

Abstract
Exploration of dynamic processes in proteins and nucleic acids by spin-locking NMR experiments has been facilitated by the development of theoretical expressions for the R1ρ relaxation rate constant covering a variety of kinetic situations. Herein, we present a generalized approximation to the chemical exchange, Rex, component of R1ρ for arbitrary kinetic schemes, assuming the presence of a dominant major site population, derived from the negative reciprocal trace of the inverse Bloch-McConnell evolution matrix. This approximation is equivalent to first-order truncation of the characteristic polynomial derived from the Bloch-McConnell evolution matrix. For three- and four-site chemical exchange, the first-order approximations are sufficient to distinguish different kinetic schemes. We also introduce an approach to calculate R1ρ for linear N-site schemes, using the matrix determinant lemma to reduce the corresponding 3N × 3N Bloch-McConnell evolution matrix to a 3 × 3 matrix. The first- and second order-expansions of the determinant of this 3 × 3 matrix are closely related to previously derived equations for two-site exchange. The second-order approximations for linear N-site schemes can be used to obtain more accurate approximations for non-linear N-site schemes, such as triangular three-site or star four-site topologies. The expressions presented herein provide powerful means for the estimation of Rex contributions for both low (CEST-limit) and high (R1ρ-limit) radiofrequency field strengths, provided that the population of one state is dominant. The general nature of the new expressions allows for consideration of complex kinetic situations in the analysis of NMR spin relaxation data.
Graphical abstract
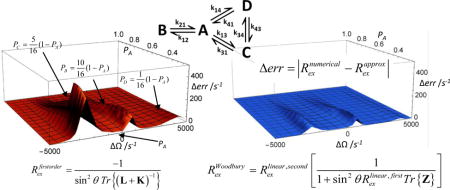
Introduction
Dynamic processes in proteins and nucleic acids can be studied using various techniques in nuclear magnetic resonance (NMR) spectroscopy [1]. Nuclear spin relaxation in the presence of a radiofrequency (rf) field is characterized by the rotating-frame R1ρ and R2ρ relaxation rate constants. Rotating-frame relaxation measurements can be used to study conformational and chemical processes that range from slow to fast on the chemical shift time scale [2]. In particular, spontaneous inter-conversion between different protein structures, ligand-binding events, chemical reaction kinetics, and protein folding processes that occur with rate constants between 10 and 105 s−1 can be probed by R1ρ experiments with rf field strengths ranging from Hz (CEST-limit) to kHz (R1ρ-limit) [2, 3]. Most analyses of experimental data have assumed two-state exchange kinetics, but examples featuring three-site [4, 5], four-site [6, 7] and other [8] topologies illustrate the need to provide theoretical insight into more complex kinetic schemes.
Exact numerical solutions for R1ρ can be obtained by numerical integration of the Bloch-McConnell differential equation or by calculation of the largest (least negative) real eigenvalue of the Bloch-McConnell evolution matrix and elegant fast numerical algorithms have been described [9]. However, analytic expressions for R1ρ facilitate understanding of the relationship between model parameters and the phenomenological relaxation rate constant and can lead to new methodological advances. For example, first-order expressions show the importance of the minor state resonance frequency when exchange is not fast [10] and the effect of differential relaxation (from processes other than chemical exchange) of the major and minor states [2, 11]. As another example, the perturbation treatment of N-site exchange mechanisms provides insight into the reduction of complex exchange schemes to simpler ones if exchange within subgroups of states is fast compared to exchange between subgroups [12]. In addition, analytic solutions allow straightforward symbolic differentiation with respect to model parameters. The obtained derivatives can be used in gradient-based minimization procedures, such as the Levenberg-Marquardt algorithm, to find optimal parameters more efficiently than possible with either numerically estimated derivatives, or non-gradient-based methods, such as grid-search or simplex routines.
Analytical solutions for R1ρ for two-state exchange outside the fast-exchange limit were obtained previously by finding the least negative (smallest magnitude) real root of the characteristic sixth-order polynomial derived from the Bloch-McConnell evolution matrix. An approximation retaining only the two lowest-order terms (first order in the eigenvalue) is valid for highly skewed site populations [10, 11]. The Laguerre approximation to a second-order truncation of the sixth-order characteristic polynomial is more accurate than the first-order treatment for site populations that are less skewed [13]. In an alternative approach, the relevant 6 × 6 Bloch-McConnell matrix for the two exchanging magnetization vectors is reduced to a 3 × 3 matrix describing evolution of the average magnetization. The average magnetization approach yields expressions of similar accuracy as the Laguerre approximation, but somewhat increased algebraic complexity [14]. The average-magnetization approach also yields analytical, although complex, expressions for R1ρ for a general N-site exchange mechanism (vide infra). Compact analytical solutions for N-site schemes can be obtained by perturbation theory applied to the 3N × 3N Bloch-McConnell evolution matrix [12]. The general solution obtained from this approach is valid only if one of the N populations is highly dominant. While most treatments of R1ρ have assumed a constant-amplitude rf field, the theoretical and experimental results have been extended to time-varying adiabatic sweeps of the rf field [9, 15–18].
Herein, we present a general approach to obtain first-order approximations for R1ρ, which are valid for any kinetic scheme in which one site is sufficiently highly populated. Furthermore, we introduce an approach to reduce the 3N × 3N Bloch-McConnell matrix for any linear exchange scheme to a 3 × 3 matrix by repetitive use of the matrix determinant lemma.
Theory
The Bloch-McConnell equation is
| (1) |
in which M(t) = [M1(t), …, MN(t)]T, Mj(t) = [Mxj(t), Myj(t), Mzj(t)]T, and N is the number of exchanging states. The evolution matrix has the form:
| (2) |
in which,
| (3) |
| (4) |
0 and I are the zero and identity matrices, respectively, with dimensions set by context (3 × 3 in Eq. 2); δj = j− ωrf is the resonance offset frequency, j is the resonance frequency, and R1j and R2j are the longitudinal and transverse relaxation rate constants in the absence of exchange, respectively, for spins in the jth site; ωrf and ω1 are the frequency and amplitude (measured in frequency units; termed the Rabi frequency) of the rf spin-lock field; and are the population-averaged longitudinal and transverse relaxation rate constants in the absence of exchange; and is assumed. The elements of the N × N kinetic rate matrix k are kjn, the (pseudo) first-order rate constant for transitions from state j to state n (j • n) and
| (5) |
The formal solution to the Bloch-McConnell equation is given by:
| (6) |
in which |ui〉 and λi are the right eigenvectors and eigenvalues of the Bloch-McConnell evolution matrix.
In a typical measurement of R1ρ, initial magnetization is aligned along a tilted rotating frame of reference, yielding Mj(0) ∝ pjv, in which pj is the equilibrium population of the jth state, v = [sinθ, 0, cosθ]T, tan θ = ω1/ΔΩ, and is the population-average resonance offset frequency. In turn, the spin-locked z-magnetization in the tilted frame of reference following the relaxation period is M′z(t) = dM(t) in which d = [v, …, v] is a 1 × 3N row vector. If p1 → 1, perturbation theory shows that |u1> → [v, 0, …, 0]T; consequently, the decay of the spin-locked magnetization is highly monoexponential with damping constant λ1 (which will be simply called λ in the following) [12]. If |u1> diverges from [v, 0, …, 0]T, then contributions from other eigenvalues contribute to the magnetization decay. In such cases, the magnetization decay contains oscillatory components, arising from complex eigenvalues, but inhomogeneity of the B1 field suppresses these contributions, particularly for long relaxation times (such as those used in CEST experiments). The ideal result that |u1> → [v, 0, …, 0]T, i.e. yielding negligible contributions from other eigenvalues, also is approached for larger values of the exchange rate constants, the resonance offset, and the spin-lock field (vide infra). The effects of dispersion of the eigenbase relative to the tilted frame are reduced if the site magnetizations are aligned with their own tilted frames, defined by tanθj = ω1/ΔΩj, as would be obtained by an adiabatic sweep shorter than the jump time between states. Broadly speaking, the assumption that the R1ρ relaxation can be described by a monoexponential decay is more accurate with off-resonance irradiation, larger exchange kinetic rate constants, larger ω1 and with relatively larger population of the dominant chemical state.
The R1ρ relaxation rate constant therefore can be defined to be the negative of the largest (least negative) real eigenvalue of the evolution matrix, i.e. R1ρ = −λ. The eigenvalues are given by the zeros of the characteristic polynomial:
| (7) |
in which |A| is the determinant and Tr[A] is the trace of the matrix A, respectively. The last line is obtained assuming that the magnitude of the desired eigenvalue is small. With the aforementioned definition of R1ρ we then obtain
| (8) |
When values of R2j are small, the above expression can be simplified to first-order in R by the following:
| (9) |
Solving this equation for λ and using Eq 4 gives,
| (10) |
in which
| (11) |
| (12) |
and is the largest negative real eigenvalue of L + K in Eq. 11. For two-state exchange, Eq. 10 is identical to Eq. 5 of Baldwin and Kay [11]. If for all j, Eq. 10 reduces to Eq. 11 in Trott and Palmer [10] and Eq. 9 in Baldwin and Kay [11]. In addition, if the population of the dominant site is large, then Eq. 12 simplifies to
| (13) |
so that
| (14) |
Equations 10–14 are the major results of this work. The approximations leading to these equations are accurate if 1/λ dominates the sum of the inverses of the other eigenvalues for L + K and L + R + K because for a matrix A, . As discussed elsewhere [19], the conditions for which this approximation holds depends in general on all the parameters in Eq. 1, but usually is met if one of the sites is sufficiently more highly populated than the other N − 1 sites. Expressions obtained from Eqs. 10–14 for N-site exchange (N > 2) are more complex than those obtained using the perturbation approach of Trott and Palmer [12], but are accurate for a larger range of site populations. Crucially, the matrix traces and inverses in these expressions do not need to be expanded algebraically for analysis of experimental data because the necessary derivatives with respect to model parameters can be obtained symbolically, as shown in Supplementary Material III (S16–S23).
The above first-order approach can be extended in accuracy by considering the evolution of the average magnetization, rather than the N individual magnetization vectors. As shown by Trott and coworkers [14], the evolution equation for the average magnetization <M>(t) under the steady-state approximation is:
| (15) |
in which 〈Lk〉 is the 3 × 3 population average of the N individual site matrices
| (16) |
and rows 2, …, N of U are orthogonal. The blocks F2, F3, and F4 have dimensions 3 × 3(N −1), 3(N −1) × 3, and 3(N − 1) × 3(N − 1), respectively. Using the above ansatz,
| (17) |
leading to:
| (18) |
Two-site chemical exchange
For two-site exchange between sites A and B, Eq. 10 yields the expression of Baldwin and Kay [11] and Eq. 11 yields the original expression (Eq. 24) of Trott and Palmer [10]:
| (19) |
in which Δω = ΩB − ΩA, , , and kex = k12 + k21. The average magnetization approach (Eq. 18) gives
| (20) |
which is identical to Eq. 74 of Trott et al. [14]. Equation 20 approaches Eq. 19 as pB → 0.
Recently, we showed that the R1ρ relaxation rate constant for two-site exchange could be reduced to the reciprocal of the trace of a 3 × 3 matrix [20]. This derivation is reproduced below, in slightly more compact form to motivate the subsequent treatment of chemical exchange in linear chains with three or more sites. The exchange matrix K is rank-deficient and can be factored as:
| (21) |
As before, the eigenvalues of L + K are determined by equating the characteristic polynomial to 0:
| (22) |
in which the last line is obtained using the matrix determinant lemma. A series of manipulations that utilize the block structure of L yields:
| (23) |
in which
| (24) |
| (25) |
The right-hand side of the last line of Eq. 23 now is the determinant of a matrix with dimension 3 × 3, rather than 6 × 6 as in the starting expression. As above, when λ is small in magnitude, the determinant can be expanded to first order in λ and solved to yield
| (26) |
Substituting Eq. 25 into Eq. 26 yields:
| (27) |
as a first-order approximation and yields a result identical to that obtained from Eq. 11 and shown in Eq. 19.
Unlike Eq. 11, the determinant of the last line of Eq. 23 can be expanded to second order in λ to give
| (28) |
in which M1(A) is the first minor of the matrix A. The Laguerre approximation of the root of Eq. 28 gives:
| (29) |
For two-site exchange schemes, X, Y and Z from Eq (25) are substituted into Eq. 29. This result can be expanded to show that this is an expression for two-site exchange identical to the second-order approximation of Miloushev and Palmer and is more accurate than Eqs. 8 or 11:
| (30) |
R1ρ can be obtained by substituting Rex from Eq. 30 into Eqs. 10 or 14. For a significantly large R1ρ contribution at = 0 (for example, for large ω1 or ), Eq. 14 is preferred.
Three-site chemical exchange
We next consider the linear three-site exchange mechanism:
| (31) |
in which the evolution matrix has the form:
| (32) |
If pA >> pB and pA >> pC, then the N-site perturbation formalism of Trott and Palmer [12] yields the compact expression:
| (33) |
The first-order expressions for Rex and R1ρ are obtained by substituting Eq. 32 into Eqs. 10–14. For completeness, the average magnetization approach defines the matrix U for a three-site scheme as:
| (34) |
from which the average magnetization expression for R1ρ is obtained from Eqs. 15–18. The resulting expanded first-order and average magnetization expressions are lengthy and not reproduced here.
In light of the above derivation for two-site exchange, the exchange matrix in Eq. 32 can be factored as:
| (35) |
By use of the matrix determinant lemma,
| (36) |
The second matrix on the last line of Eq. 36 can be factored as
| (37) |
so that a second application of the matrix determinant lemma gives:
| (38) |
The determinant of the 9 × 9 original Bloch-McConnell matrix has now been reduced to the determinant of a 3 × 3 matrix, once again. The products are expanded to obtain a polynomial in λ:
| (39) |
Keeping only the two lowest-order terms, we immediately obtain first- and second-order approximations by substituting X, Y and Z into Eqs. 26 and 29:
| (40) |
The first-order approximation is identical to that obtained from Eq. 11.
Four-site chemical exchange
The four-site linear kinetic scheme is
| (41) |
The exchange matrix again is factorizable:
| (42) |
As shown in Supplemental Material I (Eqs. S1–S5), three successive applications of the matrix determinant lemma again reduces the 12 × 12 dimensional problem to the determinant of a 3 × 3 matrix, yielding:
| (43) |
which can be substituted into Eqs. 26 and 29 to give the first- and second-order expressions for λ.
Triangular and more complex kinetic schemes
The triangular (all-with-all) three-site kinetic scheme has the kinetic matrix:
| (44) |
in which K0 is the kinetic rate matrix for the linear system. The first-order approximation of the eigenvalue according to Eq. 11 is then:
| (45) |
Additional insight is obtained by using the factorization of the matrix K. The Woodbury formula for the matrix inverse gives the result:
| (46) |
in which
| (47) |
Therefore, the first-order approximation for the triangular state is:
| (48) |
using only the first-order approximation of Rex for the linear system. If exchange between sites B and C (not part of the linear fragment) is slow, compared to exchange with site A, then the simplification
| (49) |
is obtained; this is the “weak minor site exchange” limit [12].
As shown in Supplemental Material II (Eqs. S6–S15), the modified expression:
| (50) |
which is called Woodbury approximation herein, leads to increased accuracy if the boundary condition
| (51) |
is satisfied. However, replacing both first-order approximation terms for the linear system with second-order approximations is not as accurate. The boundary condition implies that high accuracy of the linear approximations relative to that of the triangular first-order approximation yields a more accurate Woodbury approximation.
More complex kinetic schemes are also accessible using this approach because the Woodbury formula can be use in a nested manner:
| (52) |
Therefore, the Woodbury approximation for complex kinetic schemes is
| (53) |
The same boundary condition as above applies. In using the Woodbury approximation, K can only be replaced by K0 + UVT if the number of sites in K0 and K are identical. An example for a scheme where this is not immediately possible would be the star four-site scheme (three minor sites in exchange with one central major site). However, minimally occupied pseudo-sites that rapidly exchange with a site with an identical offset can be introduced as necessary (vide infra).
Results
The accuracy of the above theoretical expressions for the R1ρ relaxation rate constant was tested for selected values of the model parameters for two-, three-, and four-site kinetic schemes. Illustrative results are presented in Figures 1–3, respectively and additional figures are given in Supplemental Information.
Figure 1.
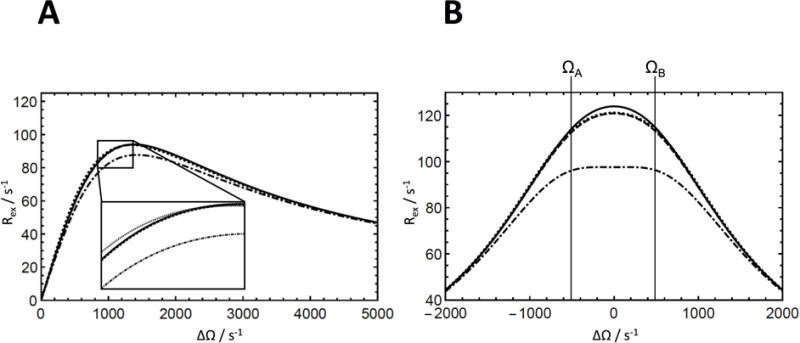
Approximations to the Rex contribution to R1ρ for two-site chemical exchange. (Solid) Numerical calculation of Rex = −λ/sin2θ from the least negative real eigenvalue of the 6 × 6 evolution matrix, (dashed) calculation from the second-order approximation from Eqs. 25 and 29, (dotted) calculation from the average magnetization approximation from Eq. 30, (dashed-dotted) calculation from the first-order approximation from Eq. 11, which is equivalent to Eq. 19. The inset exemplifies regions in which the results of the calculations differ. (a) Exchange rate, kex = k12+k21, dependence of the Rex contribution to R1ρ. Parameters used for the calculation were pA = 0.5, pB = 0.5, ω1 = 1000 s−1, ΩB − ΩA = 1000 s−1, and Ω = 1000 s−1. (b) Offset dependence of the Rex contribution to R1ρ. Parameters used for the calculations were pA = 0.5, pB = 0.5, ω1 = 1000 s−1, ΩB − ΩA = 1000 s−1, and kex = k12 + k21 = 1000 s−1.
Figure 3.
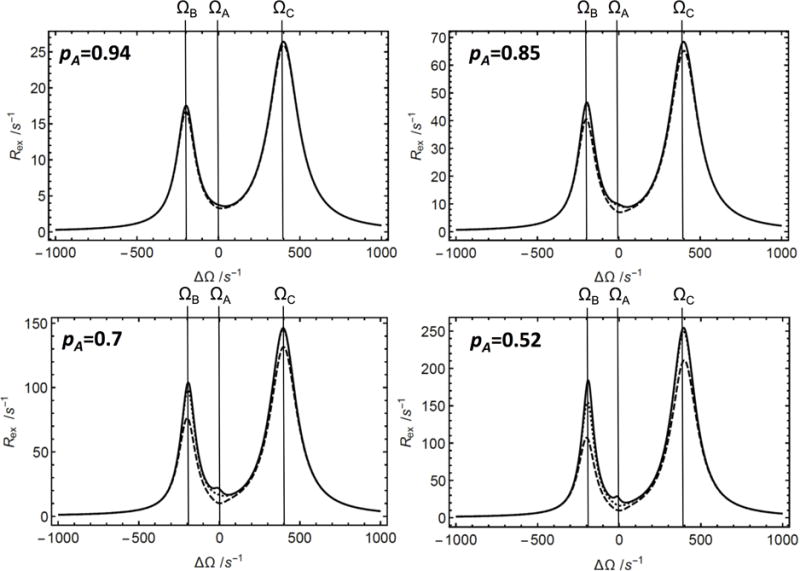
Offset dependence of the Rex contribution to R1ρ for linear three-site exchange for varying major populations in the weak field limit (pA = 0.94, pA = 0.85, pA = 0.7 and pA = 0.52, as indicated). (Solid) Numerical calculation of Rex = −λ/sin2 θ from the least negative real eigenvalue of the 9 × 9 evolution matrix, (dashed) calculation from the first-order approximation from Eq. 11, and (dotted) calculation from the second-order approximation from Eqs. 29 and 40. The inset exemplifies a region in which the results of the calculations differ. To center the graph, parameters were chosen so that δA = ΔΩ. Parameters used for the calculations were pB = 2(1− pA)/3, pC = (1−pA)/3, k12+k21 = 50 s−1, k13 +k31 = 100 s−1, ω1 = 50 s−1, ΩB − ΩA = 200 s−1, and ΩC − ΩA = −400 s−1.
The dependence of the Rex contribution to R1ρ on the chemical exchange rate constant kex = k12 + k21 and on the rf frequency offset for two-site exchange is illustrated in Figure 1 for the extreme case with pA = pB = 0.5. The accuracy of the expressions increases from the first-order result (Eqs. 11 or 19), to the average magnetization result (Eq. 20), and to the second-order Laguerre result (Eqs. 29 or 30). These results are consistent with those obtained in the original publications describing these theoretical expressions [10, 13, 14].
For the example presented in Fig. 1, the largest negative eigenvalue dominates the inverses of the other eigenvalues, as illustrated in Figure S1. However, the first-order approximations given in Eqs. 8, 10 and 14 differ in the treatment of relaxation by mechanisms other than chemical exchange. If the values of R2j vary substantially between sites, Eqs. 8 or 10 are more accurate than Eq. 14. Equation 14 is more accurate than Eqs. 8 or 10 if or for a significantly large R1ρ contribution at = 0 (for example, for large ω1 or ). Furthermore, as shown in the example in Figure 2, the higher powers of R that contribute to Eq. 8 reduce accuracy slightly compared to the truncated first-order expression of Eq. 10. Thus, Eqs. 10 or 14 are the preferred first-order approximations for practical use.
Figure 2.
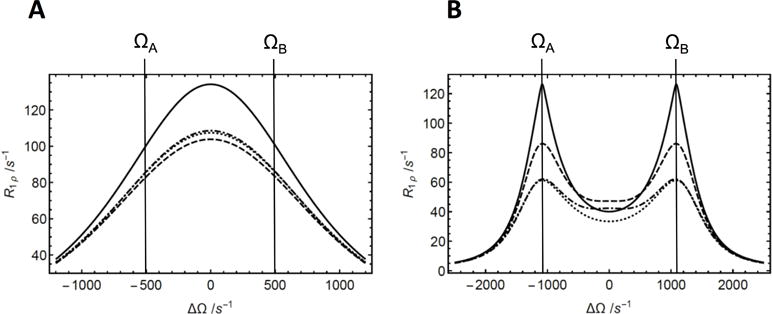
Approximations of R1ρ for two-site chemical exchange. (a) (Solid) Numerical calculation of the least negative real eigenvalue of the 6 × 6 evolution matrix, (dashed) calculation from the first-order (in) approximation, based on L + K + R (Eq. 8); (dotted) calculation from the first-order (in) approximation, based on L + K + R, and truncated to first order in R (Eq. 10), as also previously provided by Trott and Palmer [10] and Baldwin and Kay [11]; (dashed-dotted) calculation from the first-order approximation, based on L + K (Eq. 14). Parameters used for the calculations were R1 = 1.5 s−1, R2 = 11 s−1, pA = 0.5, pB = 0.5, ω1 = 1000 s−1, ΩB − ΩA = 1000 s−1, and kex = k12 + k21 = 1000 s−1. (b) (solid) Numerical calculation of the least negative real eigenvalue of the 6 × 6 evolution matrix; (dashed) calculation from the second-order approximation from Eqs. 14, 25 and 29; (dotted) calculation from the first-order (in) approximation, based on L + K + R (Eq. 8); (dashed-dotted) calculation from the first-order approximation, based on L + K (Eq. 14). Parameters used for the calculations were R1 = 1.5 s−1, R2 = 11 s−1, pA = 0.5, pB = 0.5, ω1 = 500 s−1, ΩB − ΩA = 2400 s−1, and kex = k12 + k21 = 250 s−1.
The dependence on the rf frequency offset of the Rex contribution to R1ρ is shown in Figure 3 and Supplemental Information Figures S2 – S4 for a linear three-site kinetic scheme. Results are shown for values of ω1 between 50 s−1, which corresponds to values that would be used in CEST experiments, to 1250 s−1, which corresponds to values that would be used in relaxation dispersion experiments. As shown previously using the N-site perturbation approach [12], local maxima in Rex (which would correspond to local minima in CEST experiments) are obtained when the rf frequency is resonant with the frequencies of the sparsely populated states. The widths of the profiles around the local maxima depend on the kinetic rate constants and the strength of the ω1 field [12]. Consequently, in the fast-exchange limit, discrimination between sites becomes increasingly difficult; in this limit, Rex depends only on the offset relative to the average chemical shift (Figure S2). Congruent with the results for two-site exchange, the second-order results are more accurate than the first-order ones. However, as shown in Figures 3 and S3, for skewed populations, the main region of difference between results for first- and second-order approximations is near resonance with the major species. Both first- and second-order expressions for Rex become less accurate as the site populations become less highly skewed (Fig. 2). The second-order approximation for Rex is also accurate when the time scales of the two exchange regimes in the three-site linear kinetic scheme are distinct (k12+k21 = 200 s−1, k13+k31 = 5000 s−1, Figure S3).
Not only for first-order approximations, but also for the other approximations introduced herein, R1ρ is in practice best obtained from Rex by adding Reff (from Eqs. 12 or 13) to sin2θ Rex. Figure S4 compares various second-order approximations for R1ρ, exemplified for the linear three-state kinetic scheme. Rex is obtained from Eqs. 29 and 40, and then substituted into either Eq. 10 or Eq. 14. In order to include terms that are of higher order in R, a second order approximation based on L + R + K, rather than L + R, was also used by replacing with in Eq. 40. Eq. 14 is more accurate if the values of R2k do not vary substantially between sites and if the R1ρ contribution at = 0 is large. Such large R1ρ is obtained for higher amplitude ω1 fields, much higher population of the dominant site, or . Eq. 10 is more accurate in most other cases. Using an untruncated second-order approximation based on L + R + K, which does not use Reff and includes higher-order R terms, usually is not more accurate than the second-order R1ρ approximations from Eqs. 10, 29 and 40. The second-order approximation from Eqs. 11, 29 and 40 is accurate even if no major population is present (Fig. S4, pA = pB = pC = 0.33); however, accuracy is lower than that obtained if one population is dominant. The assumption that R1ρ is identical to the largest negative eigenvalue will obtain only with the appropriate combination of other parameters, as discussed above. In the given example, the contribution of the largest negative eigenvalue to the magnetization decay t = 0 s and = 0 s−1 is only 83%, mostly due the presence of negative complex eigenvalues (using Eq. 6, similar to Fig. S1).
The dependence of the Rex contribution to R1ρ on the rf frequency offset is shown in Figure 4 for a linear four-site kinetic scheme, a four-site star-like scheme, and a four-site kite-like scheme with one dominant site population pA = 0.90 in all cases. As above, results are shown for values of ω1 between 50 s−1 and 1250 s−1. The values of Rex are very sensitive to the presence or absence of kinetic exchange between two minor sites, as illustrated in Figure 4 by comparing the star-like and kite-like topologies. In addition, as previously noted [12], fast averaging within a subset of sites can lead to a Rex profile with an apparent reduction in the number of discernable “effective sites”. This phenomenon is frequently of practical importance in reducing N-site kinetic schemes to simpler apparent topologies. For example, in studies of protein folding, fast averaging amongst conformations in the unfolded ensemble allows a two-state-like description of exchange between a folded and a “random-coil-like” effective unfolded state [21]. As another example, fast averaging amongst side chain rotameric states with different populations (a 2M-site model in which M is the number of rotamers) results in an effective two-state-like model termed population-shuffling [8].
Figure 4.
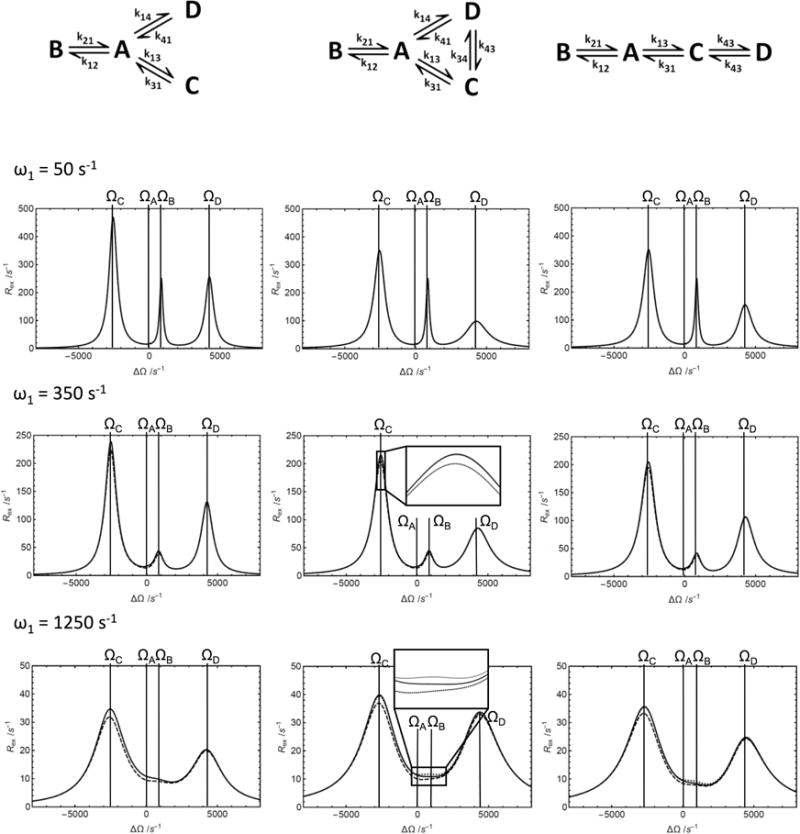
Approximations of the Rex contribution to R1ρ for 4-state chemical exchange schemes. Three different kinetic situations (illustrations at the top of the columns) have been analyzed, star-like four-state scheme (left column), kite-like four-state scheme (middle column), linear four-state scheme (right column). The calculations have been performed at the different irradiating fields, ω1 = 50 s−1 (top row), ω1 = 500 s−1 (middle row), ω1 = 1250 s−1 (bottom row). (Solid) Numerical calculation of Rex = −λ/sin2θ from the least negative real eigenvalue of the 12 × 12 evolution matrix, (dashed) calculation from the first order approximation from Eq. 11, (dotted, linear scheme) calculation from the second-order approximation from Eqs. 29 and 40, (dotted, kite scheme) calculation from the Woodbury approximation from Eq. 50. The insets exemplify regions in which the results of the calculations differ. To center the graph, parameters were chosen so that δA = ΔΩ. Parameters used for all calculations were pA = 0.95, pB = 0.05, pC = 0.025, pD = 0.005, k12+k21 = 140 s−1, k13+k31 = 350 s−1, ΩB − ΩA = −850 s−1, ΩC − ΩA = 2550 s−1, and ΩD − ΩA = −4250 s−1. Additional parameters used depending on exchange scheme: k34+k43 = 700 s−1 and k14+k41 = 350 s−1.
The Woodbury approximation can be used to obtain improved estimates of Rex for complex N-site schemes by combining second- and first-order linear N-site approximations with coupling factors for the nonlinear interchanges between sites. The Woodbury approximation is often, but not always more accurate than the first-order expression, which is exemplified by Rex profiles for the triangular three-state scheme (Figure S5). The values of the kinetic rate constants for the non-linear part of the scheme changes the relative accuracy of the two approximations. The accuracy of the Woodbury approximation also depends on which components of the kinetic scheme are chosen as the linear fragment, as demonstrated for the triangular three-state scheme (Figure 5). Excluding the slowest exchange process in the scheme from the linear fragment leads to the highest accuracy of the Woodbury approximation. If this exchange process is much slower than the processes in the linear fragment, the Woodbury approximation can be reduced further using Eq. 49. In the example given for the kite-like four-state scheme (Figure 4), the Woodbury approximation from Eq. 50 is more accurate than the first-order approximation. The Woodbury approximation also is accurate when the populations are not skewed (Figure S6). The possibility to substitute any N-site linear scheme by an (N+1)-site scheme is illustrated for the triangular scheme in Figure S7. This gives access to Woodbury approximations for N-site schemes from which a N-site linear sub-scheme cannot be readily extracted.
Figure 5.
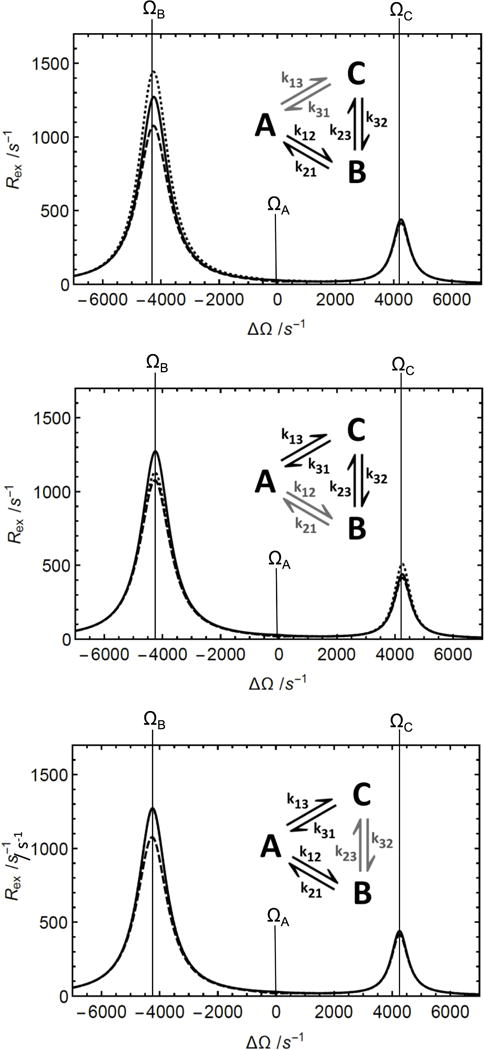
The Woodbury approximation of the Rex contribution to R1ρ in the triangular three-state chemical exchange situation depends on how the linear fragment for the approximation is chosen. (Solid) Numerical solution of Rex = −λ/sin2θ from the least negative real eigenvalue of the 9 × 9 evolution matrix, (dashed) first-order approximation (Eq. 11) and (dotted) Woodbury approximation (Eq. 50). To center the graph, parameters were chosen so that δA = ΔΩ. Parameters used for all calculations were pA = 0.9, pB = 0.05, pC = 0.05, k12 +k21 = 500 s−1, k13+k31 = 50 s−1, k23+k32 = 20 s−1, ω1= 350 s−1, ΩB − ΩA = 4250 s−1, and ΩC − ΩA = −4250 s−1. The kinetic scheme is shown in each panel (black: linear fragment; grey: non-linear fragment). Linear fragments are (top) A-B-C, (middle) A-C-B, (bottom) C-A-B. The Woodbury approximation fits best when the slowest exchange process is chosen as the nonlinear fragment.
Discussion and Conclusion
The perturbation approach of Trott and Palmer [12] provides simple closed-form approximations for R1ρ when the population of one site is much greater than the populations of the others (typically pA > 0.90, depending on other parameters). The main result obtained herein is a first-order approximation of R1ρ for any kinetic scheme. For linear kinetic schemes, the matrix determinant lemma can be used to reduce the determinant of the 3N × 3N Bloch-McConnell matrix to the determinant of a 3 × 3 matrix polynomial. This latter approach leads to both first- and second-order approximations of R1ρ. The first-order expressions of Eqs. 8, and 10 and the second-order expressions of Eq. 29 for linear chains are more accurate than the expressions obtained from the perturbation approach when the population of the major state is less dominant. The first-order approximation of the eigenvalue of the evolution matrix for the average magnetization (Eq. 18) yields expressions for R1ρ that have accuracies between the first- and second-order expressions. For these and other N-site kinetic schemes, the first-order analysis described above in Eq. 10 frequently provides analytical solutions that are accurate enough to differentiate between different kinetic scenarios provided that the site populations are skewed. Expansion of matrix trace expressions or inverse operations in these equations is not required for use in fitting procedures, as illustrated in Supplemental Material III (Eqs. S16–S23).
The above procedure based on the matrix determinant lemma can be extended to even longer N-site linear chains; however, the resulting second order expressions are increasingly complex and little insight is gained over the first-order expression of Eq. 10. The theoretical approach presented for determining a second-order approximation for R1ρ from a reduced 3 × 3 matrix yields convenient results only for linear chains. The triangular three-site exchange mechanism and the four-site kite mechanism (Figure 4) have exchange matrices that do not permit factorization to a useful form for application of the matrix determinant lemma. The four-site star mechanism (Figure 4) has an exchange matrix with a simple factorization, but commutation properties of the matrices (Lj − λ) prevent needed simplifications as obtained for linear chains.
A combination of first- and second-order R1ρ approximations for linear schemes can be used to approximate R1ρ for non-linear schemes. This approximation is obtained by the Woodbury decomposition of the inverse of the matrix L + K and can in principle be obtained for (almost) any kinetic scheme. However, the accuracy of the Woodbury approximation depends on the particular decomposition of the kinetic scheme and on the relative contributions of the various components that contribute to Rex. The best Woodbury approximation is obtained if the slowest exchange processes are not included in the linear fragment; the accuracy of the Woodbury approximation is comparably high also for unskewed populations.
In conclusion, a remarkably simple first-order analytic approximation to the R1ρ relaxation rate constant is obtained from the trace of the inverse of the Bloch-McConnell evolution matrix for N-site chemical exchange with arbitrary topology. For linear chains, second-order approximations of higher accuracy also are derived by application of the matrix determinant lemma. Second-order solutions for linear schemes can be used in many circumstances to increase the accuracy of expressions for R1ρ in related non-linear kinetic schemes. The present results extend existing results for two-state chemical exchange to complex kinetic schemes and will be powerful in interpretation of numerous spin-locking NMR experiments.
Supplementary Material
Highlights.
Analytical expressions for the R1ρ relaxation rate for arbitrary kinetic schemes
Expressions for R1ρ with increased accuracy for linear N-site kinetic schemes
Examples for linear, triangular three-site, kite- and star-like kinetic mechanisms
Acknowledgments
Supported by National Institutes of Health grant GM059273 (A.G.P.). We thank Dr. Andrew Baldwin (Oxford) for helpful discussions that clarified the relationship between the present work and earlier reports.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palmer AG, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-millisecond motions in biological macromolecules. Meth Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 2.Palmer AG. Chemical exchange in biomacromolecules: past, present, and future. J Magn Reson. 2014;241:3–17. doi: 10.1016/j.jmr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer AG, Massi F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106:1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- 4.Grey MJ, Wang C, Palmer AG. Disulfide bond isomerization in basic pancreatic trypsin inhibitor: multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J Am Chem Soc. 2003;125:14324–14335. doi: 10.1021/ja0367389. [DOI] [PubMed] [Google Scholar]
- 5.Korzhnev DM, Religa TL, Lundstrom P, Fersht AR, Kay LE. The folding pathway of an FF domain: characterization of an on-pathway intermediate state under folding conditions by 15N, 13Cα and 13C-methyl relaxation dispersion and 1H/2H-exchange NMR spectroscopy. J Mol Biol. 2007;372:497–512. doi: 10.1016/j.jmb.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Li P, Martins IR, Rosen MK. The feasibility of parameterizing four-state equilibria using relaxation dispersion measurements. J Biomol NMR. 2011;51:57–70. doi: 10.1007/s10858-011-9541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libich DS, Tugarinov V, Clore GM. Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc Natl Acad Sci USA. 2015;112:8817–8823. doi: 10.1073/pnas.1510083112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CA, Ban D, Pratihar S, Giller K, Schwiegk C, de Groot BL, Becker S, Griesinger C, Lee D. Population shuffling of protein conformations. Angew Chem Int Ed Engl. 2015;54:207–210. doi: 10.1002/anie.201408890. [DOI] [PubMed] [Google Scholar]
- 9.Chao FA, Byrd RA. Geometric approximation: A new computational approach to characterize protein dynamics from NMR adiabatic relaxation dispersion experiments. J Am Chem Soc. 2016;138:7337–7345. doi: 10.1021/jacs.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trott O, Palmer AG. R1ρ relaxation outside of the fast-exchange limit. J Magn Reson. 2002;154:157–160. doi: 10.1006/jmre.2001.2466. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin AJ, Kay LE. An R1ρ expression for a spin in chemical exchange between two sites with unequal transverse relaxation rates. J Biomol NMR. 2013;55:211–218. doi: 10.1007/s10858-012-9694-6. [DOI] [PubMed] [Google Scholar]
- 12.Trott O, Palmer AG. Theoretical study of R1ρ rotating-frame and R2 free-precession relaxation in the presence of n-site chemical exchange. J Magn Reson. 2004;170:104–112. doi: 10.1016/j.jmr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Miloushev VZ, Palmer AG. R1ρ relaxation for two-site chemical exchange: General approximations and some exact solutions. J Magn Reson. 2005;177:221–227. doi: 10.1016/j.jmr.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 14.Trott O, Abergel D, Palmer AG. An average-magnetization analysis of R1ρ relaxation outside of the fast exchange. Mol Phys. 2003;101:753–763. [Google Scholar]
- 15.Barbara TM. Nonadiabatic exchange dynamics during adiabatic frequency sweeps. J Magn Reson. 2016;265:45–51. doi: 10.1016/j.jmr.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 16.Mangia S, Liimatainen T, Garwood M, Michaeli S. Rotating frame relaxation during adiabatic pulses vs. conventional spin lock: simulations and experimental results at 4 T. Magn Reson Imaging. 2009;27:1074–1087. doi: 10.1016/j.mri.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mangia S, Traaseth NJ, Veglia G, Garwood M, Michaeli S. Probing slow protein dynamics by adiabatic R1ρ and R2ρ NMR experiments. J Am Chem Soc. 2010;132:9979–9981. doi: 10.1021/ja1038787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traaseth NJ, Chao FA, Masterson LR, Mangia S, Garwood M, Michaeli S, Seelig B, Veglia G. Heteronuclear adiabatic relaxation dispersion (HARD) for quantitative analysis of conformational dynamics in proteins. J Magn Reson. 2012;219:75–82. doi: 10.1016/j.jmr.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abergel D, Palmer AG. On the use of the stochastic Liouville equation in NMR: Application to R1ρ relaxation in the presence of exchange. Conc Magn Reson. 2003;19A:134–148. [Google Scholar]
- 20.Palmer AG. A dynamic look backward and forward. J Magn Reson. 2016;266:73–80. doi: 10.1016/j.jmr.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grey MJ, Tang Y, Alexov E, McKnight CJ, Raleigh DP, Palmer AG. Characterizing a partially folded intermediate of the villin headpiece domain under non-denaturing conditions: contribution of His41 to the pH-dependent stability of the N-terminal subdomain. J Mol Biol. 2006;355:1078–1094. doi: 10.1016/j.jmb.2005.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.