

Abstract
The primary causes of chronic kidney disease (CKD) in children differ from those of adult onset CKD. In the United States the most common diagnostic groups of CKD that manifests before 25 years of age are: i) congenital anomalies of the kidneys and urinary tract (CAKUT) (49.1%), ii) steroid-resistant nephrotic syndrome (SRNS) (10.4%), iii) chronic glomerulonephritis (8.1%), and iv) renal cystic ciliopathies (5.3 %), encompassing >70% of CKD together. Recent findings suggest that early-onset CKD is caused by mutations in any one of over 200 different monogenic genes. High-throughput sequencing has very recently rendered identification of causative mutations in this high number of genes feasible. Molecular genetic diagnostics in early onset-CKD (before the age of 25 years) will, i) provide patients and families with a molecular genetic diagnosis, ii) generate new insights into diseases mechanisms, iii) allow etiology-based classification of patient cohorts for clinical studies and, iv) may have consequences for personalized treatment and prevention of CKD. In this review, we will discuss the implications of next-generation sequencing for clinical genetic diagnostics and discovery of novel genes in early-onset CKD. We also delineate the resulting opportunities for deciphering disease mechanisms and therapeutic implications.
Keywords: genetic kidney disease, monogenic disease, clinical genetic testing, chronic kidney disease (CKD), end-stage kidney disease (ESKD)
Introduction
Chronic kidney disease (CKD) in children is defined by the presence of kidney damage or by a glomerular filtration rate that has remained below 60 ml/min/1.73 m2 for more than 3 months1. Progression of CKD to end-stage renal disease (ESRD) requires dialysis or transplantation for survival. Although the prevalence of CKD has been increasing for as yet unidentified reasons2, little is known about any of the disease mechanisms. CKD that manifests in the first 25 years of life is caused to a large degree by CAKUT, SRNS, chronic glomerulonephr-iotis and renal cystic ciliopathies (Table 1). Whereas previously many of the diagnostic groups of early-onset CKD were not viewed as being of genetic origin, recently the discovery was made that in early-onset CKD (defined as CKD manifesting before 25 years of age) a monogenic cause of disease can be detected in the surprisingly high fraction of ~20% of individuals with early-onset CKD (Table 2). Monogenic mutations are sufficient as a singular cause of disease without requiring any additional biological or environmental causes of functional damage. This mechanism of genetic disease causation is known as “full penetrance” of the mutation (see glossary).
Table 1.
Causes of chronic kidney disease (CKD) manifesting before age 25 years, and its relative frequencya.
| DIAGNOSTIC GROUPS | Total |
|---|---|
| CAKUT | 49.1% |
| Obstructive uropathy (20.7%), a/hypo/dysplastic kidney (17.3%), reflux nephropathy (8.4%), prune belly syndrome (2.7%) | |
| SRNS | 10.4% |
| FSGS (8.7%), congenital nephrotic syndrome (1.1%), membranous nephropathy (0.5%), Denys-Drash syndrome (0.1%) | |
| Chronic glomerulonephritis | 8.1% |
| SLE nephritis (1.6%), familial nephritis (Alport syndrome) (1.6%), chronic glomerulonephritis (1.2%), MPGN-Type I (1.1%), MPGN-Type II (0.4%), IgA nephritis (0.9%), idiopathic cresentic GN (0.7%), Henoch-Schonlein nephritis (0.6%) | |
| Renal cystic ciliopathies | 5.3% |
| Polycystic kidney disease (4.0%), medullary cystic kidney disease (1.3%) | |
| Hemolytic uremic syndrome | 2.0% |
| Nephrolithiasis/nephrocalcinosis | 1.6% |
| Cystinosis (1.5%), oxalosis (0.1%) | |
| Other | 20.9% |
| Renal infarct (2.2%), pyelo/interstitial nephritis (1.4%), Wilms tumor (0.5%), Other systemic immunologic diseases (0.4%), Wegener’s granulomatosis (0.4%), sickle cell nephropathy (0.2%), diabetic glomerulopathy (0.2%), other (15.6%) | |
| Unknown | 2.6% |
| Total | 100% (N=7,037) |
From NAPRTCS - NAPRTCS, North American Pediatric Renal Trials and Collaborative Studies13. CKD, chronic kidney disease; CAKUT, congenital anomalies of the kidneys and urinary tract; FSGS, focal segmental glomerulosclerosis; GN, glomerulonephritis; MPGN, membranoproliferative glomerulonephritis; SRNS steroid-resistant nephrotic syndrome.
Table 2.
Indication-driven diagnostic panels of about 200 genes identify a causative mutation in ~20% of cases with CKD that manifests before <25 years of life (www.renalgenes.org).
| Diagnostic group | Clinical indication to run a gene panel | Proportion of CKD manifesting before 21 years of lifea | Number of currently known causative genes | Fraction of causative mutations identified for the diagnostic group (multiplied by fraction of all CKD) | References |
|---|---|---|---|---|---|
| CAKUT | CAKUT evident by renal imaging | 50% | 36 | c~17% (8.5%) | 7,11,18,40,41,91 |
| Steroid-resistant nephrotic syndrome | Steroid-resistant nephrotic syndrome | 10.3% | 39 | ~30% (3%) | 49,67,92 |
| Chronic glomerulonephritisb | Evidence of proteinuria and hematuria | 8.1% | 10 | ~20% (4%) | 5 |
| Renal cystic ciliopathies | Increased echogenicity on renal US or presence of ≥2 renal cysts | 5.3% | 95 | ~70% (3.7%) | 12,21,93,94 |
| aHUS | Microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. | ~2% | 9 | ~60% (1.2%) | 95–98 |
| Nephrolithiasis/nephrocalcinosis | Known stone disease or nephrocalcinosis | 1.6% | 30 | 21% (0.4%) | 47,99 |
| Other | 23.6 | ? | ? | ||
| Total (N=7,037)a | 100% | ~219 | (~20%) |
NAPRTCS 2008100.
The estimates for chronic nephritis monogenic etiologies are based only on the relative prevalence of Alport’s syndrome and MPGN (membranoproliferative glomerulonephritis) which together account for 20% of the etiologies of chronic glomerulonephritis and for which monogenic cause has been establish in almost 100% of cases (in one of the following genes: Alport: COL4A3, COL4A4, COL4A5 and COL4A6; MPGN: Factor H, Factor I, MCP/CD46, CFHR 5 and C3)
10% of CAKUT may be caused by deleterious copy number variants.101
CKD, chronic kidney disease; CAKUT, Congenital anomalies of the kidneys and urinary tract; aHUS, atypical hemolytic uremic syndrome.
More than 200 monogenic causative genes have now been identified for the 70% most common etiologies of CKD in this age group3–12. We focus here on single-gene causes of early-onset CKD and discuss the implication of next-generation sequencing for the genetic diagnosis of early-onset CKD. We then address the discovery of novel genes that if mutated cause early-onset CKD and discuss resulting opportunities for delineating the pathomechanisms and therapeutic implications.
Epidemiology of chronic kidney disease in children
The primary causes of early-onset CKD in children differ from adult-onset CKD (Table 1). The 2008 report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS), which included data from 7,037 children and young adults with CKD,13 found the most common diagnostic groups to be (1) congenital anomalies of the kidneys and urinary tract (CAKUT) (49.1%), (2) steroid-resistant nephrotic syndrome (SRNS) (10.4%), (3) chronic glomerulonephritis (8.1%), and (4) renal cystic ciliopathies (5.3%), together encompassing over 70% of the entire pediatric CKD population (Table 1). Those diagnostic groups also represent the most common causes of early-onset CKD in developed countries outside the United States14. The etiologies of the above diagnostic groups of CKD were unknown before the past decade, when identification of many single-gene (monogenic) causes of CKD revealed their primary causes (etiologies) and providing a powerful approach to delineate the related pathomechanisms. This improved understanding of disease is exemplified, for instance, by the discovery of NPHS1 (nephrin) mutations as a cause of congenital nephrotic, thereby identifying dysfunction of the glomerular podocyte as central to the pathogenesis of steroid resistant nephrotic syndrome (SRNS)15–17.
Monogenic diseases (also referred to as Mendelian or single-gene disorders) result from mutations in a single causative gene. Patterns of Mendelian inheritance include: autosomal dominant, autosomal recessive, and X-linked. Over the past 15 years over 200 monogenic causes of early onset CKD have been identified (Table 2). Most of them were discovered in the last 5 years due to acceleration of gene discovery by modern technologies of genetic mapping and whole exome sequencing. Currently there are approximately 36 genes known to be mutated in CAKUT10,11,18,19, 39 genes in SRNS8,20 (Hildebrandt et al. 2015, NDT, in press), 10 genes in chronic glomerulonephritis, and over 95 genes in renal cystic ciliopathies4,21. These data demonstrate that in ~ 20% of patients with early-onset CKD a monogenic cause of disease can be identified by mutation analysis (Table 2).
Genetic disease causality
The degree to which causality is attributed to a certain genetic variant can be classified according to the penetrance of a given disease-causing allele. Genetic “penetrance” reflects the proportion of individuals that express a certain disease phenotype in relation to the number of individuals that carry the genetic variant(s). ”Full penetrance” means that 100% of individuals that carry a genetic variant also express the disease phenotype22. At one end of the range of genetic causality are recessive monogenic Mendelian diseases (also known as single-gene disorders), which have a tight genotype-phenotype correlation, so that the disease phenotype is almost entirely determined by disease-causing mutations in a single gene (full penetrance) (Table 3). This is the case for instance in NPHP1 mutations that cause juvenile nephronophthisis in any patient who carries mutations on both copies of the NPHP1 gene23. Those mutations inescapably cause CKD with renal fibrosis and cysts by the age of 20 years. Autosomal dominant monogenic Mendelian diseases, in contrast to recessive diseases, have reduced tightness of genotype-phenotype correlation, due to multiple characteristics of dominant diseases including (Table 3): i) age-related penetrance (with increasing age, a higher fraction of individuals that carry the causative mutation express the disease); ii) incomplete penetrance, i.e. some individuals with the mutation do not develop the diseases phenotype at all. The disease thereby appears to be skipping generations in a pedigree; iii) variable expressivity (i.e. different degrees of severity and/or organ involvement occur in different affected individuals that carry identical mutated alleles). An example of an autosomal dominant kidney disease with variable expressivity is given by HNF1B mutations that cause CAKUT, CKD and maturity-onset diabetes of the young (MODY) with variable age of onset and variable presence of MODY diabetes24,25. Variable expressivity mainly describes a complex genotype-phenotype relationship in dominant diseases. A similarly complex situation exists in recessive diseases that may exhibit ‘multiple allelism’. This phenomenon refers to the finding that different (homozygous) recessive mutations in the same gene may lead to different clinical outcomes. For instance, certain mutation in LAMB2 that cause nephrotic syndrome may lack ocular involvement26, or specific combinations of compound heterozygous mutations of NPHP2 may cause adult onset rather than childhood onset nephrotic syndrome27.
Table 3.
Degrees of genetic causality in monogenic and polygenic kidney diseases.
| Monogenic recessive diseases | Monogenic dominant diseases | Polygenic/complex Diseases, Risk alleles | |
|---|---|---|---|
| Penetrance | Full | Full or incomplete | Low |
| Predictive power of a mutation | Almost 100% | High | Low |
| Onset | Predominantly during childhood | Childhood and adulthood | Predominantly during adulthood |
| Disease frequency | Low | Low | High |
| Number of affected subjects needed for gene discovery | Few | Few | Hundreds to ten thousands |
| Gene mapping approaches include | Homozygosity mapping* or linkage analysis | Linkage analysis | Genome-wide association studies (GWAS) |
| Whole exome/genome sequencing (WES) | In consanguinity, single affecteds are sufficient | WES in distant relatives to minimize shared variants | N/A |
| Functional analysis in animal models (mice, zebrafish) | Easily feasible (gene knockdown, knockout) | Feasible | Difficult |
| Examples of genes mutated in kidney diseases | NPHP1, NPHS1 | PAX2, HNF1B | APOL1 |
Applicable to consanguineous families.
N/A, not applicable; WES, whole exome sequencing
At the other end of the spectrum of causality are more common conditions for which low-penetrance, so-called “risk alleles”, have been described22. In those conditions, which often are referred to as polygenic or complex diseases genetic variants usually exert small effects on the disease (Table 3). Therefore, usually only a small fraction of the statistical variance for a disease phenotype can be assigned to a risk allele. An exception from this situation occurs in the APOL1 gene, in which specific genetic variants, apparently in a recessive way, convey a large phenotypic risk for the development of CKD in the African American population28,29 (Table 3). An example of successful identification of disease risk alleles in kidney diseases is that of specific genotypes in the APOL1 locus that were associated with an increased risk of focal segmental glomerulosclerosis and chronic kidney disease in African-American patients.28,30–32 For instance, about 13–23% of African-Americans (compared with 0.3–1.3% of European Americans) have one out of the known two APOL1 risk alleles33,34. For African Americans carrying 2 risk alleles in trans, the risk of developing focal segmental glomerulosclerosis is increased 17-fold compared to control individuals carrying 0–1 risk allele33,34.
Finally, another aspect of genetic causality that should take into consideration is the contribution of genetic modifiers. This concept in which specific alleles are responsible for modification of disease phenotypes, have been described for monogenic forms of cystic kidney disease35 and glomerulonephritis36. Nonetheless, additional supporting evidence is needed for some of these associations in early onset CKD.
We11 as well as others37 have noted that there are many false assignments of potential disease causality at the variant level. Specifically, it has been noted that up to 30% of genetic variants published as likely disease causing and deposited in genetic databases were not confirmed as deleterious38. Consequently, any attribution of pathogenicity to a given variant should be subject to strict criteria and taking into consideration multiple levels of evidence such as amino acid sequence conservation, segregation analysis, tissue specific gene expression, functional studies, and animal models37,39. For the decision if a genetic variant qualifies as potentially disease causing we follow empiric core rules that are outlined in Box 1 for recessive monogenic diseases and in Box 2 for dominant genes. These core rules are not absolute, and provide only general guidance. Furthermore, the number of families with early CKD that have been previously reported to have a mutation in the candidate causative gene should also be considered. For instance, some of the CAKUT-causing genes were reported in only single families and therefore any generalizations regarding their role, however, must await the description and characterization of mutations in additional patients.
Box 1. Assignment of autosomal recessive mutations as being disease causing.
-
Include allele as disease causing if:
-
–
Truncating mutation (Stop, abrogation of start or stop, obligatory splice, frameshift) in an expressed gene (well annotated mRNA, sequence conservation, protein expression) or:
-
–
Missense mutation if:
Continuously conserved at least up to danio rerio (zebrafish) and:
Loss of function in human allele is supported by functional data.
-
–
-
Exclude allele as disease causing if:
-
–
Heterozygous allele frequency >1% (in EVS server: 13,000 control chromosomes) or single homozygous reported.
-
–
Non-segregation (e.g. “compound heterozygous” in cis; affected family member is without the variant; unaffected parent is with homozygous variant)
-
–
Base line assumptions: 1) Full penetrance (age related). 2) Defined clinical phenotype. 3) “Mutation” implies that an allele changes the phenotype. 4) Known genes with similar phenotype have been excluded.
Box 2. Assignment of autosomal dominant mutations as being disease causing.
-
Include allele as disease causing if:
-
–
Truncating mutation (Stop, abrogation of start or stop, obligatory splice, frame-shift) in an expressed gene (well annotated mRNA, sequence conservation, protein expression) and:
Continuously conserved to at least up to danio rerio (zebrafish) or:
-
–
Missense mutation if:
Continuously conserved to danio rerio. And:
Human allele is supported by functional data. And:
Full segregation exists And:
Known genes with similar phenotype have been excluded.
-
–
-
Exclude allele as disease causing if:
-
–
Heterozygous allele frequency >0.1%
-
–
Non-segregation – i.e. affected family member is without the allele.
-
–
Warning regarding non-segregation: if an unaffected family member is with the allele consider incomplete penetrance and variable expressivity.
-
–
Indication-driven gene panel analysis using next generation sequencing
Mutation analysis in recessive or dominant monogenic kidney diseases may reveal the primary cause (etiology) of a disease resulting from an inherited disease-causing gene. Such analyses can enable disease entities to be categorized on the basis of their genetic etiologies. A monogenic cause of the early onset CKD diagnoses listed in Table 1 may be found in a substantial portion of affected individuals who are enrolled in clinical research or drug trials4,11,40–47. Because of this, we suggest that these subjects all undergo molecular genetic diagnostics to account for subjects with “monogenic disease” in downstream epidemiologic analyses. Failure to do so may confound any conclusions. Moreover, molecular genetic diagnostics enables prenatal testing and may have prognostic and sometimes therapeutic implications.
We have developed indication-driven diagnostic exon sequencing panels45,48 for CAKUT10,11, steroid resistant nephrotic syndrome8, renal cystic ciliopathies45, glomerulonephritis, and nephrolithiasis/nephrocalcinosis (Table 2)3. These 5 diagnostic groups of CKD alone encompass 72.8% of CKD that manifest before 25 years of life (Table 1). Using a microfluidic technique (Fluidigm™) for multiplex PCR-based amplification of 600 exons of about 30 different gene known to be mutated in the respective CKD diagnostic groups, we established a cost-effective mutation analysis screen of large patient cohorts. This method includes barcoding of individual DNAs PCR product followed by next generation sequencing3,7–11,44,45. PCR products are barcoded per individual so that hundreds of PCR products can be sequenced in a single next-generation sequencing run thereby strongly reducing cost. Indications to run a diagnostic panel were kept simple (Table 2) to allow that in future applications of the panels similar results can be expected: for the CAKUT panel the indication to run the panel was any imaging study showing evidence of CAKUT (renal aplasia, renal hypodysplasia, vesicoureteral reflux or uretero-pelvic junction obstruction)11. For the proteinuria panel the indication was SRNS9,49. For the nephrolithiasis (urinary stone disease) panel indication was any history of nephrolithiasis/nephrocalcinosis47. For the glomerulonephritis panel the indication to run the panel was the presence of proteinuria and hematuria. For the renal cystic ciliopathy panel the indication was the presence of ≥2 renal cysts or increased renal echogenicity on renal sonography4,45,50 (Braun, in press 2015). Of note, the latter has over 95 known disease causing genes molecularly explaining the vast majority of cases (~70%).
CAKUT panel
Using gene panels we examined a large international cohort of 650 unrelated families with CAKUT for the presence of mutations in 17 autosomal dominant and 6 autosomal recessive known CAKUT-causing genes10,11. Our results showed that over 8% of cases with CAKUT are caused by single-gene mutations in one of the 17 genes. These results as well as results from two independent studies51,52 in which copy number variations (CNVs) were identified among 10–16% of individuals with CAKUT (most commonly involving the HNF1B or the DiGeorge/velocarodiofacial locus), suggest that CAKUT genes may already yield a monogenic cause in around 17% of affected individuals (Tables 2, 4).
Table 4.
Thirty six genes that cause monogenic-CAKUT if mutated
| Gene | Protein | Reference |
|---|---|---|
| Autosomal dominant | ||
| BMP4 | Bone Morphogenetic Protein 4 | 102 |
| CHD1L | Chromodomain Helicase DNA Binding Protein 1-Like | 103 |
| DSTYK | Dual Serine/Threonine And Tyrosine Protein Kinase | 41 |
| EYA1 | EYA Transcriptional Coactivator And Phosphatase 1 | 104 |
| GATA3 | GATA Binding Protein 3 | 105,106 |
| HNF1B | HNF1 Homeobox B | 107 |
| MUC1 | Mucin 1, Cell Surface Associated | 108 |
| PAX2 | Paired Box 2 | 109 |
| RET | Ret Proto-Oncogene | 110 |
| ROBO2 | Roundabout, Axon Guidance Receptor, Homolog 2 (Drosophila) | 111 |
| SALL1 | Spalt-Like Transcription Factor 1 | 112 |
| SIX1 | SIX Homeobox 1 | 113 |
| SIX2 | SIX Homeobox 2 | 102 |
| SIX5 | SIX Homeobox 5 | 114 |
| SOX17 | SRY (Sex Determining Region Y)-Box 17 | 115 |
| SRGAP1 | SLIT-ROBO Rho GTPase Activating Protein 1 | 116 |
| TBX18 | T-Box 18 | 19 |
| TNXB | Tenascin XB | 117 |
| UMOD | Uromodulin | 118 |
| UPK3A | Uroplakin 3A | 119 |
| WNT4 | Wingless-Type MMTV Integration Site Family, Member 4 | 120,121,122 |
| Autosomal recessive | ||
| ACE | Angiotensin I Converting Enzyme | 123 |
| AGT | Angiotensinogen (Serpin Peptidase Inhibitor, Clade A, Member 8) | 123 |
| AGTR1 | Angiotensin II Receptor, Type 1 | 123 |
| CHRM3 | Cholinergic Receptor, Muscarinic 3 | 124 |
| FGF20 | Fibroblast Growth Factor 20 | 125 |
| FRAS1 | Fraser Extracellular Matrix Complex Subunit 1 | 40,126 |
| FREM2 | FRAS1 Related Extracellular Matrix Protein 2 | 40 |
| FREM1 | FRAS1 Related Extracellular Matrix 1 | 40 |
| GRIP1 | Glutamate Receptor Interacting Protein 1 | 40 |
| HPSE2 | Heparanase 2 (Inactive) | 127 |
| ITGA8 | Integrin, Alpha 8 | 40,128 |
| LRIG2 | Leucine-Rich Repeats And Immunoglobulin-Like Domains 2 | 129 |
| REN | Renin | 123 |
| TRAP1 | TNF Receptor-Associated Protein 1 | 7 |
| X-Linked | ||
| KAL1 | Anosmin 1 | 130 |
Proteinuria panel
Mutation analysis of 27 known SRNS-causing genes in an international cohort of patients with SRNS manifesting before 25 years of age8 detected a single-gene cause in 29.5% (526/1,783) of families (Tables 2, 5). The fraction of families, in whom a single-gene cause was identified correlated inversely with age of onset. The fraction of families with detection of a single-gene cause of SRNS was 69.4%, 49.7%, 25.3%, 17.8% and 10.8% for the age groups of manifestation in the first 3 months of life, 4–12 months, 1–6 years old, 7–12 years and between 13–18 years respectively8.
Table 5.
Thirty nine monogenic genes that cause steroid-resistant nephrotic syndrome (SRNS) if mutated (marked with “*” are genes sequenced in Sadowski et al)
| Autosomal recessive | ||
|---|---|---|
| ADCK4* | AarF domain containing kinase 4 | 58 |
| ARHGDIA* | Rho GDP dissociation inhibitor (GDI) alpha | 59 |
| CD2AP* | CD2-associated protein | 80,131 |
| CFH* | Complement factor H | 132 |
| COQ2* | Coenzyme Q2 4-hydroxybenzoate Polyprenyltransferase |
85,56 |
| COQ6* | Coenzyme Q6 monooxygenase | 57 |
| CRB2 | Crumbs homolog 2 | 8 |
| CUBN* | Cubilin (intrinsic factor-cobalamin receptor) | 133 |
| DGKE* | Diacylglycerol kinase, epsilon | 134 |
| EMP2 | Epithelial membrane protein 2 | 84 |
| FAT1 | FAT tumor suppressor homolog 1 | Gee, in press |
| ITGA3* | Integrin, alpha 3 (antigen CD49C, alpha 3 subunit of VLA-3 receptor) | 135 |
| ITGB4* | Integrin, beta 4 | 136 |
| KANK1 | KN motif and ankyrin repeat domain containing protein 1 | 60,137 |
| KANK2 | KN motif and ankyrin repeat domain containing protein 2 | 60,137 |
| KANK4 | KN motif and ankyrin repeat domain containing protein 4 | 60,137 |
| LAMB2* | Laminin, β2 | 83 |
| MTTL1 | Mitochondrially encoded tRNA leucine 1 | |
| MYO1E* | Homo sapiens myosin IE (MYO1E) | 138 |
| NPHS1* | Nephrin | 15 |
| NPHS2* | Podocin | 79 |
| NUP93 | Nucleoporin 93 kDa | Braun, submitted |
| NUP107 | Nucleoporin 107 kDa | 139 |
| NUP205 | Nucleoporin 205 kDA | Braun, submitted |
| PDSS2* | Prenyl (decaprenyl) diphosphate synthase, subunit 2 | 140 |
| PLCE1* | Phospholipase C, epsilon 1 | 62 |
| PTPRO* | Protein tyrosine phosphatase, receptor type, O | 141 |
| SCARB2* | Scavenger receptor class B, member 2 | 142 |
| SMARCAL1* | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1 | 143 |
| WDR73 | WD repeat domain 73 | 144–146 |
| XPO5 | Exportin 5 | Braun, submitted |
| Autosomal dominant | ||
| ACTN4* | Actinin, alpha 4 | 81 |
| ANLN | Anillin, actin binding protein | 147 |
| ARHGAP24* | Rho GTPase activating protein 24 | 148 |
| INF2* | Inverted formin, FH2 and WH2 domain containing | 82 |
| LMX1B* | LIM homeobox transcription factor 1, beta | 149 |
| MYH9 | Myosin heavy chain 9 | 150 |
| TRPC6* | Transient receptor potential cation channel, subfamily C, member 6 | 151,152 |
| WT1* | Wilms tumor 1 | 153 |
The identification of single-gene mutations in SRNS genes may have therapeutic consequences in some cases. For instance, most individuals with a single-gene cause of SRNS will not respond to steroid treatment.53,54 WT1 mutations in patients with SRNS can predispose to certain malignancies. Consequently, the detection of WT1 mutations should trigger monitoring and further evaluation of affected individuals for associated tumors that include wilms tumor and gonadoblastoma. The latter has been mainly described with concomitant abnormal chromosomal karyotype and therefore a karyotype analysis should also be obtained.55 Furthermore, identification of the causative mutation may reveal that a potential therapy is available for some rare single-gene causes of SRNS. For example, if a mutation in a gene encoding enzymes of the coenzyme Q10 biosynthesis is detected (COQ2, COQ6, ADCK4, or PDSS2), experimental treatment with coenzyme Q10 may be warranted,56,57 because a partial response to treatment with coenzyme Q10 has been described in individuals with SRNS and mutations in COQ2,56 COQ6,57 and ADKC458. The efficacy of CoQ10 treatment has to be assessed once higher numbers of patients with mutations in genes of CoQ10 biosynthesis have become known.
Small Rho-like GTPases (RhoA/Rac1/Cdc42) are part of another pathway that has been implicated in the pathogenesis of nephrotic syndrome through the identification of mutations in the SRNS genes ARHGDI, KANK2,3, and 4 and through elucidation of the response of synaptopodin to cyclosporine A treatment in patients with SRNS59–61. Also, individuals with mutations of CUBN may be amenable to treatment with vitamin B12, and individuals with ARHGDIA may theoretically be responsive to the eplerenone treatment.59 Finally, a patient with recessive mutations in PLCE1 responded fully to treatment with steroids or cyclosporine A.62
In the future it may be advisable to initiate mutation analysis of all known nephrosis genes in any patient with an episode of proteinuria persistent for more than 3 days (urine protein greater than 4mg/m2/hour). In a first episode with gross proteinuria steroid treatment may have been commenced at the same time of initiating mutation analysis. If results from mutation analysis are returned within a few weeks, they may then guide the decision whether to complete a full course of steroid treatment or to terminate treatment, depending on whether there is enough data available for a certain mutation that would warrant discontinuation of treatment. In this way unnecessary steroid toxicity may be avoided in the near future.
Nephritis panel
In individuals with a diagnostic constellation compatible with chronic glomerulonephritis (small grade proteinuria with microscopic hematuria) exon sequencing of 10 monogenic nephritis genes may already yield a monogenic cause of nephritis in about 20% of individuals63 (Tables 2, 6).
Table 6.
Ten genes that cause monogenic chronic glomerulo-nephritis if mutated.
| Gene | Protein | Disease | Reference |
|---|---|---|---|
| Autosomal recessive | |||
| COL4A4* | Collagen, type IV, alpha 4 | Alport | 154 |
| CFH | Complement factor H | MPGN | 96 |
| Autosomal Dominant | |||
| CFI | Complement factor I | MPGN | 155 |
| CFHR5** | Complement factor H-related 5 | MPGN | 156, 98,157,158 |
| FN1 | Fibronectin 1 | GFND | 159 |
| Autosomal dominant/recessive | |||
| COL4A3* | Collagen, type IV, alpha 3 | Alport | 154 |
| CD46 | CD46 molecule, complement regulatory protein (MCP) | MPGN | 160 |
| C3 | Complmenet component 3 | MPGN | 97 |
| X-linked | |||
| COL4A5 | Collagen, type IV, alpha 5 | Alport | 161 |
| COL4A6 | Collagen, type IV, alpha 6 | Alport with LM | 162 |
Alport: Alport’s syndrome; aHUS: atypical HUS; TMA: thrombotic microangiopathy; GFND: glomerulopathy with giant fibronectin deposits; FMF: familial Mediterranean fever; LM: leiomyomatosis.
Both, COL4A3 and COL4A4 can independently lead to autosomal dominant or autosomal recessive forms of Alport syndrome163
Cystic kidney disease panel
In 50–70% of all individuals who exhibit upon renal ultrasound the presence of 2 or more cysts and/or a finding of increased echogenicity, a monogeic cause of disease can be detected by exon sequencing of one of 95 genes (Tables 2, 7).4,44,45 (Braun et al. in press). The PKD1 and PKD2 genes, which are mutated in ADPKD are not part of this panel, because their mutation analysis requires a very specialized approach64, because onset of disease in ADPKD is primarily far beyond 25 of age, and because mutation analysis is rarely requested within the PKD community as molecular diagnosis is valuable in only few specific situations mostly not for the pediatric population.
Table 7.
Sixteen frequent and 66 infrequent monogenic causes of nephronophthisis-related ciliopathies (NPHP-RC).
| Gene | Protein | References |
|---|---|---|
| NPHP1 (JBTS4) | Nephrocystin-1 | 164,165 |
| INVS (NPHP2) | Inversin | 166 |
| NPHP3 | Nephrocystin-3 | 167 |
| NPHP4 | Nephroretinin | 168,169 |
| IQCB1 (NPHP5) | IQ motif containing B1 | 170 |
| CEP290 (NPHP6) | Centrosomal protein 290 kDa | 171 |
| GLIS2 (NPHP7) | GLIS family zinc finger 2 | 172 |
| RPGRIP1L (NPHP8) | RPGRIP1-like/FTM | 173 |
| NEK8 (NPHP9) | NIMA (never in mitosis gene A)- related kinase 8 | 174 |
| SDCCAG8 (NPHP10) | Serologically defined colon cancer antigen 8 | 175 |
| TMEM67 (NPHP11) | Transmembrane protein 67 | 176 |
| TTC21B (NPHP12) | Tetratricopeptide repeat domain 21B | 177 |
| WDR19 (NPHP13) | WD repeat domain 19 | 178 |
| ZNF423 (NPHP14) | Zinc finger protein 423 | 179 |
| CEP164 (NPHP15) | Centrosomal protein 164 kDa | 179 |
| ANKS6 (NPHP16) | Ankyrin repeat and sterile alpha motif domain containing 6 | 180 |
Monogenic (recessive) mutations in the following additional 66 genes also cause the nephronophthisis-related ciliopathies (NPHP-RC) Meckel syndrome, Senior-Loken syndrome, Joubert syndrome, or Bardet-Biedl syndrome, but less frequently:
XPNPEP3, ATXN10, FAN1, SLC41A1, HNF1B, CLDN16, CLDN19, BSND, SLC12A3, CLCNKB, AGXT, GRHPR, HOGA1, PKHD1, INPP5E, TMEM216, AHI1, ARL13B, CC2D2A, OFD1, KIF7, TCTN1, TMEM237, CEP41, TSGA14, TMEM138, C5orf42, TMEM231, CSPP1, PDE6D, TBC1D32, SCLT1, MKS1, TCTN2, B9D1, B9D2, KIF14, BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, PTHB1, C21orf58, TRIM32, C4orf24, WDPCP, LZTFL1, ALMS1, IFT122, WDR35, IFT140, C14ORF179, DYNC2H1, WDR34, WDR60, IFT80, IFT172, TRAF3IP1, NEK1, POC1A, EVC, and EVC2.
Nephrolithiasis panel
Similarly, we demonstrated that 21% of cases with onset of nephrolithiasis/nephrocalcinosis before 18 years of age and 12% of cases with onset after 18 years can be explained by mutations in one of 14 genes known to cause nephrolithiasis/nephrocalcinosis3 (+Braun et al, 2015 in press) (Tables 2, 8). For this phenotype, the cystinuria gene SLC7A9 was the most frequently mutated (Table 8), being found in 15% of the cohort. Making a molecular genetic diagnosis in urinary stone disease had important implications for affected individuals as well as unaffected family members. Genetic screening of asymptomatic relatives may identify individuals who carry the same disease causing mutation. This information will guide clinicians to monitor these individuals for development of disease and to institute preventative treatment when possible. In addition, consensus guidelines recommend standard treatment for urinary stone disease such as increased fluid intake, limited sodium intake, treatment with thiazide diuretics, and potassium citrate therapy65 that may not directly address the pathophysiology of a particular molecular diagnosis. For example, clinicians should monitor for tetany and seizures, which have been reported in patients with CLDN16 mutations. We recently published such related therapeutic implications that resulted from making a molecular genetic diagnosis in urinary stone disease (Braun et al. 2015).
Table 8.
Thirty monogenic genes that cause urinary stone disease (USD) if mutated.
| Gene | Protein | Disease entity | Mode of inheritance | Reference |
|---|---|---|---|---|
| ADCY10/SAC | adenylate cyclase 10 (soluble) | Hypercalciuria, Calcium oxalate nephrolithiasis | AD | 181 |
| AGXT | alanine-glyoxylate aminotransferase | Primary hyperoxaluria, type 1 | AR | 182 |
| APRT | adenine phosphoribosyltransferase | Adenine phosphoribosyltransferase deficiency, Urolithiasis (DHA stones), renal failure | AR | 183 |
| ATP6V0A4 | ATPase, H+ transporting, lysosomal V0 subunit a4 | dRTA | AR | 184 |
| ATP6V1B1 | ATPase, H+ transporting, lysosomal 56/58kDa, V1 subunit B1 |
Distal renal tubular acidosis (dRTA) with deafness | AR | 185 |
| CA2 | carbonic anhydrase II | Osteopetrosis + d/pRTA | AR | 186 |
| CASR | calcium-sensing receptor | Hypocalcemia with Bartter syndrome/hypocalcemia, autosomal dominant | AD | 187 |
| CLCN5 | chloride channel, voltage-sensitive 5 | Dent disease/Nephrolithiasis, type 1 | XR | 188 |
| CLCNKB | chloride channel, voltage-sensitive Kb | Bartter syndrome, type 3 | AR | 189 |
| CLDN16 | claudin 16 | Familial hypomagnesemia with hypercalciuria & nephrocalcinosis, FHHNC | AR | 190 |
| CLDN19 | claudin 19 | Familial hypomagnesemia with hypercalciuria & nephrocalcinosis with ocular abnormalities | AR | 191 |
| CYP24A1 | cytochrome P450, family 24, subfamily A, polypeptide 1 | 1,25-(OH) D-24 hydroxylase deficiency, infantile Hypercalcemia |
AR | 192 |
| FAM20A | family with sequence similarity 20, member A | Enamel-Renal syndrome, amelogenesis imperfect and nephrocalcinosis | AR | 193 |
| GRHPR | glyoxylate reductase/hydroxypyruvate reductase | Primary hyperoxaluria, type 2 | AR | 194 |
| HNF4A | hepatocyte nuclear factor 4, alpha | MODY + Fanconi syndrome + Nephrocalcinosis | AD | 195 |
| HOGA1 | 4-hydroxy-2-oxoglutarate aldolase 1 | Primary hyperoxaluria, type 3 | AR | 196 |
| HPRT1 | Hypoxanthine phosphoribosyltransferase 1 | Kelley-Seegmiller syndrome, partial HPRT deficiency, HPRT-related gout | XR | 197 |
| KCNJ1 | potassium inwardly-rectifying channel, subfamily J, member 1 | Bartter syndrome, type 2 | AR | 198 |
| OCRL | oculocerebrorenal syndrome of Lowe | Lowe syndrome/Dent disease 2 | XR | 199 |
| SLC12A1 | solute carrier family 12, member 1 | Bartter syndrome, type 1 | AR | 200 |
| SLC22A12 | solute carrier family 22 (organic anion/urate transporter), member 12 | Renal hypouricemia, RHUC1 | AD/AR | 201 |
| SLC2A9 | solute carrier family 2 (facilitated glucose transporter), member 9 | Renal hypouricemia, RHUC2 | AD/AR | 202 |
| SLC34A1 | solute carrier family 34 (sodium phosphate), member 1 | Hypophosphatemic nephrolithiasis/osteoporosis-1, NPHLOP1/Fanconi renotubular syndrome 2 | AD/AR | 203 |
| SLC34A3 | solute carrier family 34 (sodium phosphate), member 3 | Hypophosphatemic rickets with hypercalciuria | AR | 204 |
| SLC3A1 | solute carrier family 3 (cystine, dibasic and neutral amino acid transporters, activator of cystine, dibasic and neutral amino acid transport), member 1 | Cystinuria, type A | AR | 205 |
| SLC4A1 | solute carrier family 4, anion exchanger, member 1 (erythrocyte membrane protein band 3, Diego blood group) | Primary distal renal tubular acidosis, dominant/recessive | AD/AR | 206 |
| SLC7A9 | solute carrier family 7 (glycoproteinassociated amino acid transporter light chain, bo,+ system), member 9 | Cystinuria, type B | AD/AR | 207 |
| SLC9A3R1 | solute carrier family 9, subfamily A (NHE3, cation proton antiporter 3), member 3 regulator 1 | Hypophosphatemic nephrolithiasis/osteoporosis-2, NPHLOP2 | AD | 208 |
| VDR | vitamin D (1,25- dihydroxyvitamin D3) receptor | Idiopathic hypercalciuria | AD | 209 |
| XDH | xanthine dehydrogenase | Xanthinuria, type 1 | AR | 210 |
AD, autosomal dominant; AR, autosomal recessive
In summary, it is expected that the use of diagnostic exon sequencing panels will expand the number of genes examined in the future for each of these groups of monogenic causes of CKD. In addition, other exon sequencing panels will be introduced into the clinical practice in order to detect monogenic causes of CKD in additional diagnostic groups of early onset CKD such as monogenic forms of hypertension. The ongoing discovery of novel genes that if mutated cause CKD, together with the continuing trend of cost reduction in exome sequencing implies that indication-driven molecular genetic diagnostics in the near future will be performed using whole exome sequencing (WES) data, which sequences all exons of all 20,000 genes in the human genome in parallel at low cost4,66. However, in this context it will be important to maintain an indication-driven “a priori” approach, in which only genes known to cause the respective disorder are evaluated for mutations, on the basis of clearly defined clinical indication criteria as mentioned above (Table 2).
Generalizability of mutation detection rates
The generalizability of the current mutation detection rates across the different early CKD etiologies and among different populations should take into consideration several important factors. First, the relative fractions in whom a molecular genetic diagnosis was made were inversely correlated with age and directly correlated to degree of consanguinity as described in the original publications for SRNS8 (Figure 1) and for urinary stone disease3. Still, it is very likely that these rates of mutation identification in SRNS (Figure 1) will hold up in other cohorts as they have been confirmed by 2 European groups20,67. Likewise, in urinary stone disease we have recently performed 2 different studies that showed a rate of detecting causative mutations in the range of 18–21% in childhood onset urinary stone disease. The rates of successful mutation identification will most likely increase as more monogenic renal genes and mutations become known.
Figure 1.
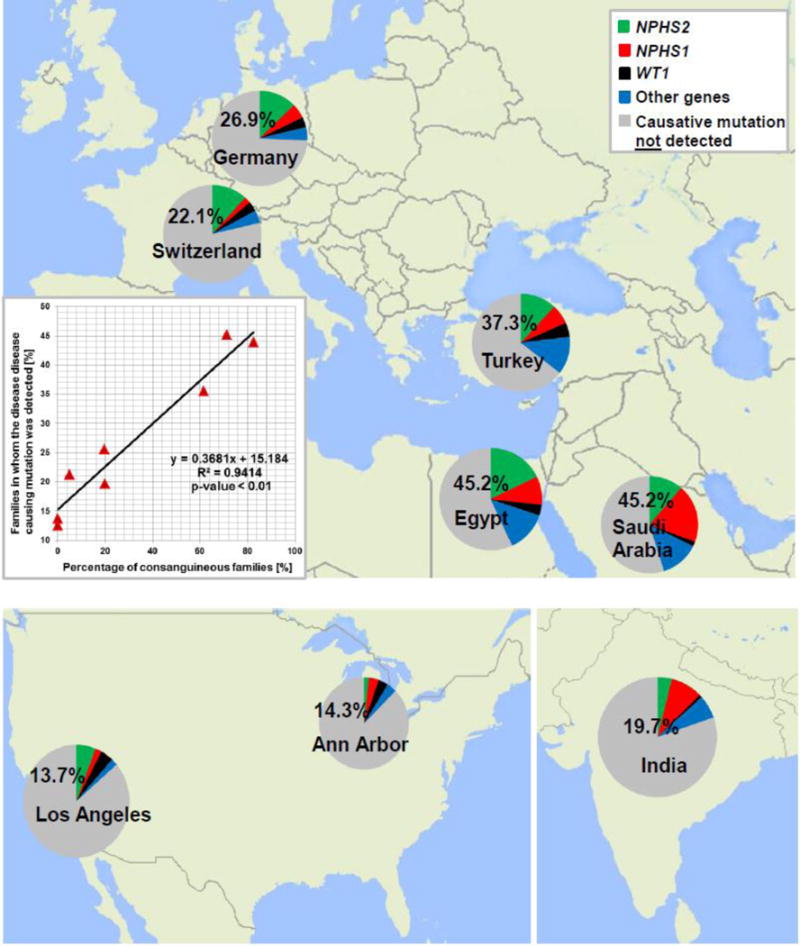
Percentage of genetic findings in SRNS families. We previously obtained samples from 1,783 SRNS families worldwide and detected the disease-causing mutation in 526 families (29.5%). For 8 centers we detected the disease causing mutations in the following fractions: (families, in whom we detected the causative mutation/total families examined from this center): Saudi-Arabia (45.2%, 28/62), Egypt (45.2%, 66/146), Turkey (37.3%, 62/169), Germany (26.9%, 123/457), Switzerland (22.1 %, 21/94), India (19.7%, 25/127,), Ann Arbor (14.3%, 8/56), and Los Angeles (13.7%, 7/51). Inset: The detection rate of the disease-causing mutations strongly correlates with the rate of consanguinity between the different centers (R2=0.9414)
Second, as more data from extensive whole exome studies are rapidly accruing on human genetic variation, question regarding incomplete penetrance of certain alleles can be address and studied. There is increasingly apparent degree of incomplete penetrance and variable expressivity especially for monogenic dominant causes of CAKUT, but also for other recessive etiologies. For instance, in early–onset disease where recessive mutation are more frequent and usually convey full penetrance, few exceptions have also been described68.
Third, the potential for false positive attribution of monogenic disease resulting from inappropriate filtering criteria or increased sequencing of patients who may have a lower probability of having monogenic disease can also hamper the true frequency of mutation detection rates. Minimization of the problem of false positive assignment of genetic variants as disease causing will be one of the most important task in the renal research area for the next 10 years to come. These data will be generated using cell-based functional assays, animal models, and large data bases on genetic variants in large populations around the world. Fourth, for dominantly inherited conditions, the presence of familial cases will often also positively influence the mutation detection rate69.
Finally, one of the potential adverse outcomes of mutation analysis in monogenic disease genes may result from mutational screening of unaffected family members. This may be particularly detrimental to an individual if there is incomplete penetrance or variable expressivity for a disease allele, leading prognostication of an unfavorable health condition that may never manifest. In this context it is important to observe the recommendation by the American College of Medical Genetics and Genomics70 that discourages mutation analysis in individuals that have not manifested with symptoms of disease. Nonetheless, there are certain circumstances where clinical judgment should be applied in which a disease can be “silent” or “subtle” and apparently “unaffected” persons are affected but are asymptomatic (e.g. asymptomatic nephrocalcinosis or asymptomatic renal hypodysplasia).
Novel gene discoveries using whole exome sequencing (WES)
The “exome” describes the entirety of all exons-encoding sequences in the human genome. Although the exon represents only one percent of the human genome, it represents the protein-encoding sequences. WES offers also a powerful approach towards identification of novel monogenic causes of disease. Detailed description of the WES technique can be found elsewhere71,72. Briefly, genomic DNA is mechanically broken into random short fragments, which then are hybridized (bound by sequence matching) to oligonucleotides that represent all human exons. The unbound fragments are washed off (99% of the genome), and the exon-bound DNA fragments are eluted specifically and then loaded onto a next generation sequencer for whole exome sequencing. The millions of sequencing reads that are thus generated are aligned for comparison with a “normal reference sequence” of the human genome. Finally, WES data output file is generated containing all genetic variants from reference sequence found in the tested individual’s DNA. If a genetic sequence variant leads to a phenotypic change of an organism, for instance causes disease, that sequence variant is called a “mutation”. Other sequence variants are called “variance of unknown significance”.
The detection of mutations in novel disease-causing genes using WES can reveal new medical conditions that were not previously recognized. For instance, it was that mutations in genes regulating coenzyme Q10 biosynthesis may cause steroid resistant nephrotic syndrome (CQO2, COQ6, ADCK4, PDSS2)56–58. However, the utility of WES for novel gene discovery is hampered by the fact that a large number of genetic variants results when comparing the exome sequences of the studied individual to the normal genome reference sequence. On average in WES data of an individual there are between 2,000 to 4,000 non-synonymous variants. This limitation can be overcome by restricting sequence variant calling to smaller regions of interest that are generated for instance by homozygosity mapping or linkage analysis73, or by analyzing only shared variants across several affected individuals within the same family. These approaches enable one to exclude DNA variants from further consideration and allow an a priori restriction for the pool of potentially causal mutations. Finally, WES can result in identification of incidental findings - that are results not related to the indication for performing WES but may still be of medical importance to the patient. A policy statement with recommendations regarding the utility and reporting of incidental findings were published by the American College of Medical Genetics (ACMG).74 Considering to strictly consenting the patient for the purpose of only identifying the molecular cause of the kidney disease in question should minimize this problem.
Using WES, disease-causing genes may be detected that were not suspected from the patient’s clinical presentation. For instance, by combining homozygosity mapping with WES in 10 sibling pairs with renal cystic ciliopathies, we detected the causative gene in 7 out of the 10 families studied. In 5 families we identified mutations of known renal cystic ciliopathies genes, however, in 2 additional families we found mutations in other known CKD-causing genes, specifically SLC4A1 (a causative gene for distal renal tubular acidosis) and AGXT (the causative gene for hyperoxaluria type 1). Neither diagnosis had been made clinically and represented phenocopies for renal cystic ciliopathies4. Similar results regarding phenocopies have been described for other non-renal conditions75,76.
Exome sequencing reveals pathogenic pathways: The example of nephrotic syndrome
Nephrotic syndrome (NS) is a chronic kidney disease defined by proteinuria, that causes hypoalbuminemia, edema and hyperlipidemia. The condition is categorized by the patient’s clinical response to steroid therapy as “steroid-sensitive” (SSNS) vs. “steroid-resistant” (SRNS). SRNS is the second most frequent cause of CKD in children and young adults (Table 1). The disease mechanisms are poorly understood and no curative treatment is available. The most frequent renal histological feature of SRNS is focal-segmental glomerulosclerosis (FSGS), which carries a 33% risk of recurrence in a kidney transplant, thereby leading again to end-stage kidney disease53. For SRNS, the primary etiology and pathomechanisms have been obscure until recently. However, identification of genes that, if mutated, cause recessive or dominant monogenic forms of SRNS has dramatically changed this picture by providing the first fundamental insight into disease mechanisms of SRNS15–17,77. The discovery of novel SRNS genes has led to the understanding that the renal glomerular podocyte represents the cell type at which disease mechanisms of SRNS converge (Figure 2)16,78. At this juncture there are over 39 genes known to cause SRNS if mutated (Table 5). Those genes encode proteins that can currently be grouped into the following four major categories (Figure 2): (1) Proteins that are associated with the glomerular slit membrane, e.g. Nephrin (NPHS1)15, Podocin (NPHS2)79, and CD2-associated protein (CD2AP)80; (2) proteins that are involved in actin binding and regulation and hence affect the cytoskeleton of the podocyte, e.g. ACTN481, INF282, and ARHGDIA59; (3) proteins associated with focal adhessions that tether the sole of the podocyte to the underlying glomerular basement membrane, e.g. LAMB283 and EMP284, and (4) proteins involved in the biosynthesis of coenzyme Q10 (CoQ10), e.g. CoQ285, CoQ686 and ADCK487 (Figure 2).
Figure 2. Proteins involved in single-gene causes and pathogenic pathways of steroid resistant nephrotic syndrome.
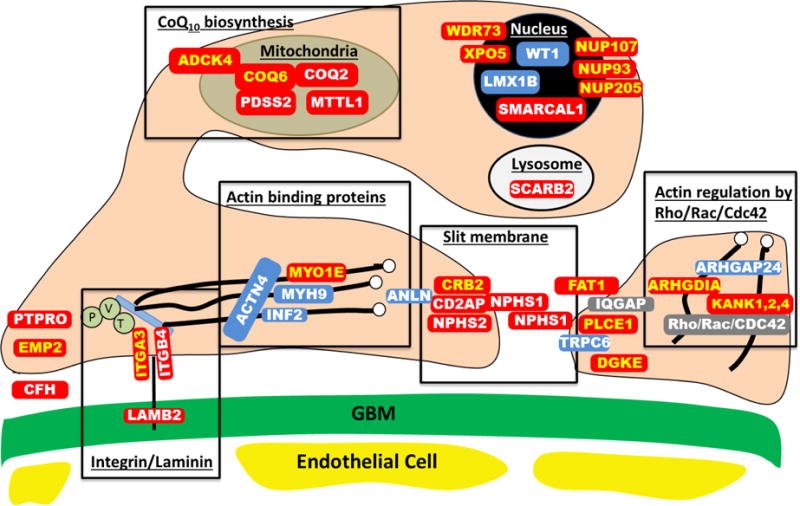
Identification of single-gene (monogenic) causes of steroid resistant nephrotic syndrome has revealed the renal glomerular epithelial cell, the podocyte, as the center of action in the pathogenesis of SRNS, because all of the related genes are highly expressed in podocytes. In this way identification of genes that, if mutated, cause SRNS revealed certain proteins and functional pathways as essential for glomerular function, because a mutation in any single one of them is sufficient to cause SRNS.
This figure depicts a simplified cross section through two neighboring podocyte foot processes, that attach to the glomerular basement membrane (GBM) via laminin-integrin receptors. Proteins that if mutated cause recessive monogenic forms of SRNS in red, and proteins that if mutated cause dominant forms of SRNS in blue. These SRNS-related proteins were found to be part of protein-protein interaction complexes that participate in defined structural components or signaling pathways of podocyte function (black frames). These proteins include: laminin/integrin receptors (focal adhessions), actin binding proteins, glomerular slit membrane-associated components, actin regulating small GTPases of the Rho/Rac/Cdc42 family, lyposomal proteins, nuclear transcription factors, and proteins involved in coenzyme Q10 biosynthesis. IQGAP, IQ motif containing GTPase activating protein 1; P, Paxillin; V, Vinculin and T, Talin.
Proteins that are encoded by recessive SRNS genes are marked in red:
ADCK4, AarF domain containing kinase 4; ARHGDIA, Rho GDP dissociation inhibitor (GDI) alpha; CD2AP, CD2-associated protein; CFH, Complement factor H; COQ2, coenzyme Q2 4-hydroxybenzoate polyprenyltransferase; COQ6, coenzyme Q6 monooxygenase 6; CRB2, Crumbs family member 2; DGKE, Diacylglycerol kinase, epsilon; EMP2, epithelial membrane protein 2; FAT1, FAT tumor suppressor homolog 1; GBM, glomerular basement membrane. ITGA3, integrin, alpha 3; ITGB4, integrin, beta 4; KANK, KN otif And Ankyrin Repeat Domains 1/2/4; LAMB2, laminin, β2; MTTL1, mitochondrial tRNA leucine 1; MYO1E, homo sapiens myosin 1e; NPHS1, nephrin; NPHS2, podocin; NUP93, Nucleoporin 93 kDa; NUP107, Nucleoporin 107 kDa; NUP205, Nucleoporin 205 kDA; PDSS2, prenyl (decaprenyl) diphosphate synthase, subunit 2; PLCE1, phospholipase C, epsilon 1; PTPRO, protein tyrosine phosphatase, receptor type, O; SCARB2, scavenger receptor class B, member 2; SMARCAL1, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1.
WDR73, WD repeat domain 73; XPO5, Exportin 5.
Proteins that encoded by dominant SRNS genes are marked in blue:
ACTN4, actinin, alpha 4; ANLN, anillin; ARHGAP24, Rho GTPase activating protein 24; INF2, inverted formin, FH2 and WH2 domain containing; LMX1B, LIM homeobox transcription factor 1-beta; MYH9, Myosin, heavy chain 9; TRPC6, transient receptor potential cation channel, subfamily C, member 6; WT1, Wilms tumor 1.
Consequences for therapy
Early CKD diagnosis should trigger clinicians to consider genetic analysis for their patients. Molecular analysis of early CKD-causing genes using experimental known genes panels are becoming increasingly avilable. Following identification of early CKD-causing mutations, the patient should be referred to a CLIA (Clinical Laboratory Improvement Amendments) certified clinical laboratory, as well as for genetic counselling. Optimally, the care for patients with monogenic CKD should be provided by a multidisciplinary team of nephrologist, urologists, and clinical geneticists.
Identification of the causative mutation of an individual with SRNS, for example, may have expiramnetal therapeutic consequences in some forms of SRNS, because patients harboring mutations in genes of the CoQ10 biosynthesis pathway can be treated with CoQ10 supplementation86,87. The discovery that CoQ10 treatment is beneficial for patients with SRNS due to mutations in the CoQ10 biosynthetic genes opened a window of opportunity for treatment with CoQ10 especially since CoQ10, is an innocuous food supplement with a high safety profile. It has been suggested that reactive oxygen species production and accumulation may play a role in the pathophysiology of many mitochondrial diseases and associated renal damage leading to nephrotic syndrome88. Treatment with CoQ10 supplementation for CoQ10 deficiency was first described for CoQ10 deficiency caused by CoQ2 recessive mutations56. Initially several case reports were published89,90 which showed improvement in the neurologic symptoms but failed to show any benefit on renal function, since advanced chronic renal failure had already developed. Subsequently, a case report study by Salviati et al56 suggested that early initiation of the treatment, immediately after the onset of renal symptoms, was beneficial in resolution of the proteinuria in a patient with nephrotic syndrome secondary to COQ2 mutations. This form of monogenic SRNS, for which further study is needed, provides one of the first examples how identification of monogenic causes of SRNS may revealed the possibility to treat this disease, for which currently no efficient treatment exists.
Identifying a monogenic caouse for disease provides a better disease categorization for clinical trials that study outcome of diseases. Nontheless, in addition to research and future implications, identifying a monogenic cause for patients with early CKD has already several imidiate clinical implications which include: 1) providing the patient and family with the definitive cause of their disease; 2) placing the clinical phenotype into context by gene specific stratification and delivery of personalized medicine; 3) allowing precise genetic counseling for family planning; 4) detection of previously unrecognized affected family members; 5) avoiding unnecessary diagnostic procedures, tests and treatments; 6) early detection and treatment initiation of asymptomatic (or subtle) extra renal manifestations; 7) providing guidance for monitoring of potential future complications and 8) guiding advanced medical management on a gene specific basis. Box 3 outlines specific examples of monogenic early CKD causes for each one of those implications.
Box 3. Clinical implications of genetic testing of early onset chronic kidney diseases.
|
| |
| • | Providing the patient and family with the definitive cause of their disease |
|
| |
| • | Placing the clinical phenotype into context by gene specific stratification and delivery of personalized medicine. This may have both immediate as well as future clinical implications. e.g. (1) it is increasingly recognized that heterozygous contiguous gene deletions in the 17q12 region (which includes the gene HNF1B) can result in congenital anomalies of the kidney and urinary tract (CAKUT) with a neurologic phenotype such as autism spectrum disorder or schizophrenia; (2) Future possible implications include allele specific drug treatments as it has been established for other genetic diseases like cystic fibrosis* |
|
| |
| • | Allows precise genetic counseling for family planning. e.g. (1) prediction of disease recurrence; (2) providing the option for preimplantation genetic diagnosis. |
|
| |
| • | Detection of previously unrecognized affected family members. e.g. (1) patients with dominantly inherited CAKUT can be asymptomatic in early disease stages. For instance, index patients with CAKUT secondary to PAX2 or GATA3 mutations may have affected parent/child or sibling with overlooked CAKUT which can only be detected by recognizing the genetic nature of the disease which can apparently present as “sporadic” case. This should trigger renal ultrasonographic screening for CAKUT in other family members; (2) identification of asymptomatic individuals harboring heterozygous COL4A4 or COL4A5 mutations, who should be monitored yearly for proteinuria and hypertension. Both of which may be the first sign of evolving chronic kidney disease. |
|
| |
| • | Avoiding unnecessary diagnostic procedures, tests and treatments. e.g. (1) avoiding renal biopsy. For instance, in patients with congenital or infantile nephrotic syndrome who has established genetic diagnosis secondary to NPHS1 or NPHS2 mutations or for patients with characteristic nephronophthisis phenotype and NPHP1 mutations; (2) avoiding aggressive anti- recurrence treatment for FSGS in kidney transplant patients with FSGS secondary to NPHS2 mutations. The latter has been shown to have low recurrence risk; (3) patients with CAKUT secondary to HNF1B mutations may have elevated liver function tests. Acknowledging this as part of the HNF1B-mutation related phenotype can prevent unnecessary invasive investigation (such as liver biopsy for “idiopathic elevated LFTs”). |
|
| |
| • | Early detection and treatment initiation of asymptomatic (or subtle) extra renal manifestations. e.g. (1) heterozygous mutations in HNF1B may cause “isolated CAKUT” or “syndromic CAKUT” that is associated with one or more of the following extra renal manifestations: maturity onset diabetes of the young (MODY type 5), hyperuricemia and hypomagnesaemia. Early identification of those conditions can lead to early monitoring and treatment; (2) similarly, deafness has been associated with three other CAKUT-causing mutations in EYA1, SALL1 or PAX2; (3) patients with CAKUT secondary to GATA3 mutations may have hypoparathyroidism which can be asymptomatic in early disease stages however should be recognized and treated. |
|
| |
| • | Providing guidance for monitoring of potential future complications. e.g. (1) patients with nephrotic syndrome secondary to WT1 are at increased risk for WT1-related Wilms tumor; (2) patients with WT1 mutations in the donor splice site of intron-9, resulting in the splice form +KTS are at risk for gonadoblastoma; (3) patients with nephronophthisis secondary to NPHP5 mutations are at risk for progressive blindness secondary to retinitis pigmentosa (i.e. Senior-Løken syndrome) |
|
| |
| • | Guiding advanced medical management on a gene specific basis. e.g. (1) recessive mutations in CTNS establish the diagnosis of cystinosis and should trigger treatment with cystine-depleting agents; (2) considering CoQ10 supplements for patients with nephrotic syndrome harboring mutations in genes of the CoQ10 biosynthesis pathway such as CoQ2, CoQ4 and ADCK4; (3) guiding thrombocytopenia management for patients with nephrotic syndrome secondary to MYH9 mutations. |
|
| |
| * Lumacaftor and ivacaftor therapy have been specifically shown to have clinical efficacy in patients who are homozygous for the Phe508del CFTR mutation211. | |
| “One author declares competing financial interests: F.H. receives royalties on a mutation analysis panel licensed to Claritas (Cambridge).” | |
In summary, mutation analysis by WES and indication-driven analysis of relevant gene panels can currently be recommended for all individuals who manifest with one of the following before age 25 years: CKD, steroid-resistant nephrotic syndrome, renal ultrasound showing increased echogenicity or 2 or more cysts, urinary stone disease, CAKUT, or chronic glomerulonephritis. The likelihood of identifying a causative monogenic mutation is estimated to be ~20% in this setting currently. It will rise in the future as more disease genes and causative mutations become known. Clinical consequences from these findings are currently emerging (Braun et al. cJASN, in revision). The literature on clinical consequences from identification of monogenic mutations will rapidly accumulate as genotype-phenotype correlations and relationships between genotype and clinical consequences will accrue over the next years.
Conclusions and future directions
Two thirds of early-onset CKD is due to CAKUT, SRNS, renal cystic ciliopathies or chronic glomerulonephritis. Recently, over 200 genes, that if mutated cause monogenic forms of these disorders have been identified.
High throughput exon sequencing using exon panels or WES now allows identification of the causative mutation in a high proportion (~20%) of individuals with early onset CKD (Table 2). Molecular genetic diagnostics can be planned in a well defined clinical indication-driven way for SRNS, cystic kidney diseases (presence of >/=2 cysts or increased echogenicity, presence of CAKUT, glomerulonephritis, or nephrolithiasis/nephrocalcinosis (history of at least one stone or nephrocalcinosis).
Indication-driven gene panel analysis with the use of next generation sequencing is an emerging tool, which will continue to be introduced into clinical research and practice75,76. Despite several challenges it is expected to be further implemented for clinical use in the near future. Novel gene identification will allow establishing an molecular genetic diagnosis, etiologic classification of disease for therapeutic trials and development of animal models of disease, as well as small molecule screening for therapeutic purposes.
Furthermore, the progress in high-throughput sequencing will ensure that additional CKD-causing genes will be detected in the near future. This may lead to more relevant etiologic categorization of disease entities than can be provided by ultrasound imaging or histopathology alone. Lastly, detection of monogenic causes of CKD already has implications for genetic consulting as well as for clinical management of patients with CKD (Box 3).
Key points.
~20% of CKD that manifest before 25 years of age are caused by single gene mutations in more than 200 different genes
Molecular genetic diagnostics can provide patients with molecular diagnosis, and can generate new insights into diseases mechanisms
Molecular genetic diagnostics may also have consequences for personalized treatment and prevention of CKD.
Indication driven mutation analysis panels are available for early-onset CKD e.g. CAKUT, SRNS, ciliopathies and nephrolithiasis (www.renalgenes.org).
Acknowledgments
F.H. is an Investigator of the Howard Hughes Medical Institute, a Doris Duke Distinguished Clinical Scientist, and the Warren E. Grupe Professor of Pediatrics. This research was supported by grants from the National Institutes of Health (to FH; R01-DK088767) and by the March of Dimes Foundation (6FY11-241). A.V. is a recipient of the Fulbright Post-doctoral Scholar Award for 2013. A.V. is also supported by grants from the Talpiot Medical Leadership Program, Chaim Sheba Medical Center, Tel-Hashomer, Israel and by the Manton Center Fellowship Program, Boston Children’s Hospital, Boston, MA.
GLOSSARY OF GENETIC TERMS
- Allele
Specific DNA sequence variant in a given gene. Alleles can be designated according to their frequency as common or rare alleles
- Exon
The protein coding part of a gene. Exons are spliced together following gene transcription to form messenger RNA, which is translated into protein
- Exome
The protein coding sequences of the entire genome (about 1% of the human genome)
- Expressivity
Variation of the expression of the phenotype among affected individuals with the same genotype. Variable expressivity refers to different degrees of severity and/or organ involvement in different affected individuals that carry identical mutation
- Genotype
The set of alleles (variants of genes) that structure an individual’s genetic makeup
- Homozygosity
The presence of identical alleles in the two copies of a gene or locus. The presence of different alleles is referred to as heterozygosity
- Homozygosity mapping
A technique in which the homozygous region across the genome are identified. This is an effective strategy for the discovery of autosomal recessive monogenic diseases genes in consanguineous families
- Next generation sequencing
This is a DNA sequencing method, also known as massively parallel sequencing, which allows to simultaneously sequence multiple DNA segments in a high-throughput manner
- Phenotype
The observable characteristics of an individual as a morphological, clinical or biochemical trait. A phenotype can also be the presence or absence of a disease
- Penetrance
The proportion of individuals that express a certain phenotype in relation to the number of individuals that carry the pathogenic variant(s). It can be age dependent. Incomplete penetrance refers to the observation that some individuals with the mutation do not develop the diseases phenotype at all
- Sanger sequencing (first generation sequencing)
DNA sequencing method (invented by Frederick Sanger) that involves termination of polymerized DNA strands at the position of specific labeled nucleotides
- Variant filtering
Variant filtering refers to the process of excluding variants between the individual examined and a “normal reference individual” from further consideration as disease causing. For instance, very common variants and variants which do not alter the protein sequence are excluded
- Variant
A difference in a DNA sequence as compared to normal reference sequence. A variant may be benign, i.e. single nucleotide polymorphism (SNP) or disease causing (i.e. mutation)
- Whole exome sequencing
Targeted capture and sequencing of the exome (exons of all genes) using next generation sequencing. This method offers a powerful approach towards identification monogenic disease causing genes
References
- 1.Inker LA, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2014;63:713–735. doi: 10.1053/j.ajkd.2014.01.416. [DOI] [PubMed] [Google Scholar]
- 2.Coresh J, et al. Prevalence of chronic kidney disease in the United States. Jama. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 3.Halbritter J, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. Journal of the American Society of Nephrology: JASN. 2015;26:543–551. doi: 10.1681/ASN.2014040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gee HY, et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney international. 2014;85:880–887. doi: 10.1038/ki.2013.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devuyst O, et al. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383:1844–1859. doi: 10.1016/S0140-6736(14)60659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014 doi: 10.1007/s00467-013-2684-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saisawat P, et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney international. 2014;85:1310–1317. doi: 10.1038/ki.2013.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebarasi L, et al. Defects of CRB2 Cause Steroid-Resistant Nephrotic Syndrome. American journal of human genetics. 2014;96:153–161. doi: 10.1016/j.ajhg.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovric S, et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clinical journal of the American Society of Nephrology: CJASN. 2014;9:1109–1116. doi: 10.2215/CJN.09010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohl S, et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology: JASN. 2014;25:1917–1922. doi: 10.1681/ASN.2013101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang DY, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney international. 2014;85:1429–1433. doi: 10.1038/ki.2013.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hildebrandt F. Genetic kidney diseases. Lancet. 2010;375:1287–1295. doi: 10.1016/S0140-6736(10)60236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Pediatric transplantation. 2007;11:366–373. doi: 10.1111/j.1399-3046.2007.00704.x. [DOI] [PubMed] [Google Scholar]
- 14.Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. 2012;27:363–373. doi: 10.1007/s00467-011-1939-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kestila M, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Molecular cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 16.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney international. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 17.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 18.Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatric nephrology. 2014;29:695–704. doi: 10.1007/s00467-013-2684-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivante A, et al. Mutations in TBX18 Cause Dominant Urinary Tract Malformations via Transcriptional Dysregulation of Ureter Development. American journal of human genetics. 2015;97:291–301. doi: 10.1016/j.ajhg.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trautmann A, et al. Spectrum of Steroid-Resistant and Congenital Nephrotic Syndrome in Children: The PodoNet Registry Cohort. Clinical journal of the American Society of Nephrology: CJASN. 2015;10:592–600. doi: 10.2215/CJN.06260614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaki M, et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney international. 2011;80:1239–1245. doi: 10.1038/ki.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palumbo P, et al. Variable phenotype in 17q12 microdeletions: clinical and molecular characterization of a new case. Gene. 2014;538:373–378. doi: 10.1016/j.gene.2014.01.050. [DOI] [PubMed] [Google Scholar]
- 25.Chen YZ, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chinese medical journal. 2010;123:3326–3333. [PubMed] [Google Scholar]
- 26.Hasselbacher K, et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney international. 2006;70:1008–1012. doi: 10.1038/sj.ki.5001679. [DOI] [PubMed] [Google Scholar]
- 27.Tory K, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. 2014;46:299–304. doi: 10.1038/ng.2898. [DOI] [PubMed] [Google Scholar]
- 28.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzur S, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human genetics. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freedman BI, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21:1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genovese G, et al. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney international. 2010;78:698–704. doi: 10.1038/ki.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dummer PD, et al. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Seminars in nephrology. 2015;35:222–236. doi: 10.1016/j.semnephrol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopp JB, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khanna H, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28:2946–2960. doi: 10.1093/ndt/gft253. [DOI] [PubMed] [Google Scholar]
- 37.MacArthur DG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bell CJ, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Science translational medicine. 2011;3:65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohl S, et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology: JASN. 2014;25:1917–1922. doi: 10.1681/ASN.2013101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanna-Cherchi S, et al. Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med. 2013;369:621–629. doi: 10.1056/NEJMoa1214479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. N Engl J Med. 2012;366:1119–1131. doi: 10.1056/NEJMra1108178. [DOI] [PubMed] [Google Scholar]
- 43.Sadowski CE, et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. Journal of the American Society of Nephrology: JASN. 2014 doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halbritter J, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. Journal of medical genetics. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 45.Halbritter J, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Human genetics. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:654–666. doi: 10.1056/NEJMra1312353. [DOI] [PubMed] [Google Scholar]
- 47.Halbritter J, et al. Fourteen Monogenic Genes Account for 15% of Nephrolithiasis/Nephrocalcinosis. Journal of the American Society of Nephrology: JASN. 2014 doi: 10.1681/ASN.2014040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halbritter J, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. Journal of medical genetics. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 49.Sadowski CE, et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. Journal of the American Society of Nephrology: JASN. 2014;26:1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halbritter J, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. Journal of medical genetics. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 51.Sanna-Cherchi S, et al. Copy-number disorders are a common cause of congenital kidney malformations. American journal of human genetics. 2012;91:987–997. doi: 10.1016/j.ajhg.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber S, et al. Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2011;26:136–143. doi: 10.1093/ndt/gfq400. [DOI] [PubMed] [Google Scholar]
- 53.Ruf RG, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15:722–732. doi: 10.1097/01.asn.0000113552.59155.72. [DOI] [PubMed] [Google Scholar]
- 54.Weber S, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney international. 2004;66:571–579. doi: 10.1111/j.1523-1755.2004.00776.x. [DOI] [PubMed] [Google Scholar]
- 55.Chernin G, et al. Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clinical journal of the American Society of Nephrology: CJASN. 2010;5:1655–1662. doi: 10.2215/CJN.09351209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358:2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 57.Heeringa SF, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. The Journal of clinical investigation. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of clinical investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gee HY, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. The Journal of clinical investigation. 2013;123:3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gee HY, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. The Journal of clinical investigation. 2015;125:2375–2384. doi: 10.1172/JCI79504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nature medicine. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hinkes B, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 63.Moriniere V, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. 2014;25:2740–2751. doi: 10.1681/ASN.2013080912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nature reviews. Nephrology. 2010;6:197–206. doi: 10.1038/nrneph.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearle MS, et al. Medical management of kidney stones: AUA guideline. The Journal of urology. 2014;192:316–324. doi: 10.1016/j.juro.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 66.Otto EA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nature genetics. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giglio S, et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. 2015;26:230–236. doi: 10.1681/ASN.2013111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boyer O, et al. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. Journal of medical genetics. 2010;47:445–452. doi: 10.1136/jmg.2009.076166. [DOI] [PubMed] [Google Scholar]
- 69.Lipska BS, et al. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney international. 2013;84:206–213. doi: 10.1038/ki.2013.93. [DOI] [PubMed] [Google Scholar]
- 70.Ross LF, et al. Technical report: Ethical and policy issues in genetic testing and screening of children. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15:234–245. doi: 10.1038/gim.2012.176. [DOI] [PubMed] [Google Scholar]
- 71.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in genetics: TIG. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 72.Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155:27–38. doi: 10.1016/j.cell.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hildebrandt F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS genetics. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Green RC, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee H, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama. 2014;312:1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nature genetics. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 78.Shibazaki S, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nature genetics. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 80.Lowik MM, et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney international. 2007;72:1198–1203. doi: 10.1038/sj.ki.5002469. [DOI] [PubMed] [Google Scholar]
- 81.Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 82.Brown EJ, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42:72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zenker M, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 84.Gee HY, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. American journal of human genetics. 2014;94:884–890. doi: 10.1016/j.ajhg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diomedi-Camassei F, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 86.Heeringa SF, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. The Journal of clinical investigation. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of clinical investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ozaltin F. Primary coenzyme Q10 (CoQ 10) deficiencies and related nephropathies. Pediatr Nephrol. 2014;29:961–969. doi: 10.1007/s00467-013-2482-z. [DOI] [PubMed] [Google Scholar]
- 89.Salviati L, et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65:606–608. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 90.Rotig A, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 91.Saisawat P, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney international. 2012;81:196–200. doi: 10.1038/ki.2011.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trautmann A, et al. Spectrum of Steroid-Resistant and Congenital Nephrotic Syndrome in Children: The PodoNet Registry Cohort. Clinical journal of the American Society of Nephrology: CJASN. 2015 doi: 10.2215/CJN.06260614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. Centrosomes and cilia in human disease. Trends in genetics: TIG. 2011;27:307–315. doi: 10.1016/j.tig.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lemaire M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531–536. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neumann HP, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. Journal of medical genetics. 2003;40:676–681. doi: 10.1136/jmg.40.9.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Noris M, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clinical journal of the American Society of Nephrology: CJASN. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Westra D, et al. Atypical hemolytic uremic syndrome and genetic aberrations in the complement factor H-related 5 gene. Journal of human genetics. 2012;57:459–464. doi: 10.1038/jhg.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goldfarb DS. The search for monogenic causes of kidney stones. J Am Soc Nephrol. 2015;26:507–510. doi: 10.1681/ASN.2014090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hamiwka LA, Midgley JP, Wade AW, Martz KL, Grisaru S. Outcomes of kidney transplantation in children with nephronophthisis: an analysis of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Registry. Pediatric transplantation. 2008;12:878–882. doi: 10.1111/j.1399-3046.2008.00942.x. [DOI] [PubMed] [Google Scholar]
- 101.Sanna-Cherchi S, et al. Copy-number disorders are a common cause of congenital kidney malformations. American journal of human genetics. 2012;91:987–997. doi: 10.1016/j.ajhg.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weber S, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol. 2008;19:891–903. doi: 10.1681/ASN.2006111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brockschmidt A, et al. CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT) Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27:2355–2364. doi: 10.1093/ndt/gfr649. [DOI] [PubMed] [Google Scholar]
- 104.Abdelhak S, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 105.Pandolfi PP, et al. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995;11:40–44. doi: 10.1038/ng0995-40. [DOI] [PubMed] [Google Scholar]
- 106.Van Esch H, et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422. doi: 10.1038/35019088. [DOI] [PubMed] [Google Scholar]
- 107.Lindner TH, et al. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–2008. doi: 10.1093/hmg/8.11.2001. [DOI] [PubMed] [Google Scholar]
- 108.Kirby A, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45:299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995;4:2183–2184. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- 110.Allison SJ. Basic research: Ret signaling reveals insights into the pathogenesis of CAKUT. Nature reviews. Nephrology. 2012;8:432. doi: 10.1038/nrneph.2012.119. [DOI] [PubMed] [Google Scholar]
- 111.Lu W, et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. American journal of human genetics. 2007;80:616–632. doi: 10.1086/512735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet. 1998;18:81–83. doi: 10.1038/ng0198-81. [DOI] [PubMed] [Google Scholar]
- 113.Ruf RG, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101:8090–8095. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoskins BE, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. American journal of human genetics. 2007;80:800–804. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gimelli S, et al. Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Human mutation. 2010;31:1352–1359. doi: 10.1002/humu.21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hwang DY, et al. Mutations of the SLIT2-ROBO2 pathway genes SLIT2 and SRGAP1 confer risk for congenital anomalies of the kidney and urinary tract. Human genetics. 2015;134:905–916. doi: 10.1007/s00439-015-1570-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gbadegesin RA, et al. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–1322. doi: 10.1681/ASN.2012121148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hart TC, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. Journal of medical genetics. 2002;39:882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jenkins D, et al. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J Am Soc Nephrol. 2005;16:2141–2149. doi: 10.1681/ASN.2004090776. [DOI] [PubMed] [Google Scholar]
- 120.Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N Engl J Med. 2004;351:792–798. doi: 10.1056/NEJMoa040533. [DOI] [PubMed] [Google Scholar]
- 121.Mandel H, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. American journal of human genetics. 2008;82:39–47. doi: 10.1016/j.ajhg.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vivante A, et al. Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol. 2013;24:550–558. doi: 10.1681/ASN.2012010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gribouval O, et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet. 2005;37:964–968. doi: 10.1038/ng1623. [DOI] [PubMed] [Google Scholar]
- 124.Weber S, et al. Muscarinic Acetylcholine Receptor M3 Mutation Causes Urinary Bladder Disease and a Prune-Belly-like Syndrome. American journal of human genetics. 2011;89:668–674. doi: 10.1016/j.ajhg.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barak H, et al. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Developmental cell. 2012;22:1191–1207. doi: 10.1016/j.devcel.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McGregor L, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003;34:203–208. doi: 10.1038/ng1142. [DOI] [PubMed] [Google Scholar]
- 127.Bulum B, et al. HPSE2 Mutations in Urofacial Syndrome, Non-Neurogenic Neurogenic Bladder and Lower Urinary Tract Dysfunction. Nephron. 2015;130:54–58. doi: 10.1159/000381465. [DOI] [PubMed] [Google Scholar]
- 128.Humbert C, et al. Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. American journal of human genetics. 2014;94:288–294. doi: 10.1016/j.ajhg.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stuart HM, et al. LRIG2 mutations cause urofacial syndrome. American journal of human genetics. 2013;92:259–264. doi: 10.1016/j.ajhg.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hardelin JP, et al. X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc Natl Acad Sci U S A. 1992;89:8190–8194. doi: 10.1073/pnas.89.17.8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shih NY, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286:312–315. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 132.Sethi S, Fervenza FC, Zhang Y, Smith RJ. Secondary focal and segmental glomerulosclerosis associated with single-nucleotide polymorphisms in the genes encoding complement factor H and C3. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2012;60:316–321. doi: 10.1053/j.ajkd.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ovunc B, et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J Am Soc Nephrol. 2011;22:1815–1820. doi: 10.1681/ASN.2011040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ozaltin F, et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol. 2013;24:377–384. doi: 10.1681/ASN.2012090903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Has C, et al. Integrin alpha3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012;366:1508–1514. doi: 10.1056/NEJMoa1110813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kambham N, et al. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2000;36:190–196. doi: 10.1053/ajkd.2000.8293. [DOI] [PubMed] [Google Scholar]
- 137.Gee HY, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. The Journal of clinical investigation. 2015;125:2375–2384. doi: 10.1172/JCI79504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mele C, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miyake N, et al. Biallelic Mutations in Nuclear Pore Complex Subunit NUP107 Cause Early-Childhood-Onset Steroid-Resistant Nephrotic Syndrome. American journal of human genetics. 2015;97:555–566. doi: 10.1016/j.ajhg.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lopez LC, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. American journal of human genetics. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ozaltin F, et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. American journal of human genetics. 2011;89:139–147. doi: 10.1016/j.ajhg.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berkovic SF, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. American journal of human genetics. 2008;82:673–684. doi: 10.1016/j.ajhg.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boerkoel CF, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 144.Colin E, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. American journal of human genetics. 2014;95:637–648. doi: 10.1016/j.ajhg.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vodopiutz J, et al. WDR73 mutations cause infantile neurodegeneration and variable glomerular kidney disease. Human mutation. 2015 doi: 10.1002/humu.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jinks RN, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain: a journal of neurology. 2015 doi: 10.1093/brain/awv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gbadegesin RA, et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol. 2014;25:1991–2002. doi: 10.1681/ASN.2013090976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Akilesh S, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. The Journal of clinical investigation. 2011;121:4127–4137. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Philippe A, et al. A missense mutation in podocin leads to early and severe renal disease in mice. Kidney international. 2008;73:1038–1047. doi: 10.1038/ki.2008.27. [DOI] [PubMed] [Google Scholar]
- 150.Heath KE, et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. American journal of human genetics. 2001;69:1033–1045. doi: 10.1086/324267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Winn MP, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 152.Reiser J, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jeanpierre C, et al. Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. American journal of human genetics. 1998;62:824–833. doi: 10.1086/301806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mochizuki T, et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet. 1994;8:77–81. doi: 10.1038/ng0994-77. [DOI] [PubMed] [Google Scholar]
- 155.Fremeaux-Bacchi V, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. Journal of medical genetics. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McRae JL, et al. Human factor H-related protein 5 (FHR-5). A new complement-associated protein. The Journal of biological chemistry. 2001;276:6747–6754. doi: 10.1074/jbc.M007495200. [DOI] [PubMed] [Google Scholar]
- 157.Gale DP, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Athanasiou Y, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clinical journal of the American Society of Nephrology: CJASN. 2011;6:1436–1446. doi: 10.2215/CJN.09541010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Castelletti F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A. 2008;105:2538–2543. doi: 10.1073/pnas.0707730105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Noris M, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362:1542–1547. doi: 10.1016/S0140-6736(03)14742-3. [DOI] [PubMed] [Google Scholar]
- 161.Knebelmann B, et al. Spectrum of mutations in the COL4A5 collagen gene in X-linked Alport syndrome. American journal of human genetics. 1996;59:1221–1232. [PMC free article] [PubMed] [Google Scholar]
- 162.Anker MC, et al. Alport syndrome with diffuse leiomyomatosis. American journal of medical genetics. Part A. 2003;119A:381–385. doi: 10.1002/ajmg.a.20019. [DOI] [PubMed] [Google Scholar]
- 163.Marcocci E, et al. Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24:1464–1471. doi: 10.1093/ndt/gfn681. [DOI] [PubMed] [Google Scholar]
- 164.Hildebrandt F, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 165.Saunier S, et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet. 1997;6:2317–2323. doi: 10.1093/hmg/6.13.2317. [DOI] [PubMed] [Google Scholar]
- 166.Otto EA, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Olbrich H, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 168.Otto E, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. American journal of human genetics. 2002;71:1167–1171. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Delous M, et al. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet. 2009;18:4711–4723. doi: 10.1093/hmg/ddp434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Otto EA, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 171.Sayer JA, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 172.Attanasio M, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–1024. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 173.Delous M, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 174.Otto EA, et al. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Otto EA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nature genetics. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Otto EA, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) Journal of medical genetics. 2009;46:663–670. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- 177.Davis EE, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Coussa RG, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clinical genetics. 2013;84:150–159. doi: 10.1111/cge.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chaki M, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hoff S, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet. 2013;45:951–956. doi: 10.1038/ng.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Reed BY, et al. Identification and characterization of a gene with base substitutions associated with the absorptive hypercalciuria phenotype and low spinal bone density. The Journal of clinical endocrinology and metabolism. 2002;87:1476–1485. doi: 10.1210/jcem.87.4.8300. [DOI] [PubMed] [Google Scholar]
- 182.Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Natl Acad Sci U S A. 1991;88:10900–10904. doi: 10.1073/pnas.88.23.10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hidaka Y, Palella TD, O’Toole TE, Tarle SA, Kelley WN. Human adenine phosphoribosyltransferase. Identification of allelic mutations at the nucleotide level as a cause of complete deficiency of the enzyme. The Journal of clinical investigation. 1987;80:1409–1415. doi: 10.1172/JCI113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stover EH, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. Journal of medical genetics. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Karet FE, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 186.Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His––Tyr): complete structure of the normal human CA II gene. American journal of human genetics. 1991;49:1082–1090. [PMC free article] [PubMed] [Google Scholar]
- 187.Pearce SH, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335:1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- 188.Lloyd SE, et al. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 189.Simon DB, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 190.Simon DB, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 191.Konrad M, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. American journal of human genetics. 2006;79:949–957. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schlingmann KP, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365:410–421. doi: 10.1056/NEJMoa1103864. [DOI] [PubMed] [Google Scholar]
- 193.Jaureguiberry G, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron. Physiology. 2012;122:1–6. doi: 10.1159/000349989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999;8:2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 195.Hamilton AJ, et al. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a beta cell phenotype. Journal of medical genetics. 2014;51:165–169. doi: 10.1136/jmedgenet-2013-102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Belostotsky R, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. American journal of human genetics. 2010;87:392–399. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Davidson BL, et al. Identification of 17 independent mutations responsible for human hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. American journal of human genetics. 1991;48:951–958. [PMC free article] [PubMed] [Google Scholar]
- 198.Simon DB, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 199.Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Tightly linked flanking markers for the Lowe oculocerebrorenal syndrome, with application to carrier assessment. American journal of human genetics. 1988;42:748–755. [PMC free article] [PubMed] [Google Scholar]
- 200.Simon DB, et al. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13:183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 201.Enomoto A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 202.Matsuo H, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. American journal of human genetics. 2008;83:744–751. doi: 10.1016/j.ajhg.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Prie D, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med. 2002;347:983–991. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 204.Lorenz-Depiereux B, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. American journal of human genetics. 2006;78:193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Calonge MJ, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 1994;6:420–425. doi: 10.1038/ng0494-420. [DOI] [PubMed] [Google Scholar]
- 206.Bruce LJ, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. The Journal of clinical investigation. 1997;100:1693–1707. doi: 10.1172/JCI119694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Feliubadalo L, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet. 1999;23:52–57. doi: 10.1038/12652. [DOI] [PubMed] [Google Scholar]
- 208.Karim Z, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008;359:1128–1135. doi: 10.1056/NEJMoa0802836. [DOI] [PubMed] [Google Scholar]
- 209.Scott P, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol. 1999;10:1007–1013. doi: 10.1681/ASN.V1051007. [DOI] [PubMed] [Google Scholar]
- 210.Ichida K, et al. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. The Journal of clinical investigation. 1997;99:2391–2397. doi: 10.1172/JCI119421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wainwright CE, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]