

ABSTRACT
Solid tumors contain numerous regions with insufficient oxygen concentrations, a condition termed hypoxia. Tumor hypoxia is significantly associated with metastasis, refractory to conventional cancer therapies, and poor patient survival. Therefore, eradication of hypoxic tumor cells will likely have significant impact on the overall progression-free patient survival. This article reports a new discovery that Benznidazole, a bioreductive drug currently used to treat Chagas disease caused by the parasitic protozoan Trypanosoma cruzi, is activated by hypoxia and can kill clonogenic tumor cells especially those under severe hypoxic conditions (≤0.1 % O2). This type of hypoxia selectivity is important in that severely hypoxic tumor microenvironment is where tumor cells exhibit the strongest resistance to therapy. Mechanistically, activation of Benznidazole coincides with the stabilization of the Hypoxia-Inducible Factor 1α (HIF-1α), suggesting that Benznidazole is activated after tumor cells have entered into a fully hypoxic state. Under such hypoxic conditions, Benznidazole induces the formation of 53BP1 foci, a hallmark of DNA double-stranded breaks that can cause clonogenic inhibition or cell death. These results demonstrate that Benznidazole is a hypoxia-activated cytotoxin with the potential to specifically eliminate hypoxic tumor cells.
KEYWORDS: 53BP1, Benznidazole, clonogenic growth, DNA damage, hypoxia, nuclear foci, PARP cleavage
Introduction
Hypoxia or insufficient cellular oxygenation is a hallmark of tumor microenvironment and is commonly observed in solid tumors. The development of hypoxia is largely caused by abnormal angiogenesis, defective vascular structures, and compromised hemodynamic functions.1-3 Hypoxia occurs randomly throughout a solid tumor with oxygen (O2) concentrations reaching 0 mmHg pO2 in areas adjacent to necrosis. In addition to relatively stable or sustaining hypoxia, it can also be highly dynamic with fluctuating pO2.1,4 Importantly, hypoxia can strongly influence tumor malignant progression and treatment outcomes. Many critical intracellular pathways are potentially regulated by hypoxia, including cell cycle progression,5 multiple aspects of cellular metabolism,6,7 apoptosis,8 tumor invasion and metastasis,9,10 DNA damage repair and maintenance of genomic integrity,11,12 as well as maintenance of cancer stem cell characteristics.13,14 Furthermore, tumor hypoxia poses a significant challenge to conventional cancer therapies.15 Clinical studies have shown that tumor hypoxia is an independent prognostic factor that predicts advanced disease progression and poor patient survival.16-20 Therefore, eradication of hypoxic tumor cells will have significant positive impact on both the control of malignant progression and the patient survival.
Although hypoxia poses a formidable challenge to conventional cancer therapies, it also provides a unique opportunity for clinical intervention because of its nearly ubiquitous nature in solid tumors independent of tumor types. The idea of developing drugs that specifically target hypoxic tumor cells was largely inspired by the early studies on the bioreductive compound Mitomycin C.21 In the ensuing years, many studies including both experimental and clinical studies have been devoted to the development of hypoxia-activated prodrugs.22,23 However, the struggle continues to find an effective drug against hypoxic tumor cells.
In this study, we report that Benznidazole (N-Benzyl-2-nitro-1H-imidazole-1-acetamide) can function as a hypoxia-activated cytotoxin to specifically inhibit the growth of clonogenic tumor cells under hypoxic conditions. Benznidazole is especially effective at killing clonogenic tumor cells under severely hypoxic conditions (≤0.1 % O2) with insignificant toxicity toward cells at ≥0.5% O2. One potential mechanism may involve DNA double-stranded breaks induced by Benznidazole under hypoxic conditions. Benznidazole is best known as a widely used drug to treat Chagas disease caused by the parasitic protozoan Trypanosoma cruzi.24,25 It has also been investigated as a radiosensitizer or a chemosensitizer,26,27 but its anti-cancer activities remain poorly understood. Our results have revealed a previously unrecognized anti-cancer function of Benznidazole with hypoxia-activated cytotoxic effects against clonogenic tumor cells. These findings suggest that Benznidazole is a promising drug candidate that has the potential to specifically target the hypoxic tumor microenvironment in solid tumors.
Results
Benznidazole specifically inhibits clonogenic growth of tumor cells under hypoxic conditions
Benznidazole is a nitroheterocyclic compound (Fig. 1) and becomes an active cytotoxic agent upon biological reduction mediated by nitroreductases.28,29 Although it had been studied for radiosensitization and chemosensitization,26,27 it remains to be fully evaluated whether Benznidazole can directly kill tumor cells, especially those with clonogenic potentials. Because the cytotoxicity of Benznidazole against the parasitic protozoan Trypanosoma cruzi results from its emzymatic reduction,24,25 we hypothesized that hypoxia may also lead to bioreduction of Benznidazole and formation of chemically reactive intermediates toxic to hypoxic tumor cells. Toward this end, we examined whether Benznidazole could specifically kill hypoxic cells using the clonogenicity assay for stringent assessment of clonogenic potentials. First, we treated SK-N-ER neuroblastoma cells and MDA-MB-231 breast cancer cells with 500 µM Benznidazole for 24 hours either at normoxia (20% O2) or severe hypoxia (approximately 0% O2). For the clonogenicity assay, the drug-treated cells were washed in PBS, plated in 6-well plates at 3,000 and 300 cells/well, respectively, and further incubated under the conventional (normoxic) tissue culture condition. The two cell-seeding densities chosen represent a clonal density at 300 cells/well or approximately 0.3 cells/mm230 and non-clonal or high density at 3,000 cells/well or approximately 3 cells/mm2. The seeding at the high density was to determine whether Benznidazole was capable of killing clonogenic tumor cells under hypoxic conditions even when paracrine communication among cells were not a limiting factor. Using Trypan Blue, we found that Benznidazole (≤500 µM) did not induce significant cell death under either normoxic or hypoxic conditions. Consistently, Calvo et al. reported no significant apoptosis of acute leukemia cells treated with up to 1 mM Benznidazole for up to 48 hrs.31
Figure 1.
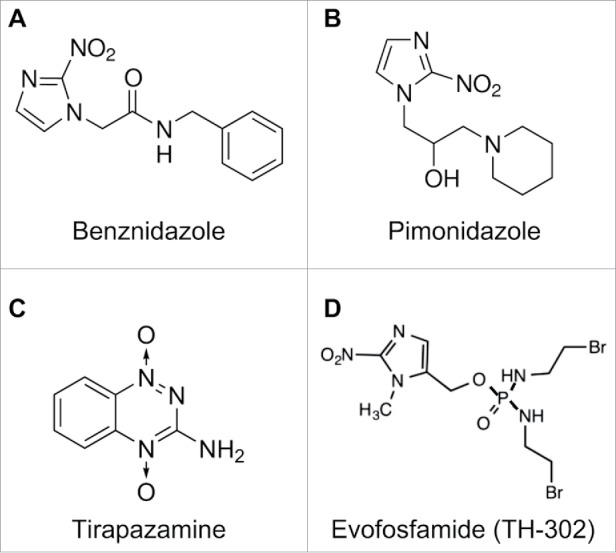
Chemical structure of Benznidazole (A) in comparison to representative bioreductive nitroheterocyclic compounds including the radiosensitizer Pimonidazole (B) and the hypoxia-activated prodrugs Tirapazamine (C) and Evofosfamide (TH-302) (D).
As shown in Fig. 2, both the SK-N-ER and MDA-MB-231 tumor cells treated by Benznidazole under the hypoxic condition completely lost their clonogenic potential even at the high seeding density of 3 cells/mm2. In contrast, tumor cells treated by Benznidazole at the same concentration under the normoxic condition still maintained their clonogenic potential. Similar results were obtained using Benznidazole at ≥200 µM. It is worth noting that clonogenic inhibition is much more stringent than the commonly used cell growth/viability assays to reliably assess anti-cancer functions of experimental drugs. Furthermore, our data clearly demonstrate that Benznidazole can function as a hypoxia-activated cytotoxin to specifically eliminate clonogenic tumor cells under hypoxic conditions without significant toxicity toward non-hypoxic cells, which is important for reducing toxic side effects in normal tissues.
Figure 2.

Benznidazole specifically inhibits clonogenic growth of hypoxic tumor cells. MDA-MB-231 breast cancer cells (A & B) and SK-N-ER neuroblastoma cells (C & D) were incubated with 500 µM Benznidazole (BZDZ) or DMSO under aerobic (20% O2) and anoxic (0% O2) conditions, respectively. Treated cells were seeded in triplicates at 300 and 3000 cells per well, respectively, in 6-well plates for clonogenic growth. (A & C) Representative images of colonies. (B & D) Plating efficiencies (mean ± SD, n = 3). These experiments were independently confirmed 3 times.
To determine whether the synergistic effects of Benznidazole and hypoxia could apply to other types of tumor cells, we examined 4 additional cancer cell lines: HCT116 colon cancer cells, C33A cervical cancer cells, KNS42 glioma cells, and LN-18 glioma cells. As shown in Fig. 3, all 4 cell lines showed strictly hypoxia-dependent loss of clonogenic growth upon treatment with Benznidazole. Compared to HCT116 and C33A cells, KNS42 and LN-18 cells appeared relatively less sensitive to Benznidazole under hypoxia. This is likely due to cell type-dependent differences in the expression of oxidoreductases or overall intracellular oxidoreductive potentials. Nevertheless, these data collectively demonstrate that Benznidazole has the ability to preferentially eradicate clonogenic tumor cells under hypoxic conditions but independent of tumor types.
Figure 3.

Hypoxia-dependent clonogenic inhibition by Benznidazole is independent of tumor cell types. Human tumor cell lines were incubated with Benznidazole (BZDZ, 100 µM) or DMSO at normoxia (20% O2) or anoxia (0% O2). (A) Images of colony growth for tumor cells plated at 3,000 cells/well in 6-well plates to demonstrate the synergistic effects of BZDZ and hypoxia. (B) Relative surviving fraction (mean ± SD, n = 3) was calculated using data from the plates seeded at the clonal density of 300 cells/well. The data from DMSO-treated cells at 20% O2 were used as control. These results were validated by at least 2 independent experiments.
Dose- and pO2-dependent clonogenic inhibition by benznidazole
In solid tumor microenvironment, distribution of O2 concentrations is highly variable, ranging from anoxia in necrosis to physiological levels of O2 concentrations, with the mean pO2 values of around 9 mmHg or 1.2% O2.32 At this mean tumor pO2, the Hypoxia-Inducible Factor 1α (HIF-1α) and/or 2α (HIF-2α) proteins become robustly stabilized,33 a canonical mechanism of cellular response to hypoxia,34 suggesting a true state of cellular hypoxia.
We examined the ability of Benznidazole to inhibit clonogenic growth of SK-N-ER and MDA-MB-231 cells at normoxia (20% O2 at the conventional tissue culture condition) and different levels of hypoxia (1, 0.5, 0.1 and 0% O2, respectively). As shown in Fig. 4, the clonal inhibitory effects of Benznidazole did not significantly change over the dose range of 0–500 µM at 20% O2. However, the clonogenic inhibition by Benznidazole increased dramatically with at <0.1% O2 (Fig. 4). SK-N-ER cells appeared to be more sensitive to Benznidazole than MDA-MB-231 cells under the same conditions. Nevertheless, these results clearly demonstrate that Benznidazole is capable of preferentially killing clonogenic tumor cells under severe hypoxic conditions. It is worth noting that tumor cells residing in the severely hypoxic tumor microenvironment are likely to be highly resistant to conventional therapies.15,23 Thus, Benznidazole could become a viable drug candidate for elimination of these hypoxic and therapy-resistant tumor cells. Furthermore, because normal tissue O2 concentrations are often >2% O2,32 the cytotoxic side-effects of Benznidazole in normal tissues are likely to be very limited.
Figure 4.
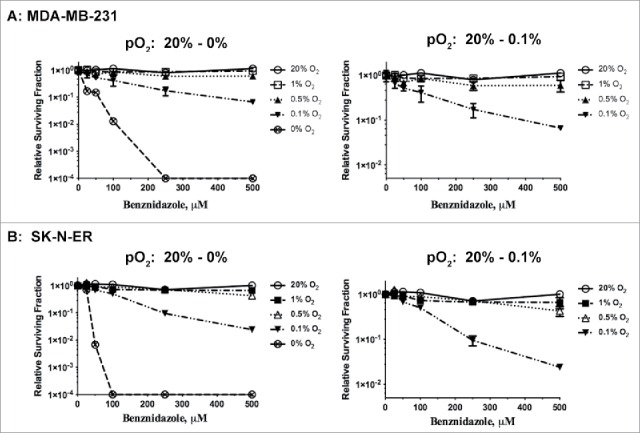
Clonogenic inhibition by Benznidazole is inversely correlated with pO2. MDA-MB-231 breast cancer cells (A) and SK-N-ER neuroblastoma cells (B) were incubated for 24 hrs with Benznidazole (BZDZ) at different concentrations under normoxia (20% O2) or hypoxia (1%, 0.5%, 0.1%, or 0% O2). Relative survival fractions (mean ± SD, n = 3) were calculated with DMSO treatment at each pO2 as control. These experiments were independently confirmed more than 3 times.
To further appreciate the hypoxia-dependent activation of Benznidazole, we determined when Benznidazole became cytotoxically active upon exposure to hypoxia. We found that clonogenic inhibition by Benznidazole became robustly strong after 6–8 hr incubation under anoxia (Fig. 5A & C). Incidentally, the stabilization of the hypoxia-inducible factor 1α (HIF-1α) protein, a canonical event of hypoxia response,34 occurred after 2 hr of exposure to anoxia (Fig. 5B & D). These data suggest that Benznidazole becomes activated after cells are in a completely hypoxic state, which is also consistent with the general mechanisms of activation of hypoxia-activated cytotoxins and prodrugs.23
Figure 5.
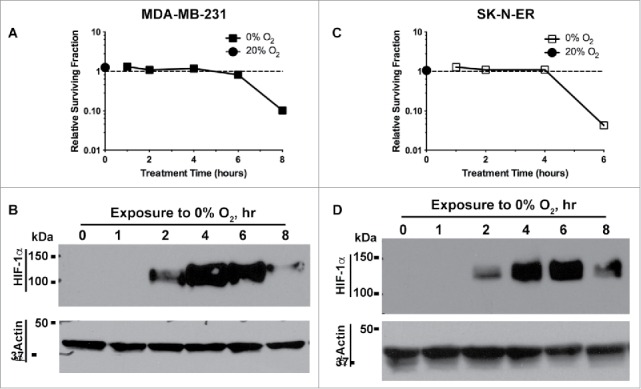
Hypoxia-induced activation of Benznidazole correlates with HIF-1α stabilization. MDA-MB-231 breast cancer cells (A) and SK-N-ER neuroblastoma cells (C) were incubated with Benznidazole (BZDZ, 500 µM) for 1, 2, 4, 6, or 8 hrs at 0% O2. As reference, MDA-MB-231 cells, for 8 hours at 20% O2 (A) and SK-N-ER cells were incubated with BZDZ for 6 hours at 20% O2 (D), respectively. (A & C) Relative survival fractions (mean ± SD, n = 3) were normalized to dimethyl sulfoxide (DMSO) treatment at 20% O2. (B & D) Stabilization of HIF-1α protein was examined by Western blots with β-actin as the internal loading control. These experiments were independently confirmed more than 3 times.
Hypoxia-activated benznidazole induces DNA double-stranded breaks
To understand the mechanisms of Benznidazole-mediated clonogenic inhibition under hypoxic conditions, we investigated whether Benznidazole could induce apoptosis. Using proteolytic cleavage of PARP1, a characteristic result of activation of cell death proteases including caspases,35 as a readout of apoptosis, we found that Benznidazole did not induce proteolytic cleavage of PARP1 under either normoxia or anoxia. However, PARP1 cleavage did occur in both MDA-MB-231 and SK-N-ER cells in response to etoposide, a DNA-damaging anticancer agent, suggesting that the PARP1 cleavage pathway is functional in these cells (lane 1, Fig. 6A & B). Furthermore, Benznidazole-treated SK-N-ER cells, but not MDA-MB-231 cells or DMSO-treated cells, exhibited PARP1 cleavage during reoxygenation (lanes 6–7, Fig. 6A & B), suggesting that the PARP1 cleavage pathway is regulated by both cell- and stress-dependent mechanisms. Nevertheless, these observations suggest that the Benznidazole-induced clonogenic inhibition of hypoxic tumor cells does not directly involve acute apoptotic cell death. These results are also consistent with a previous report showing that Benznidazole, when used at up to 1 mM, does not induce significant apoptosis of acute leukemia cells.31
Figure 6.
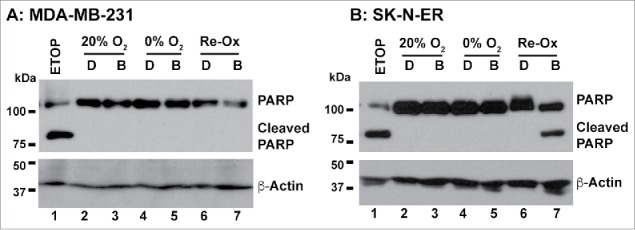
Benznidazole treatment does not induce PARP1 cleavage. MDA-MB-231 breast cancer cells (A) and SK-N-ER neuroblastoma cells (B) were incubated for 24 hours with Benznidazole (abbreviated as B, 100 µM) or dimethyl sulfoxide (abbreviated as D) under aerobic (20% O2) and anoxic (0% O2) conditions, respectively. For the reoxygenation treatment, the hypoxia-treated cells were placed in the aerobic incubator for 24 hours. Etoposide (ETOP, 10 µM) was used as positive control. PARP1 cleavage was examined by Western blot analysis with β-actin as the internal loading control. These results were confirmed by more than 3 independent experiments.
The chemically active metabolic products of hypoxia-activated cytotoxic agents can potentially react with a wide range of cellular macromolecules. As a hallmark of their cytotoxicity, hypoxia-activated cytotoxins induce DNA damages and double-stranded breaks.22,23 DNA double-stranded breaks lead to the formation of punctate aggregates of the nuclear protein 53BP1.36,37 Using the nuclear 53BP1 foci assay, we found that, specifically under severe hypoxia, Benznidazole significantly induced 53BP1 foci formation in both SK-N-ER and MDA-MB-231 cells compared to DMSO-treated cells (Fig. 7). In contrast, Benznidazole did not significantly induce 53BP1 foci formation under normoxia. As a positive control, etoposide induced 53BP1 foci formation in the presence (normoxia) or absence of O2. These results suggest that, consistent with other hypoxia-activated cytotoxins, Benznidazole can produce cytotoxic intermediates upon bioreductive activation under hypoxia to induce DNA double-stranded breaks or other genotoxic events.
Figure 7.
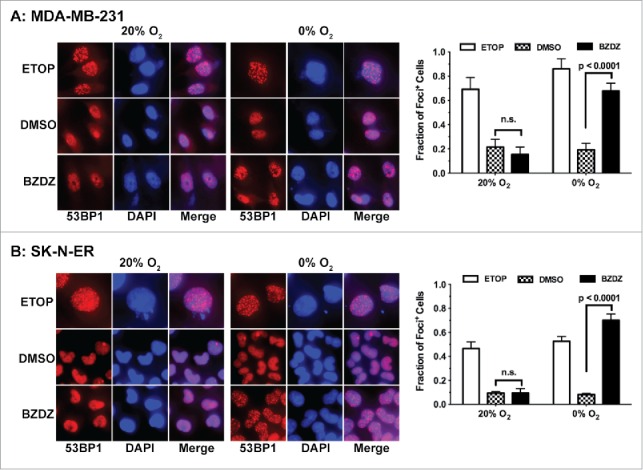
Benznidazole induces DNA double-stranded breaks under hypoxia. MDA-MB-231 breast cancer cells (A) and cells SK-N-ER neuroblastoma (B) were incubated for 24 hours with Benznidazole (BZDZ, 100 µM), etoposide (ETOP, 1 µM) or dimethyl sulfoxide (DMSO) under aerobic (20% O2) and anoxic (0% O2) conditions, respectively. The nuclear foci containing the tumor suppressor p53 binding protein 1 (53BP1) were examined by immunofluorescence using antibodies specifically against 53BP1. Cells with more than 5 53BP1+ foci were counted in 5 random fields (200x magnification) under microscope. Total number of cells counted per experimental group: >500 cells for SK-N-ER and >200 cells for MDA-MB-231, except that total numbers of ETOP-treated cells counted were approximately 200 for each cell type.
Discussion
Benznidazole is perhaps best known now as a drug to treat American Trypanosomiasis or the Chagas disease caused by the protozoan parasite Trypanosoma cruzi.24,25 In 1980s, it was also studied as an adjuvant to increase tumor cell sensitivity to ionizing radiation or chemotherapeutic drugs including nitroureas.26,27 However, clinical studies of Benznidazole as a chemosensitizer of CCNU, a nitrourea compound, failed to clearly demonstrate beneficial effects.38,39 Perhaps, it is, at least in part, due to a lack of clear understanding of its anti-cancer activities. In light of our current findings, the anti-cancer potential of Benznidazole could potentially be fully realized in patients with hypoxic tumors.
In this study, we have used the clonogenic assay, a gold standard assay used in the field of radiation biology and stem cell biology, to determine the ability of Benznidazole to eradicate the clonogenic tumor cells. The clonogenic assay offers a more robust assessment of the clonogenic survival and expansion of tumor cells under very stringent conditions by seeding cells at clonal density of 1–2 cells/mm2 or less.30 We have found that the cytotoxic effects of Benznidazole are strongly potentiated under severely hypoxic conditions. Benznidazole has the ability to preferential kill clonogenic tumor cells under these hypoxic conditions, while showing no significant inhibition of clonogenic growth at or above 0.5% O2 at least at ≤500 μM. It is worth noting that preclinical studies have shown the effective concentrations of Benznidazole found in this study can be easily reached in vivo.40,41 Because clonogenicity in vitro often strongly correlates with tumorigenicity in vivo and cancer stem cell characteristics, our results suggest that Benznidazole can significantly contribute to the overall tumor control by specifically targeting tumor-initiating or stem-like tumor cell located in the hypoxic tumor microenvironment.
Similar to other hypoxia-activated cytotoxins, Benznidazole becomes activated upon biological reduction.22,23 Its chemically active metabolites can react with many cellular macromolecules and potentially affect multiple intracellular pathways. We have found that Benznidazole does not appear to activate apoptosis based on PARP1 cleavage, which is also consistent with the literature.31 Nevertheless, we could not rule out the possibilities that Benznidazole could trigger other forms or pathways of cell death, including mitotic catastrophe and/or necrosis, either directly by itself or via its reduced metabolites. As a hallmark of their cell-killing effects, hypoxia-activated cytotoxins cause DNA double-stranded breaks likely via free radicals generated by the one-electron reduction, which can lead to cell death.22,23 Here, we have found that Benznidazole can also induce DNA double-stranded breaks specifically upon hypoxia-dependent activation, as shown by the formation of 53BP1-positive nuclear foci. Consistent with our findings, it has recently been reported that Benznidazole can cause potentially lethal double-stranded breaks in Trypanosoma cruzi DNA.42 Therefore, it is highly likely that Benznidazole-induced double-stranded DNA breaks could be a leading cause of clonogenic inhibition.
Tumor hypoxia poses a significant challenge to cancer treatment because hypoxic tumor cells are highly aggressive and resistant to all conventional therapies.15 Relapse-free survival of cancer patients will inevitably hinge upon successful elimination of tumorigenic or stem cell-like tumor cells, especially those localized in the severely hypoxic microenvironment. Discovery and development of a drug that specifically target hypoxic tumor cells will undoubtedly make a significant impact on cancer therapy. As our data have demonstrated, Benznidazole is an excellent candidate as a hypoxia-selective anticancer drug. Its clinical potential could be fully realized in a combination therapy with either standard chemotherapy or radiation therapy.
Materials and methods
Cell culture and hypoxia
MDA-MB-231 breast carcinoma cells, HCT116 colorectal carcinoma cells, C33A cervical carcinoma cells, and LN-18 glioma cells were from American Type Culture Collection (ATCC). KNS42 glioma cells were from RIKEN, Japan. SK-N-ER neuroblastoma cells were obtained from Dr. Nai-Kong V. Cheung of Memorial Sloan-Kettering Cancer Center. For routine culture, SK-N-ER cells were maintained in MEM and F12 (1:1). MDA-MB-231 cells were grown in RPMI. HCT116, LN-18, and KNS42 cells were cultured in DMEM. C33A cells were maintained in MEM. All cell culture media were supplemented with 10% fetal bovine serum and 20 mM HEPES. The hypoxia experiments at 1%, 0.5%, or 0.1 % O2 were performed in InVivo2 400 Hypoxia Workstation (Ruskinn Technology) and those at 0% O2 were performed in Bactron Anaerobic Chamber (Sheldon Manufactures, Inc.). Aerobic (20% O2) experiments were carried out in a conventional CO2 incubator with ambient air.
Drug treatment
N-Benzyl-2-nitro-1H-imidazole-1-acetamide (Benznidazole, catalog #419656, Sigma-Aldrich) and etoposide (E1383, Sigma-Aldrich) were dissolved in dimethyl sulfoxide (DMSO) and diluted in culture media to final concentrations. Cells were incubated at different drug concentrations under different pO2 conditions for up to 24 hrs. After the drug treatment, the monolayer cultures were washed with phosphate-buffer saline (PBS) and trypsinized to single cell suspension for the subsequent experiments. Cell viability was routinely examined using Trypan Blue.
Clonogenicity assay
Tumor cells were plated in 6-well plates at a seeding density of 300 or 3,000 cells per well in complete culture media. Cells were incubated in a conventional incubator with ambient air for up to 14 days. The resultant tumor cell colonies were fixed and stained with crystal violet. The colonies (≥50 cells per colony) were counted manually under a magnifying glass. Plates with large number of colonies were examined using a digital colony counter equipped with a VRmagic camera mounted on Kaiser Copylizer eVision exe.cutive stand and ProtoCOL SR software. Plating Efficiency (PE) = #colonies formed divided by #seeded cells per well × 100%. Surviving Fraction (SF) = PE of drug-treated cells divided by PE of control cells.
Western blot
The monolayer cell culture was washed in ice-cold PBS and lysed on ice in a lysis buffer containing 25 mM HEPES buffer at pH 7.4, 1% Nonidet P-40, 150 mM NaCl, 2 mM EDTA, and protease inhibitors (Complete™, Roche Diagnostics). After centrifugation, the supernatants were collected as whole cell lysates. Equal amounts of proteins were loaded per lane and separated in a SDS-polyacrylamide gel under reducing conditions. For Western blots, proteins were electrotransferred onto nitrocellulose membranes and were probed with rabbit anti-PARP-1 monoclonal antibody (46D11, 1:1000, Cell Signaling Technology, Product Number 9532), rabbit anti-HIF-1α polyclonal antibody (1:1000, Cell Signaling Technology) or mouse monoclonal anti-β-actin (1:20,000, Sigma-Aldrich), followed by incubation with horseradish peroxidase (HRP)-conjugated secondary IgG. Protein bands were visualized using ECL substrates (ThermoFisher Scientific, #34080) and imaged on Kodak X-OMAT 2000A.
Immunofluorescence
Cells were seeded in 48-well plates and incubated for 24 hours before they were treated with Benznidazole or Etoposide for another 24 hours. After treatment, cells were washed twice with ice-cold PBS, fixed in a solution containing 2% paraformaldehyde and 1% sucrose for 15 minutes at room temperature, and permeabilized with ice-cold methanol and acetic acid (1:1) for 20 minutes at −20°C. After being washed in PBS, the cells were incubated in a blocking buffer (4% BSA with 0.2% Triton X100 in PBS) for 30 minutes. Incubation with rabbit polyclonal anti-53BP1 antibody (1:200, Santa Cruz, sc22760) was carried out at room temperature for 1 hour or at 4°C for overnight, followed by incubation with Alexa 555–conjugated anti-rabbit IgG (1:500; Invitrogen) for 1 hour at room temperature in the dark. Nuclei were counterstained with DAPI (0.2 μg/mL, Sigma-Aldrich). Immunofluorescence were examined and pictures taken using the EVOS-FL fluorescence microscope (ThermoFisher Scientific). Nuclei with >5 foci were counted. Fraction of foci+ cells = number of foci+ cells divided by total number of cells counted.
Statistics
Two group comparison were analyzed by 2-tailed, unpaired Student t test. Significant difference was declared if p < 0.05.
Disclosure of potential conflicts of interests
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Nai-Kong V. Cheung of Memorial Sloan-Kettering Cancer Center for SK-N-ER cells. We also thank Drs. Ranjit Bindra, Joseph Contessa, Peter Glazer, and Faye Rogers of the Department of Therapeutic Radiology, Yale School of Medicine for reagents.
Funding
This work was supported by a grant from the National Institutes of Health to ZY (R01CA178254). Q.L. was supported by a fellowship from the China Scholarship Council.
Authors contributions
Conception and Design: Q. Lin and Z. Yun; Development of Methodology: Q. Lin and Z. Yun; Acquisition of Data: Q. Li and Q. Lin; Analysis and Interpretation of Data: Q. Li, Q. Lin, and Z. Yun; Writing, Review, and/or Revision of the Manuscript: Q. Li, Q. Lin, and Z. Yun; Administrative, Technical, or Material Support: Q. Lin and Z. Yun; Study Supervision: Z. Yun. Quhuan Li is a visiting scholar from School of Bioscience and Bioengineering, South China University of Technology, Guangzhou 510006, China.
Reference
- 1.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008; 8:425-37; PMID:18500244; http://dx.doi.org/ 10.1038/nrc2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayer C, Vaupel P. Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther Onkol 2012; 188:616-27; PMID:22454045; http://dx.doi.org/ 10.1007/s00066-012-0085-4 [DOI] [PubMed] [Google Scholar]
- 3.Ljungkvist AS, Bussink J, Kaanders JH, van der Kogel AJ. Dynamics of tumor hypoxia measured with bioreductive hypoxic cell markers. Radiat Res 2007; 167:127-45; PMID:17390721; http://dx.doi.org/ 10.1667/RR0719.1 [DOI] [PubMed] [Google Scholar]
- 4.Brown JM. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br J Radiol 1979; 52:650-6; PMID:486895; http://dx.doi.org/ 10.1259/0007-1285-52-620-650 [DOI] [PubMed] [Google Scholar]
- 5.Evans SM, Hahn SM, Magarelli DP, Koch CJ. Hypoxic heterogeneity in human tumors: EF5 binding, vasculature, necrosis, and proliferation. Am J Clin Oncol 2001; 24:467-72; PMID:11586098; http://dx.doi.org/ 10.1097/00000421-200110000-00011 [DOI] [PubMed] [Google Scholar]
- 6.Eales KL, Hollinshead KE, Tennant DA. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016; 5:e190; PMID:26807645; http://dx.doi.org/ 10.1038/oncsis.2015.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillies RJ, Gatenby RA. Metabolism and its sequelae in cancer evolution and therapy. Cancer J 2015; 21:88-96; PMID:25815848; http://dx.doi.org/ 10.1097/PPO.0000000000000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996; 379:88-91; PMID:8538748; http://dx.doi.org/ 10.1038/379088a0 [DOI] [PubMed] [Google Scholar]
- 9.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science 2016; 352:175-80; PMID:27124451; http://dx.doi.org/ 10.1126/science.aaf4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subarsky P, Hill RP. The hypoxic tumour microenvironment and metastatic progression. Clin Exp Metastasis 2003; 20:237-50; PMID:12741682; http://dx.doi.org/ 10.1023/A:1022939318102 [DOI] [PubMed] [Google Scholar]
- 11.Yuan J, Narayanan L, Rockwell S, Glazer PM. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res 2000; 60:4372-6; PMID:10969780 [PubMed] [Google Scholar]
- 12.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 1998; 2:259-65; PMID:9734364; http://dx.doi.org/ 10.1016/S1097-2765(00)80137-9 [DOI] [PubMed] [Google Scholar]
- 13.Lin Q, Yun Z. Impact of the hypoxic tumor microenvironment on the regulation of cancer stem cell characteristics. Cancer biology & therapy 2010; 9:949-56; PMID:20581454; http://dx.doi.org/ 10.4161/cbt.9.12.12347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 2008; 9:285-96; PMID:18285802; http://dx.doi.org/ 10.1038/nrm2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Lin Q, Yun Z. Cellular and molecular mechanisms underlying oxygen-dependent radiosensitivity. Radiat Res 2015; 183:487-96; PMID:25938770; http://dx.doi.org/ 10.1667/RR13959.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nordsmark M, Overgaard J. Tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol 2004; 43:396-403; PMID:15303502; http://dx.doi.org/ 10.1080/02841860410026189 [DOI] [PubMed] [Google Scholar]
- 17.Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, Dewhirst MW. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res 1996; 56:941-3; PMID:8640781 [PubMed] [Google Scholar]
- 18.Brizel DM, Dodge RK, Clough RW, Dewhirst MW. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiother Oncol 1999; 53:113-7; PMID:10665787; http://dx.doi.org/ 10.1016/S0167-8140(99)00102-4 [DOI] [PubMed] [Google Scholar]
- 19.Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 1996; 56:4509-15; PMID:8813149 [PubMed] [Google Scholar]
- 20.Young SD, Marshall RS, Hill RP. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci U S A 1988; 85:9533-7; PMID:3200838; http://dx.doi.org/ 10.1073/pnas.85.24.9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sartorelli AC. Therapeutic attack of hypoxic cells of solid tumors: presidential address. Cancer Res 1988; 48:775-8; PMID:3123053 [PubMed] [Google Scholar]
- 22.Hunter FW, Wouters BG, Wilson WR. Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br J Cancer 2016; 114(10):1071-7; PMID:27070712; http://dx.doi.org/21606941 10.1038/bjc.2016.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011; 11:393-410; PMID:21606941; http://dx.doi.org/ 10.1038/nrc3064 [DOI] [PubMed] [Google Scholar]
- 24.Bermudez J, Davies C, Simonazzi A, Real JP, Palma S. Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Trop 2016; 156:1-16; PMID:26747009; http://dx.doi.org/ 10.1016/j.actatropica.2015.12.017 [DOI] [PubMed] [Google Scholar]
- 25.Malik LH, Singh GD, Amsterdam EA. The Epidemiology, Clinical Manifestations, and Management of Chagas Heart Disease. Clin Cardiol 2015; 38:565-9; PMID:25993972; http://dx.doi.org/ 10.1002/clc.22421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siemann DW, Morrissey S, Wolf K. In vivo potentiation of 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea by the radiation sensitizer benznidazole. Cancer Res 1983; 43:1010-3; PMID:6825075 [PubMed] [Google Scholar]
- 27.Twentyman PR, Workman P. The effect of radiosensitizer pretreatment on the response of the RIF-1 mouse sarcoma to cytotoxic drugs. Int J Radiat Oncol Biol Phys 1982; 8:611-3; PMID:7107382; http://dx.doi.org/ 10.1016/0360-3016(82)90695-2 [DOI] [PubMed] [Google Scholar]
- 28.Maya JD, Cassels BK, Iturriaga-Vasquez P, Ferreira J, Faundez M, Galanti N, Ferreira A, Morello A. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol A Mol Integr Physiol 2007; 146:601-20; PMID:16626984; http://dx.doi.org/ 10.1016/j.cbpa.2006.03.004 [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A 2008; 105:5022-7; PMID:18367671; http://dx.doi.org/ 10.1073/pnas.0711014105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maciag T, Hoover GA, Stemerman MB, Weinstein R. Serial propagation of human endothelial cells in vitro. J Cell Biol 1981; 91:420-6; PMID:7309790; http://dx.doi.org/ 10.1083/jcb.91.2.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calvo KL, Ronco MT, Noguera NI, Garcia F. Benznidazole modulates cell proliferation in acute leukemia cells. Immunopharmacol Immunotoxicol 2013; 35:478-86; PMID:23855487; http://dx.doi.org/ 10.3109/08923973.2013.811597 [DOI] [PubMed] [Google Scholar]
- 32.Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxidants & redox signaling 2007; 9:1221-35; PMID:17536958; http://dx.doi.org/ 10.1089/ars.2007.1628 [DOI] [PubMed] [Google Scholar]
- 33.Lin Q, Cong X, Yun Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol Cancer Res 2011; 9:757-65; PMID:21571835; http://dx.doi.org/ 10.1158/1541-7786.MCR-11-0053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 2000; 88:1474-80; PMID:10749844 [DOI] [PubMed] [Google Scholar]
- 35.Duriez PJ, Shah GM. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol 1997; 75:337-49; PMID:9493956; http://dx.doi.org/ 10.1139/o97-043 [DOI] [PubMed] [Google Scholar]
- 36.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/ 10.1038/nrm3719 [DOI] [PubMed] [Google Scholar]
- 37.Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA damage foci: Meaning and significance. Environ Mol Mutagen 2015; 56:491-504; PMID:25773265; http://dx.doi.org/ 10.1002/em.21944 [DOI] [PubMed] [Google Scholar]
- 38.Bleehen NM, Freedman LS, Stenning SP. A randomized study of CCNU with and without benznidazole in the treatment of recurrent grades 3 and 4 astrocytoma. Report to the Medical Research Council by the Brain Tumor Working Party. Int J Radiat Oncol Biol Phys 1989; 16:1077-81; PMID:2539345; http://dx.doi.org/ 10.1016/0360-3016(89)90920-6 [DOI] [PubMed] [Google Scholar]
- 39.Bleehen NM, Roberts JT, Newman HF. A phase II study of CCNU with benznidazole for metastatic malignant melanoma. Int J Radiat Oncol Biol Phys 1986; 12:1401-3; PMID:3759564; http://dx.doi.org/ 10.1016/0360-3016(86)90181-1 [DOI] [PubMed] [Google Scholar]
- 40.Walton MI, Workman P. Nitroimidazole bioreductive metabolism. Quantitation and characterisation of mouse tissue benznidazole nitroreductases in vivo and in vitro. Biochem Pharmacol 1987; 36:887-96; PMID:3105539; http://dx.doi.org/ 10.1016/0006-2952(87)90181-X [DOI] [PubMed] [Google Scholar]
- 41.Workman P, White RA, Walton MI, Owen LN, Twentyman PR. Preclinical pharmacokinetics of benznidazole. Br J Cancer 1984; 50:291-303; PMID:6466543; http://dx.doi.org/ 10.1038/bjc.1984.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajao MA, Furtado C, Alves CL, Passos-Silva DG, de Moura MB, Schamber-Reis BL, Kunrath-Lima M, Zuma AA, Vieira-da-Rocha JP, Garcia JB, et al.. Unveiling benznidazole's mechanism of action through overexpression of DNA repair proteins in Trypanosoma cruzi. Environ Mol Mutagen 2014; 55:309-21; PMID:24347026; http://dx.doi.org/ 10.1002/em.21839 [DOI] [PubMed] [Google Scholar]