medium, and to form tumors in immunodeficient mice.35, 36 Conversely, the ectopic expression of EWSR1/ETS proteins in primary cells induces growth arrest or cell death, rather than promoting cellular transformation, suggesting that the cellular context is critical for the oncogenic potential of EWSR1/ETS fusions.13, 14 Despite the rare associations of EWSR1/ETS fusion genes with human leukemia,16, 17 the development of leukemia was observed in transgenic mouse models of EWSR1/FLI1 18 and EWSR1/ERG.19 Torchia et al.18 emphasized that a high expression level of EWS/FLI1, driven by the retroviral long terminal repeat (LTR), was toxic to murine hematopoietic cells. This might also be the case in our experiments, because retroviral transduction did not induce leukemia even after 12 months in our model. We speculate that the retroviral LTR‐driven EWSR1/ELF5 expression system may not be suitable for leukemogenesis study in murine model, as has been observed in EWSR1/FLI1. Under the light of the finding that EWSR1 is essential for stem cell quiescence and the maintenance of hematopoietic stem cells,37 endogenous EWSR1 promoter dependent expression of EWSR1/FLI1 might be essential for the leukemogenic transformation.
The ETS transcription factor family is a diverse group of proteins cooperating with other factors to regulate a wide range of cellular processes, such as proliferation, differentiation, apoptosis and senescence; members also possess oncogenic or tumor suppressive activities.38 Chromosomal translocations involving ETS family members are associated with several forms of human cancers. Among the various EWSR1 fusion partners, only EWSR1/ZNF384 (CIZ/NMP4) has been identified in acute leukemia.12 ZNF384 is known to fuse with various partner genes, including EWSR1, TAF15, TCF3 (E2A) and EP300, mainly in acute lymphoblastic leukemia.39, 40, 41 ZNF384 exerts its function as a transcription factor and regulates matrix‐related proteins and/or represses PTH‐induced bone formation.42, 43 However, the role of this gene in the hematopoietic lineage has not been fully elucidated yet, although fusion genes have been identified in leukemia. As for ZNF384, the function of ELF5 in hematopoietic lineage is also uncertain. The protein encoded by ELF5 belongs to an epithelium‐specific subclass of ETS and regulates the later stages of terminal differentiation in keratinocytes, and participates in the regulation of cell fate, beginning with the specification of the trophectoderm in the blastocyst.44, 45 Most ETS genes appear to be functional and expressed during hematopoiesis. However, ELF5 is not expressed in hematopoietic cells. In normal human tissues, ELF5 is expressed in the kidney, prostate, lung, mammary gland, salivary gland, placenta and stomach. However, in cancer tissue, dysregulated expression of ELF5 has been reported, including in leukemia/lymphoma.46 Regarding the fusion gene, only the ZFPM2‐ELF5 fusion gene in multicystic mesothelioma has been reported.47 The only similarity between ZNF384 and FLF5 is the DNA binding transcription factor. These results suggest that an EWSR1 fusion with any DNA binding transcription factor in the hematopoietic lineage may be sufficient for leukemogenesis.
The fusion of ELF5 with EWSR1 in hematopoietic cell lineages makes it difficult to explain the leukemogenic mechanism on its own. Further study is needed to show whether ELF5 aberrantly expressed by a chimeric protein may induce tumorigenesis, or this fusion may play a role in malignant transformation in the hematopoietic system.
In this study we demonstrated that the novel EWSR1/ELF5 fusion gene disrupts the p53/p21‐dependent pathway, similar to the case of sarcoma genesis induced by other members of the EWSR1/ETS family. In most primary cells, the ectopic expression of EWSR1/ETS leads to cell cycle arrest or apoptosis, whereas an inhibition of p53 activity may rescue the cells from EWSR1/ETS‐induced toxicity and facilitate tumorigenesis.13, 14 This observation suggests that the p53/p21‐dependent pathway plays an important role in the prevention of tumorigenesis by EWSR1/ETS. However, 90% of the tumors of EFT express wild‐type p53, suggesting that the function of p53 is blocked in the downstream of p53 by EFT‐specific cancer‐related molecular changes. In the EFT cell line, EWSR1/FLI1 was reported to modulate p21 activity by several mechanisms.32, 33, 34 The expression of p21 is induced by wild‐type p53 in the presence of DNA damage, leading to cell cycle arrest at the G1 checkpoint. The activity of the p21 promoter is negatively regulated by the EWSR1/FLI1 fusion protein through ETS‐binding sites located within the promoter region of p21.32 In addition, EWSR1/FLI1 interacted with the cotransactivator of p300 and suppressed its histone acetyltransferase activity. The epigenetic regulation of the p21 promotor by EWSR1/FLI1 also contributed to the dysregulation of p21 transactivation. In the present study, the AML patient did not show any mutation in TP53 (data not shown), suggesting that the same interfering mechanism was exerted by EWSR1/ELF5 (Fig. 7). In fact, p21 was reported to inhibit the proliferation of leukemic cells, and the suppression of p21 expression fostered the progression of leukemia.48
Although EWSR1/ETS is rarely detected in hematopoietic diseases, the homologue FUS/ERG was identified in several cases of acute myeloid or lymphoblastic leukemia and in Ewing tumors.17, 49, 50, 51 There is considerable amino acid sequence homology (56%) between FUS and EWSR1.52 Incidentally, EWSR1 and FUS are functionally interchangeable in the context of fusion oncogenes, as shown by the identification of FUS/ERG gene fusions in Ewing tumors and FUS/CHOP fusions in myxoid liposarcoma.51, 52 Therefore, it is not surprising that rare EWSR1/ETS family members, such as EWSR1/ELF5, may contribute to leukemic development. Unfortunately, our young patient died after the second round of HSCT. Likewise, a poor outcome was reported for FUS/ERG‐positive leukemia patients.53 As the present study is based on a single patient, additional patients expressing this novel EWSR1/ELF5 fusion gene are required to evaluate the relationship between the presence of the chimeric gene and patient outcome in leukemia. A novel therapy based on the understanding of molecular mechanism may be required for this EWSR1/ETS positive group.
In conclusion, EWSR1/ELF5, a novel EWSR1/ETS chimeric gene, was identified in a patient diagnosed with refractory AML, suggesting a potential role of leukemogenesis in rare cases of AML. This fusion gene is very likely to exhibit oncogenic potential by interfering with the p53/p21‐dependent pathway.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. The EWSR1/ELF5 fusion transcript does not stimulate colony formation in murine bone marrow cells.
Table S1. Top 200 significantly upregulated genes in acute myeloid leukemia (AML) cells compared to Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL).
Table S2. Top 200 significantly down regulated genes in acute myeloid leukemia (AML) cells compared to Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL).
Data S1 Mutation or silenced gene candidates in leukemic cell.
Data S2 Gene ontology terms list of mutations.
Data S3 Pathway list of mutations.
Data S4 Gene ontology terms list of gene silencing.
Data S5 Pathway list of gene silencing.
Acknowledgments
This work was supported by a Grant‐in‐aid for Scientific Research (25461580) from the Ministry of Education, Culture, Sport, Science and Technology of Japan. The pG13 plasmid was a kind gift from Dr Vert Vogelstein, John Hopkins University. The pTM816‐2 plasmid was a kind gift from Dr Toshiaki Miyashita, Kitazato University.
Cancer Sci 107 (2016) 1745–1754
Funding Information
Ministry of Education, Culture, Sport, Science, and Technology of Japan, (Grant/Award Number: ‘25461580’).
References
- 1. Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships: EWS‐FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clin Cancer Res 2010; 16: 4077–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cantile M, Marra L, Franco R et al Molecular detection and targeting of EWSR1 fusion transcripts in soft tissue tumors. Med Oncol 2013; 30: 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delattre O, Zucman J, Plougastel B et al Gene fusion with an ETS DNA‐binding domain caused by chromosome translocation in human tumours. Nature 1992; 359: 162–5. [DOI] [PubMed] [Google Scholar]
- 4. Sorensen PH, Lessnick SL, Lopez‐Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS‐family transcription factor, ERG. Nat Genet 1994; 6: 146–51. [DOI] [PubMed] [Google Scholar]
- 5. Jeon IS, Davis JN, Braun BS et al A variant Ewing's sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995; 10: 1229–34. [PubMed] [Google Scholar]
- 6. Kaneko Y, Yoshida K, Handa M et al Fusion of an ETS‐family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosom Cancer 1996; 15: 115–21. [DOI] [PubMed] [Google Scholar]
- 7. Peter M, Couturier J, Pacquement H et al A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997; 14: 1159–64. [DOI] [PubMed] [Google Scholar]
- 8. Ladanyi M, Gerald W. Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res 1994; 54: 2837–40. [PubMed] [Google Scholar]
- 9. Clark J, Benjamin H, Gill S et al Fusion of the EWS gene to CHN, a member of the steroid/thyroid receptor gene superfamily, in a human myxoid chondrosarcoma. Oncogene 1996; 12: 229–35. [PubMed] [Google Scholar]
- 10. Zucman J, Delattre O, Desmaze C et al EWS and ATF‐1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 1993; 4: 341–5. [DOI] [PubMed] [Google Scholar]
- 11. Panagopoulos I, Hoglund M, Mertens F, Mandahl N, Mitelman F, Aman P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996; 12: 489–94. [PubMed] [Google Scholar]
- 12. Martini A, La Starza R, Janssen H et al Recurrent rearrangement of the Ewing's sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res 2002; 62: 5408–12. [PubMed] [Google Scholar]
- 13. Deneen B, Denny CT. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene 2001; 20: 6731–41. [DOI] [PubMed] [Google Scholar]
- 14. Lessnick SL, Dacwag CS, Golub TR. The Ewing's sarcoma oncoprotein EWS/FLI induces a p53‐dependent growth arrest in primary human fibroblasts. Cancer Cell 2002; 1: 393–401. [DOI] [PubMed] [Google Scholar]
- 15. Riggi N, Suva ML, Stamenkovic I. Ewing's sarcoma origin: from duel to duality. Exp Rev Anticancer Ther 2009; 9: 1025–30. [DOI] [PubMed] [Google Scholar]
- 16. Hawkins JM, Craig JM, Secker‐Walker LM, Prentice HG, Mehta AB. Ewing's sarcoma t(11;22) in a case of acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1991; 55: 157–62. [DOI] [PubMed] [Google Scholar]
- 17. Jakovljevic G, Nakic M, Rogosic S et al Pre‐B‐cell acute lymphoblastic leukemia with bulk extramedullary disease and chromosome 22 (EWSR1) rearrangement masquerading as Ewing sarcoma. Pediatr Blood Cancer 2010; 54: 606–9. [DOI] [PubMed] [Google Scholar]
- 18. Torchia EC, Boyd K, Rehg JE, Qu C, Baker SJ. EWS/FLI‐1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol Cell Biol 2007; 27: 7918–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Codrington R, Pannell R, Forster A et al The Ews‐ERG fusion protein can initiate neoplasia from lineage‐committed haematopoietic cells. PLoS Biol 2005; 3: e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pelloquin F, Lamelin JP, Lenoir GM. Human B lymphocytes immortalization by Epstein‐Barr virus in the presence of cyclosporin A. In vitro Cell Dev Biol 1986; 22: 689–94. [DOI] [PubMed] [Google Scholar]
- 21. Steidl C, Shah SP, Woolcock BW et al MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011; 471: 377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper‐free packaging cell line. Nucleic Acids Res 1990; 18: 3587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morita S, Kojima T, Kitamura T. Plat‐E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 2000; 7: 1063–6. [DOI] [PubMed] [Google Scholar]
- 24. Welford SM, Hebert SP, Deneen B, Arvand A, Denny CT. DNA binding domain‐independent pathways are involved in EWS/FLI1‐mediated oncogenesis. J Biol Chem 2001; 276: 41977–84. [DOI] [PubMed] [Google Scholar]
- 25. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence‐associated beta‐galactosidase assay. Methods Mol Biol 2007; 371: 21–31. [DOI] [PubMed] [Google Scholar]
- 26. Piao J, Sakurai N, Iwamoto S et al Functional studies of a novel germline p53 splicing mutation identified in a patient with Li‐Fraumeni‐like syndrome. Mol Carcinog 2013; 52: 770–6. [DOI] [PubMed] [Google Scholar]
- 27. Im YH, Kim HT, Lee C et al EWS‐FLI1, EWS‐ERG, and EWS‐ETV1 oncoproteins of Ewing tumor family all suppress transcription of transforming growth factor beta type II receptor gene. Cancer Res 2000; 60: 1536–40. [PubMed] [Google Scholar]
- 28. Thompson AD, Teitell MA, Arvand A, Denny CT. Divergent Ewing's sarcoma EWS/ETS fusions confer a common tumorigenic phenotype on NIH3T3 cells. Oncogene 1999; 18: 5506–13. [DOI] [PubMed] [Google Scholar]
- 29. Tomita O, Iijima K, Ishibashi T et al Sensitivity of SNX2‐ABL1 toward tyrosine kinase inhibitors distinct from that of BCR‐ABL1. Leuk Res 2014; 38: 361–70. [DOI] [PubMed] [Google Scholar]
- 30. Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX‐ENL. EMBO J 1997; 16: 4226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 32. Nakatani F, Tanaka K, Sakimura R et al Identification of p21WAF1/CIP1 as a direct target of EWS‐Fli1 oncogenic fusion protein. J Biol Chem 2003; 278: 15105–15. [DOI] [PubMed] [Google Scholar]
- 33. Li Y, Tanaka K, Fan X et al Inhibition of the transcriptional function of p53 by EWS‐Fli1 chimeric protein in Ewing Family Tumors. Cancer Lett 2010; 294: 57–65. [DOI] [PubMed] [Google Scholar]
- 34. Li Y, Li X, Fan G et al Impairment of p53 acetylation by EWS‐Fli1 chimeric protein in Ewing family tumors. Cancer Lett 2012; 320: 14–22. [DOI] [PubMed] [Google Scholar]
- 35. May WA, Gishizky ML, Lessnick SL et al Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA‐binding domain encoded by FLI1 for transformation. Proc Nat Acad Sci USA 1993; 90: 5752–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. May WA, Arvand A, Thompson AD, Braun BS, Wright M, Denny CT. EWS/FLI1‐induced manic fringe renders NIH 3T3 cells tumorigenic. Nat Genet 1997; 17: 495–7. [DOI] [PubMed] [Google Scholar]
- 37. Cho J, Shen H, Yu H et al Ewing sarcoma gene Ews regulates hematopoietic stem cell senescence. Blood 2011; 117: 1156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou J, Ng AY, Tymms MJ et al A novel transcription factor, ELF5, belongs to the ELF subfamily of ETS genes and maps to human chromosome 11p13‐15, a region subject to LOH and rearrangement in human carcinoma cell lines. Oncogene 1998; 17: 2719–32. [DOI] [PubMed] [Google Scholar]
- 39. Gocho Y, Kiyokawa N, Ichikawa H et al A novel recurrent EP300‐ZNF384 gene fusion in B‐cell precursor acute lymphoblastic leukemia. Leukemia 2015; 29: 2445–8. [DOI] [PubMed] [Google Scholar]
- 40. Nyquist KB, Thorsen J, Zeller B et al Identification of the TAF15‐ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12). Cancer Genet 2011; 204: 147–52. [DOI] [PubMed] [Google Scholar]
- 41. Zhong CH, Prima V, Liang X et al E2A‐ZNF384 and NOL1‐E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia 2008; 22: 723–9. [DOI] [PubMed] [Google Scholar]
- 42. Bidwell JP, Childress P, Alvarez MB et al Nmp4/CIZ closes the parathyroid hormone anabolic window. Crit Rev Eukaryot Gene Expr 2012; 22: 205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bidwell JP, Torrungruang K, Alvarez M et al Involvement of the nuclear matrix in the control of skeletal genes: the NMP1 (YY1), NMP2 (Cbfa1), and NMP4 (Nmp4/CIZ) transcription factors. Crit Rev Eukaryot Gene Expr 2001; 11: 279–97. [PubMed] [Google Scholar]
- 44. Oettgen P, Kas K, Dube A et al Characterization of ESE‐2, a novel ESE‐1‐related Ets transcription factor that is restricted to glandular epithelium and differentiated keratinocytes. J Biol Chem 1999; 274: 29439–52. [DOI] [PubMed] [Google Scholar]
- 45. Donnison M, Beaton A, Davey HW, Broadhurst R, L'Huillier P, Pfeffer PL. Loss of the extraembryonic ectoderm in Elf5 mutants leads to defects in embryonic patterning. Development 2005; 132: 2299–308. [DOI] [PubMed] [Google Scholar]
- 46. Piggin CL, Roden DL, Gallego‐Ortega D, Lee HJ, Oakes SR, Ormandy CJ. ELF5 isoform expression is tissue‐specific and significantly altered in cancer. Breast Cancer Res 2016; 18: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Panagopoulos I, Gorunova L, Davidson B, Heim S. Novel TNS3‐MAP3K3 and ZFPM2‐ELF5 fusion genes identified by RNA sequencing in multicystic mesothelioma with t(7;17)(p12;q23) and t(8;11)(q23;p13). Cancer Lett 2015; 357: 502–9. [DOI] [PubMed] [Google Scholar]
- 48. Roman‐Gomez J, Castillejo JA, Jimenez A et al 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 2002; 99: 2291–6. [DOI] [PubMed] [Google Scholar]
- 49. Ichikawa H, Shimizu K, Hayashi Y, Ohki M. An RNA‐binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res 1994; 54: 2865–8. [PubMed] [Google Scholar]
- 50. Kanazawa T, Ogawa C, Taketani T, Taki T, Hayashi Y, Morikawa A. TLS/FUS‐ERG fusion gene in acute lymphoblastic leukemia with t(16;21)(p11;q22) and monitoring of minimal residual disease. Leuk Lymphoma 2005; 46: 1833–5. [DOI] [PubMed] [Google Scholar]
- 51. Shing DC, McMullan DJ, Roberts P et al FUS/ERG gene fusions in Ewing's tumors. Cancer Res 2003; 63: 4568–76. [PubMed] [Google Scholar]
- 52. Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA‐binding protein in human myxoid liposarcoma. Nature 1993; 363: 640–4. [DOI] [PubMed] [Google Scholar]
- 53. Kong XT, Ida K, Ichikawa H et al Consistent detection of TLS/FUS‐ERG chimeric transcripts in acute myeloid leukemia with t(16;21)(p11;q22) and identification of a novel transcript. Blood 1997; 90: 1192–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The EWSR1/ELF5 fusion transcript does not stimulate colony formation in murine bone marrow cells.
Table S1. Top 200 significantly upregulated genes in acute myeloid leukemia (AML) cells compared to Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL).
Table S2. Top 200 significantly down regulated genes in acute myeloid leukemia (AML) cells compared to Epstein–Barr virus‐transformed lymphoblastoid cell line (EB‐LCL).
Data S1 Mutation or silenced gene candidates in leukemic cell.
Data S2 Gene ontology terms list of mutations.
Data S3 Pathway list of mutations.
Data S4 Gene ontology terms list of gene silencing.
Data S5 Pathway list of gene silencing.