

ABSTRACT
Vibrio cholerae is the causative bacteria of the diarrheal disease cholera, but it also persists in aquatic environments, where it displays an expression profile that is distinct from that during infection. Upon entry into the host, a tightly regulated circuit coordinates the induction of two major virulence factors: cholera toxin and a toxin-coregulated pilus (TCP). It has been shown that a set of bile salts, including taurocholate, serve as host signals to activate V. cholerae virulence through inducing the activity of the transmembrane virulence regulator TcpP. In this study, we investigated the role of calcium, an abundant mental ion in the gut, in the regulation of virulence. We show that whereas Ca2+ alone does not affect virulence, Ca2+ enhances bile salt-dependent virulence activation for V. cholerae. The induction of TCP by murine intestinal contents is counteracted when Ca2+ is depleted by the high-affinity calcium chelator EGTA, suggesting that the calcium present in the gut is a relevant signal for V. cholerae virulence induction in vivo. We further show that Ca2+ enhances virulence by promoting bile salt-induced TcpP-TcpP interaction. Moreover, fluorescence recovery after photobleaching (FRAP) analysis demonstrated that exposure to bile salts and Ca2+ together decreases the recovery rate for fluorescently labeled TcpP, but not for another inner membrane protein (TatA). Together, these data support a model in which physiological levels of Ca2+ may result in altered bile salt-induced TcpP protein movement and activity, ultimately leading to an increased expression of virulence.
KEYWORDS: calcium, bile salts, virulence gene expression, TcpP, dimerization, Vibrio cholerae, virulence regulation
INTRODUCTION
Vibrio cholerae is the etiologic agent of the diarrheal disease cholera. This bacterium typically lives in brackish environments. Indeed, there are many strains of V. cholerae that are not considered pathogenic. Typically, only strains possessing two major virulence factors are considered to cause epidemic disease (1). The first of these factors is cholera toxin (CT), which is sufficient to promote the hallmark “rice water stool” associated with cholera. The second is the toxin-coregulated pilus (TCP), which is essential for colonization (2). These two virulence factors, among a range of other virulence associated accessory proteins, are tightly regulated in response to environmental conditions and signals (3). In the aquatic environment, V. cholerae is often associated with chitinous surfaces in a biofilm mode of growth, which can be important for initial infection (4–6).
When V. cholerae is ingested by the host through contaminated food or water, the bacteria will encounter drastic changes in environmental composition and signaling molecules. Oxygen tension decreases and passage through the stomach exposes the bacteria to acidic conditions. In the intestines the bacteria encounter bile, bicarbonate, and an array of other immune defenses, including mucin, antimicrobial peptides, and antibodies (7–9). To achieve the diarrheal disease cholera, V. cholerae coordinates the regulation of the major virulence factors CT and TCP (3). The genes encoding proteins that make up the toxin (ctxAB) and pilus (tcpA-F) are directly activated by ToxT (10). The expression of ToxT is achieved when ToxR and TcpP regulators, in conjunction with ToxS and TcpH, respectively, bind upstream of the toxT coding sequence (11). An additional level of regulation occurs at the point of TcpP induction. AphA, AphB, and OhrR transcription factors work together to activate TcpP in response to environmental stimuli, including low-oxygen tension (12–15). AphA also links quorum sensing and virulence regulation in V. cholerae (16, 17).
A number of environmental conditions, such as oxygen concentration and bicarbonate, have been shown to influence V. cholerae virulence gene expression (15, 18). In addition, bile, which is released into the proximal small intestine, has several components that are known to affect virulence in V. cholerae. Bile salts (BS), such as taurocholate and glycocholate, stimulate virulence factor production, whereas fatty acids inhibit ToxT activity by directly binding to the protein (19–21). Since mammalian gastrointestinal tracts are complex environments, we explore here whether there are additional host-produced compounds that modulate virulence gene expression. We examined calcium, which is part of the biliary secretion released into the proximal intestine following a meal and is abundant in the intestine (22). The importance of calcium signaling and regulation has been well defined in eukaryotic cells (23, 24). More recently, bacterial responses to and regulation of calcium homeostasis have been investigated. Calcium regulates processes that include cell division, biofilm formation, and pathogenesis (25–27). In the gut, calcium is present as free Ca2+ but can also be bound to bicarbonate and to bile salts (22, 28). We found that calcium enhances bile salt-induced virulence gene expression through modulating TcpP protein movement and activity. Our study adds to the growing body of work suggesting that calcium signaling is an important part of bacterial physiology as it is for eukaryotic cells. The model we propose further sheds light on the ways in which pathogens can co-opt host-resident signals for more efficient colonization.
RESULTS
Calcium enhances bile salt-induced virulence.
To examine the possible effect of calcium on V. cholerae virulence gene expression, we measured the expression of tcpA, which encodes the major virulence determinant (29), in the presence of calcium and a known activator, taurocholate (TC). We found that calcium alone did not induce tcpA (Fig. 1A). When calcium was supplied with TC, we observed an increase in TcpA expression greater than that of TC without added calcium (Fig. 1A). Other ions, including K+ and Mg2+, had no effect on virulence expression with or without TC (Fig. 1A), suggesting that this enhanced virulence induction is not due to a general salt or ion effect but is specific to calcium. We then tested whether calcium-enhanced virulence is specific for TC. We have previously shown that the bile salts TC and glycocholate (GC), but not deoxycholate (DC), promote virulence induction (21). We found that calcium promoted tcpA induction only for known inducers TC and GC in a dose-dependent manner (Fig. 1B). These data suggest that calcium acts together with the bile salts to promote the activation of virulence for V. cholerae. To confirm that calcium and TC also affect virulence factor production, we performed a Western blot analysis of TCP. Calcium greatly enhanced TCP expression in the presence of TC but had no effect alone (Fig. 1C).
FIG 1.
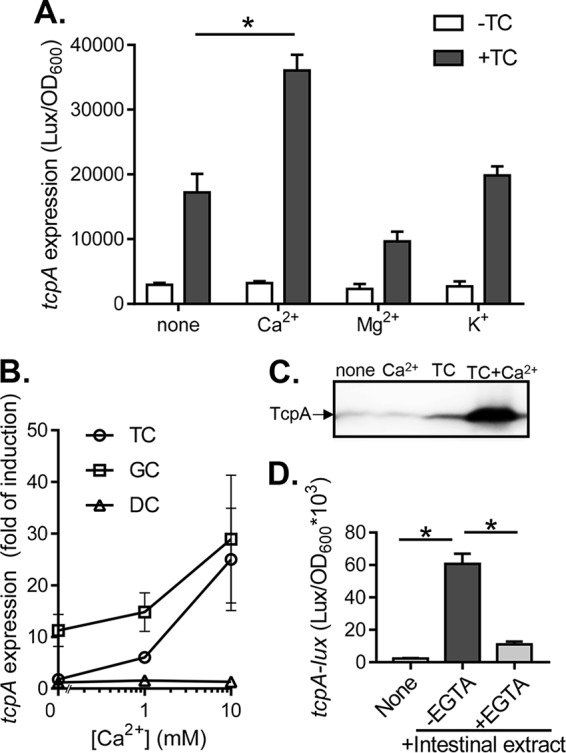
Contribution of Ca2+ to virulence gene expression in V. cholerae. (A) tcpA expression in the presence of taurocholate (TC) and different mental ions. V. cholerae containing PtcpA-luxCDABE plasmids were grown in LB medium containing 10 mM ion without or with 0.1 mM TC and grown microaerobically until reaching mid-log phase (OD600 ∼0.2). The luminescence was measured and normalized for growth against the OD600. (B) Bile salt effects. The tcpA expression was measured for cultures grown in 0.1 mM TC, 0.1 mM glycocholate (GC), and 0.1 mM deoxycholate (DC) in the presence of indicated calcium concentration. (C) TcpA production. Wild-type strains were grown in the absence or in the presence of 0.1 mM TC and 10 mM CaCl2 microaerobically at 37°C until reaching mid-log phase. The lysed cells were normalized by protein concentration (200 μg/sample) and subjected to SDS-PAGE and immunoblotting with anti-TcpA antiserum. (D) Intestinal extract. tcpA expression was measured without intestinal extract, with intestinal extract, or with intestinal extract previously incubated with EGTA. Intestinal extracts were incubated for 12 h with or without EGTA and added to cultures at a final EGTA concentration of 0.25 mM. The data shown are from three independent experiments. *, P < 0.05 (Student t test).
To explore whether calcium-enhanced virulence gene expression is relevant in the gut environment, we tested virulence activation of intestinal extracts that were treated with the high-affinity calcium chelator EGTA. V. cholerae incubated with intestinal extracts expressed high levels of tcpA (Fig. 1D). When cultures were incubated with EGTA-treated intestinal extracts, virulence expression declined drastically (Fig. 1D). This decrease in virulence induction was not due to the EGTA effects on bacterial growth since the amount of EGTA added in the intestinal extracts did not affect V. cholerae growth (data not shown). Together, these results imply that intestinally derived calcium contributes to virulence gene expression in vitro and suggest that calcium may affect V. cholerae virulence expression in vivo.
Ca2+ promotes TcpP induction of ToxT.
Multiple regulatory networks converge to achieve virulence in V. cholerae. The genes and proteins involved in this process are well-characterized and are known to act in a highly regulated manner (3) (Fig. 2A). To understand how calcium can promote virulence gene expression in V. cholerae, we examined whether calcium affects the expression of the known virulence regulators. When incubated with calcium alone or with calcium plus TC, the expression of aphB, aphA, tcpP, and toxR was unaffected, as measured by using promoter-luciferase transcriptional fusion reporters (Fig. 2B). Calcium alone also did not alter the expression of toxT (Fig. 2B), encoding the master virulence regulator (10). The addition of TC induced toxT, a finding consistent with previous reports (21). Importantly, the addition of both calcium and TC further enhanced the expression of toxT (Fig. 2B), suggesting that calcium-promoted virulence induction acts upstream of ToxT. Since toxT is regulated by TcpP and ToxR, it is possible that calcium affects the activity of either or both TcpP and ToxR. Alternatively, calcium may posttranslationally act on the ToxT protein. To test these hypotheses, we first examined whether calcium affects ToxT activity independent of expression. When we used a ΔtoxT mutant strain with a ToxT expression vector (pBAD-toxT) to control for the level of ToxT protein, we found that calcium, TC, or both did not affect tcpA induction by ToxT (see Fig. S1 in the supplemental material), indicating that calcium does not affect ToxT activity. We then tested whether calcium affects ToxR or TcpP activity in Escherichia coli. We found that calcium alone, TC, and TC+Ca2+ did not alter ToxR-induced ctxA expression (Fig. 2C), whereas the addition of calcium and TC enhanced TcpP-dependent toxT expression in E. coli (Fig. 2D). These data suggest that calcium may modulate TcpP activity to enhance bile salt-activated virulence gene expression.
FIG 2.
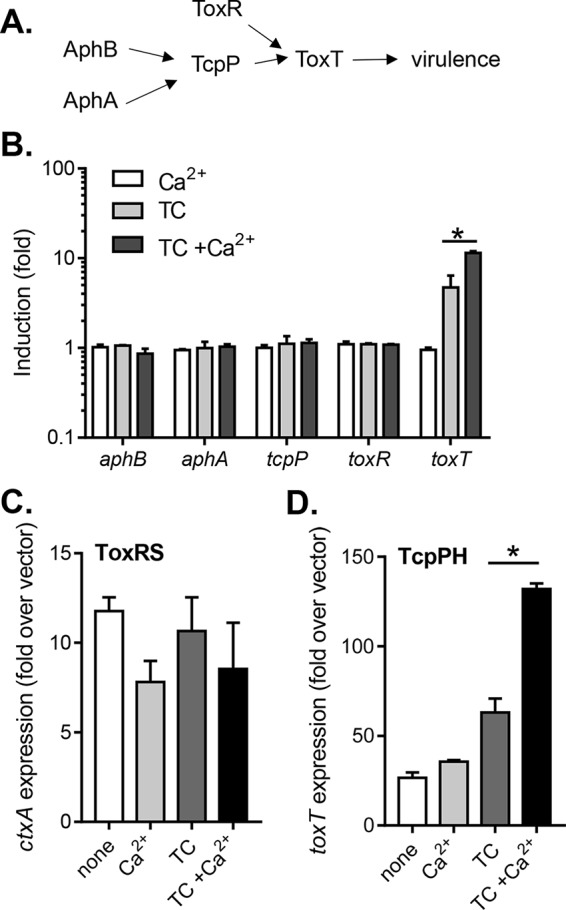
Effect of Ca2+ on the expression of virulence regulatory genes. (A) Schematic of virulence regulation in V. cholerae. AphA and AphB promote expression of TcpPH, which acts coordinately with ToxRS to induce ToxT and activate virulence. (B) Expression of toxT, toxR, aphB, aphA, and tcpP genes. Wild type containing the indicated promoter-lux reporters was grown at 37°C microaerobically until reaching mid-log phase, and the luminescence was measured. When indicated, 10 mM CaCl2 or 0.1 mM TC was included in the medium. (C and D) ToxR and TcpP. E. coli containing pBAD-toxRS or pBAD-tcpPH when exposed to TC, CaCl2, or both together. (C) Induction of ctxA by ToxR (pBAD-toxRS) in E. coli when exposed to TC, CaCl2, or both together. (D) Induction of toxT by TcpP (pBAD-tcpPH) in E. coli when exposed to TC, CaCl2, or both together. The data shown are from three independent experiments. *, P < 0.05 (Student t test).
Ca2+ promotes TC-dependent TcpP interaction and activity.
To confirm that calcium affects the activity of TcpP to induce virulence, we constitutively expressed tcpPH in a ΔtcpPH ΔtoxR mutant strain and measured tcpA expression with calcium and/or EGTA. Calcium alone did not alter tcpA expression; however, the same concentration of calcium enhanced tcpA expression in the presence of TC (Fig. 3A). To verify that this effect was due to calcium, we added different amounts of EGTA in the medium. We found that EGTA could eliminate the enhanced virulence expression (Fig. 3A). Taken together, these data suggest that calcium affects virulence at the level of TcpP in V. cholerae. To further dissect the possible role of calcium on TcpP activity, we examined TcpP cysteine mutants. Previously, we discovered that two cysteine residues in the periplasmic domain of TcpP are critical for TcpP activity (21). To test whether calcium may affect the activity of TcpP cysteine mutants, we compared the effects of calcium on induction of tcpA by TcpPWT, TcpPC207S, and TcpP218S. We found that unlike the wild type, calcium displayed little effects on the activity of TcpPC207S (null activity) and TcpP218S (constitutive activity) (Fig. 3B). Since these two cysteine residues are involved in TcpP-TcpP intermolecular disulfide bond formation (21), these data suggest that calcium may modulate TcpP interaction.
FIG 3.
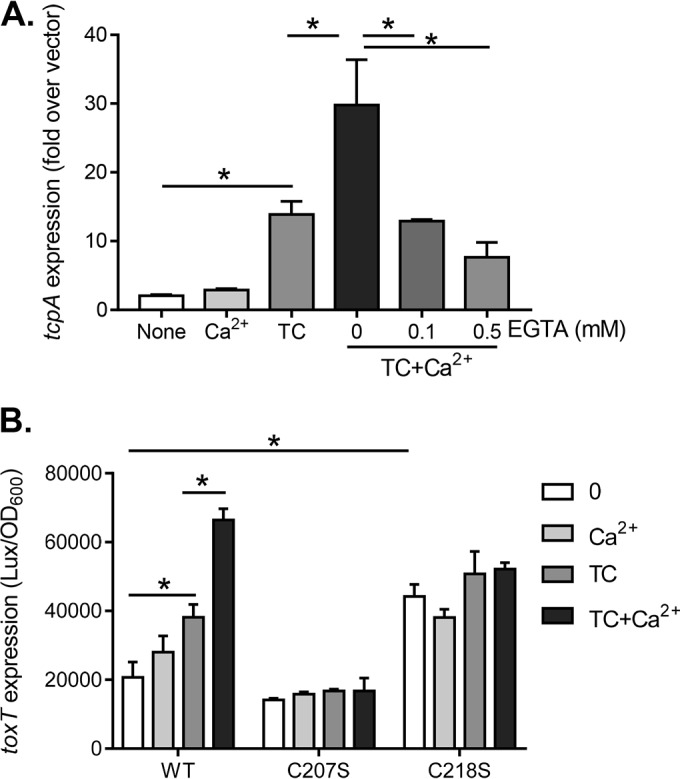
Calcium effects on TcpP induction of virulence. (A) Effect of Ca2+ on TcpP-mediated tcpA expression. The expression of tcpA with controlled levels of TcpP (ΔtcpPH; ΔtoxR with pBAD-tcpPH) when exposed to 0.1 mM TC, 10 mM CaCl2, or both together in the presence or absence of the calcium-specific chelator, EGTA, at the final concentrations indicated was assessed. (B) Activation of ToxT by TcpP and its cysteine mutant derivatives. The expression of toxT by wild-type (WT) TcpP and mutants with TcpPC207S (null activity) or TcpP218S (constitutive activity) was evaluated in the presence of 0.1 mM TC, 10 mM CaCl2, or both together. Cells were grown at 37°C microaerobically until reaching mid-log phase, and the luminescence was measured. The data shown are from three independent experiments. *, P < 0.05 (Student t test).
To further investigate how calcium affects TcpP activity, we examined whether calcium alters TcpP interaction using a bacterial two-hybrid system (30). We found that whereas calcium alone had little effect, in the presence of TC, calcium greatly enhanced TcpP-TcpP interaction (Fig. 4A). To ensure that this calcium effect does not represent a general effect on any membrane-bound protein, we measured ToxR-ToxR interaction as a control. We found that ToxR-ToxR interaction was unaffected by calcium under all the conditions tested (Fig. 4B). These data suggest that calcium may act to enhance virulence activation in the presence of TC by increasing Tcp-TcpP interaction, which may lead to increased induction of downstream virulence factors.
FIG 4.
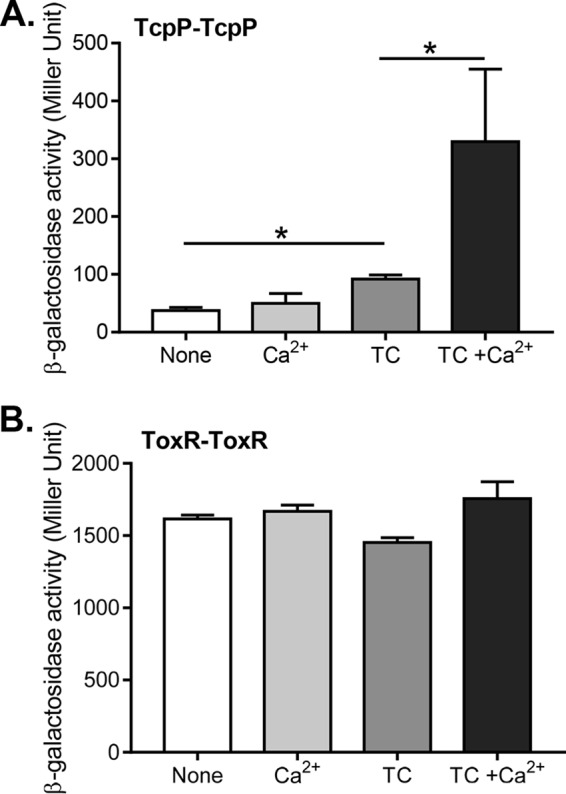
Effect of Ca2+ and TC on TcpP-TcpP interaction. E. coli cells harboring the adenylate cyclase two-hybrid constructs for TcpP or ToxR were grown microaerobically until reaching mid-log phase and then assessed for β-galactosidase activity, reported in Miller Units. (A) TcpP interaction in the presence of 1 mM TC, 10 mM CaCl2, or both together. (B) ToxR interaction in the presence of 1 mM TC, 10 mM CaCl2, or both together. Data shown are from three independent experiments. *, P < 0.05 (Student t test).
Calcium affects TcpP membrane diffusion.
We next sought to elucidate the mechanism by which calcium and TC affect TcpP activity. We first examined whether calcium may enhance bacterial uptake of bile salts, thereby increasing TcpP activity. We incubated 14C-labeled TC with wild-type V. cholerae in the absence or presence of calcium and measured the amount of 14C-labeled TC in cells after 30 min. We did not observe any difference of TC uptake between cultures with and without calcium (see Fig. S2 in the supplemental material). To avoid the possible effect of effluxing of bile salts by efflux pumps, we performed the same uptake assay using ΔtolC mutants, which abolish efflux pump activity in V. cholerae (31). Again, calcium did not affect TC uptake in ΔtolC mutant strains (see Fig. S2 in the supplemental material). These data suggest that at least under the conditions tested, calcium does not affect bacterial bile salt uptake.
TcpP is a membrane-bound regulatory protein, and the transmembrane domain of this protein is required for virulence activation (21, 32). Bile salts are known to be detergents and to interact with membranes (33–35). We thus considered the possibility that calcium may affect the interactions between bile salts, TcpP proteins, and/or the bacterial inner membrane. In attempt to quantify these relationships, we performed fluorescence recovery after photobleaching (FRAP) experiments (36). We expressed green fluorescent protein-TcpP (GFP-TcpP) in V. cholerae and monitored fluorescence recovery on a confocal microscope following bleaching of cell portions with an argon laser. To calculate recovery, equal areas of the bleached (A1) and unbleached (A2) portions of the cell were measured and the recovery rate was calculated (37). As seen in Fig. S3A in the supplemental material and Fig. 5A, cells grown in the presence of both TC and Ca2+ showed a decreased rate of recovery compared to controls. Those grown with calcium or TC alone did not have significantly different recovery from control (see Fig. S3B in the supplemental material). FRAP is typically used as a proxy to measure membrane fluidity by measuring fluorescent proteins within the membrane (38). We considered the possibility that membrane fluidity was decreased in the presence of TC and Ca2+ together for V. cholerae. To further explore this possibility, we performed FRAP on V. cholerae cells harboring fluorescently labeled TatA. TatA is an inner membrane protein with homologues in several bacterial species that is commonly used for FRAP due to the high tolerance for this protein in the inner membrane (36, 39). Surprisingly, cells expressing fluorescent TatA showed no difference in recovery between untreated cells and those grown in the presence of both TC and Ca2+ (Fig. 5B). These data imply that TC and Ca2+ may not affect overall membrane fluidity but may instead affect the membrane movement of TcpP. Ongoing work is under way to better assess the nature of the membrane alteration and mechanism by which TcpP dimerization is affected by bile salts and calcium.
FIG 5.
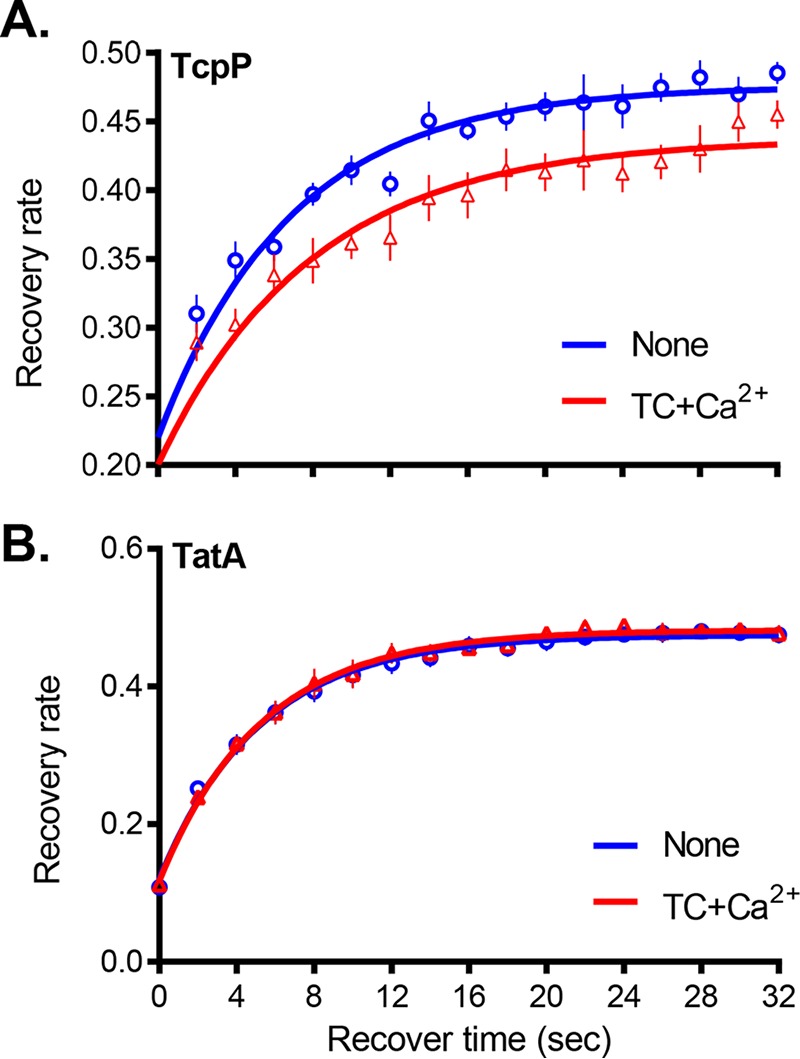
Fluorescence recovery after photobleaching (FRAP) of fluorescently labeled membrane proteins. Cells were grown to mid-log phase in the presence of arabinose with 1 mM TC and 10 mM CaCl2 where indicated. After near-complete photobleaching with an argon laser, the fluorescence intensity was recorded within two equally sized circular regions, one in the middle of the bleached compartment A1(t) and the other in the middle of the unbleached compartment A2(t), for a given time. The fluorescence recovery data were normalized to the total remaining fluorescence. (A) GFP-TcpP recovery; (B) TatA recovery. The data shown are from three independent experiments and over 10 individual cells.
DISCUSSION
V. cholerae must integrate many signals in the host environment to coordinate the proper timing and level of expression of virulence genes to achieve efficient colonization and cause disease. Decades of research have provided a great understanding of the genes involved in V. cholerae virulence regulation and the signals that alter this response in vitro. In this study, we find that physiologically relevant levels of calcium promote bile salt-dependent virulence activation (24, 40). This occurs through enhanced interaction of TcpP proteins concurrent with altered TcpP protein membrane diffusion. Our data suggest that TcpP may have more complex regulation than previously appreciated.
Both TcpP and ToxR contribute to maximal induction of ToxT. However, detailed studies have shown that TcpP is the direct activator of ToxT and that ToxR facilitates TcpP binding at the toxT promoter (3). Our experiments showing that TcpP is sufficient for toxT induction are consistent with this idea. Both ToxR and TcpP have a membrane-spanning region, a periplasmic region, and a cytoplasmic region containing a DNA binding domain. For proteins not coordinately regulated by TcpP, the ToxR cytoplasmic DNA-binding domain is sufficient to promote induction (41). However, membrane localization of ToxR is required for its role in promoting ToxT expression (41). All three domains of TcpP are required for virulence activation, whereas the membrane-spanning and periplasmic regions are sufficient for TcpP dimerization (21). In addition, TcpP and ToxR interact at the membrane and at the toxT promoter, but this interaction is abolished when the TcpP membrane-spanning region is swapped with the ToxR membrane region (32, 41). These findings suggest that the interaction of ToxR and TcpP at the membrane is important for proper virulence induction. This is consistent with the hypothesis that alteration of the cellular membrane affecting TcpP could lead to changes in TcpP activity and therefore the activation of downstream genes such as toxT. Our data demonstrate that calcium and bile salts may affect TcpP membrane movement and may play an important role in regulating TcpP activity.
Increased virulence in the presence of calcium was specific and not replicated by other similar metal ions such as Mg2+. This finding, paired with the observation that calcium does not seem to act on a new pathway but rather to enhance all known effects of bile salts such as TC, suggests that these molecules may act together as one bound molecule. The binding of Ca2+ to bile salts in the gut, particularly conjugated bile salts such as TC and GC, has long been appreciated. Bile acids are considered to be protective against gallstones resulting from calcium precipitation by buffering intraluminal Ca2+ (28). Numerous biochemical studies have described the preferential binding of bile salts (BS) to Ca2+ at a 2:1 ratio, resulting in a CaBS2 molecule with unique biochemical properties (28, 42, 43). Thus, it is likely that V. cholerae and other intestinal pathogens encounter not just bile salts in the gut, but bile salts in various forms of binding to calcium and other complexes. We synthesized calcium taurocholate (CaTC2) and found that it induced virulence at a significantly higher level than TC or TC with additional calcium (see Fig. S4 in the supplemental material). This suggests that CaTC2 may be physiologically relevant in vivo. CaTC2 has different properties than the ionic form of TC (TC−), which would likely predominate in the gut environment compared to the protonated form (44). For example, ionized bile salts preferentially partition in the outer hemileaflet of the phospholipid bilayer. Calcium-bound bile salts, such as CaTC2, instead partition between the two hemileaflets, residing within the hydrophobic lipid core (33, 43). The difference in charge and placement of CaTC2 compared to TC− could inherently change the interaction between bile salt, membrane, and perhaps TcpP protein(s). Further studies are required to fully elucidate the effect of these complex bile salt-based molecules on bacterial physiology and the role they play in vivo.
The contribution of protein diffusion to pathogenesis has been explored in the opportunistic fugal pathogen Candida albicans (45), where mutants lacking the ability to modulate protein diffusion are defective for virulence. In V. cholerae, the diffusion of TcpP within the inner membrane has also been investigated. Using single molecule tracking (46), Haas et al. concluded that the presence of ToxR and the presence of a toxT promoter both affect diffusion of a fluorescently labeled TcpP molecule. These researchers also noted that a portion of TcpP proteins were immobile and suggested that this halting was due to TcpP binding at the toxT promoter. A mutant strain lacking the toxT promoter region had fewer immobile TcpP events (46). In our experiments, TcpP mobility was shifted under conditions that are both strongly virulence inducing and physiologically relevant (9, 24, 47).
The idea that bile salts can affect bacterial membranes, and therefore likely affect membrane proteins as well, is in fact not new. Bile salts can cause damage membranes directly as detergents and indirectly by promoting redox stress (34). In addition, in response to bile, some bacteria can alter their membranes. For example, when exposed to bile, Bifidobacterium animalis displays decreased membrane fluidity due to changes in lipid composition, as well as an altered ratio of protein to phospholipids (48). Bacteria can also respond to bile in other ways, including the upregulation of efflux pumps and DNA repair enzymes, among other things (31, 34). Many pathogenic bacteria have also adapted to integrate exposure to bile acids into their virulence regulation mechanisms. In Campylobacter jejuni, many virulence genes are positively regulated by bile (49). Deoxycholate specifically promotes the expression of major virulence factors, including invasion antigens, leading to more rapid invasion of epithelial cells. Bile salts consistently repress virulence-associated genes in Salmonella. In particular, bile decreased the invasion of epithelial cells in vivo (50). V. cholerae has mixed responses to bile; fatty acids repress virulence, while certain bile salts promote the expression of virulence genes (19–21). In our study, we have expanded the list of molecules that V. cholerae likely senses and responds to in the gut to include Ca2+ and possibly calcium bound to taurocholate. These findings reinforce the notion that pathogens are exquisitely adapted to the environments and niches that they colonize. Further research will better elucidate whether the calcium-bile salt signal is sensed by other enteric pathogens and whether there are other specific signals used that exist in the complex intestinal milieu.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All V. cholerae strains used in this study were derived from El Tor C6706 and were propagated in Luria-Bertani (LB) medium containing appropriate antibiotics at 37°C (51). In-frame deletions were constructed by cloning the regions flanking the target genes into the suicide vector pWM91 containing a sacB counterselectable maker (52). The transcriptional luxCDABE reporters of the promoter regions of aphA, aphB, tcpP, toxR, toxT, ctxA, and tcpA have been described previously (15). Briefly, the ∼500 bp upstream of the coding region for each gene was amplified from genomic V. cholerae DNA and cloned into the pBBR-lux plasmid (53) directly upstream of the luxCDABE sequence to prepare transcriptional fusion reporters. Plasmids for overexpressing virulence regulators were described previously (54). For GFP-TcpP and TatA-GFP constructs, PCR-amplified fragments containing gfp-tcpPH or tatA-gfp coding sequences were cloned into pBAD24 (55), and the resulting plasmids were introduced into V. cholerae by electroporation.
Measurement of virulence gene expression and virulence factor production.
Overnight cultures of E. coli or V. cholerae strains containing promoter luxCDABE transcriptional fusions were subcultured at a dilution of 1:1,000 in LB medium with the indicated compounds and grown microaerobically (stationary and covered to limit oxygen availability) until reaching mid-log phase (optical density at 600 nm [OD600] ∼0.2). Luminescence was measured by using a Bio-Tek Synergy H1 spectrophotometer and normalized for growth against the OD600. Luminescence expression is reported as light units/OD600 unit. TCP production was measured by Western blotting with an anti-TcpA polyclonal antibody (54). Briefly, mid-log-phase cultures of wild type grown microaerobically in the absence or in the presence of 0.1 mM TC and 10 mM CaCl2 were collected. The cells were lysed by sonication, and samples were normalized by protein concentration (Pierce bicinchoninic acid protein assay kit; Thermo Scientific). Portions (200 μg) of proteins were loaded and separated by using SDS-PAGE with a 10% polyacrylamide gel. The gel was then transferred to a polyvinylidene difluoride membrane and immunoblotted with anti-TcpA antiserum and horseradish peroxidase-labeled goat anti-rabbit IgG antibody.
Purification of intestinal extracts and calcium depletion.
All animal studies were carried out in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of University of Pennsylvania. Intestinal extracts were purified as described previously (21). Briefly, fragments of small intestines from 5-week-old CD-1 mice were cut open and flushed with double-distilled H2O (ddH2O). The intestinal flush was then autoclaved and extracted with phenol-chloroform and subsequently ethyl acetate. The aqueous phase was then precipitated with 70% (vol/vol) ethanol. The supernatant was dried using a rotary evaporator and resuspended with ddH2O. EGTA was incubated with intestinal extracts for 12 h prior to use in experiments.
Preparation of CaTC2.
A solution of sodium taurocholate was acidified to ∼pH 3 with 1 M HCl and then frozen and lyophilized to dryness. The resulting white powder was suspended in acetone and filtered through a pad of silica gel to remove sodium chloride by-products. This material was evaporated to dryness with air, yielding taurocholic acid as a white film that was then dissolved in water. An aqueous solution of calcium hydroxide (0.5 eq, 1-mg/ml solution) was added, and the mixture was stirred for 2 days. This mixture was filtered through a pad of Celite to remove insoluble particulates, frozen, and lyophilized to dryness to give a white solid.
Bacterial two-hybrid system to determine TcpP-TcpP interaction.
Full-length tcpP or toxR fragments were cloned into pUT18C and pKT25 (30) vector as described previously (21, 32). Overnight cultures of tcpP and toxR reporters were subcultured at a dilution of 1:100 in LB medium containing 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) with or without 1 mM taurocholate and/or 10 mM CaCl2. TcpP-TcpP and ToxR-ToxR strains were then incubated without shaking at 30°C for 6 h. The cultures were then assayed for β-galactosidase activity, and the results were reported as Miller units as previously described (56).
FRAP analysis.
Overnight cultures of V. cholerae strains containing either PBAD-tcpP-gfp or PBAD-tatA-gfp fusions were subcultured at a dilution of 1:1,000 in LB medium with 0.1 or 0.05% arabinose, respectively, and grown aerobically to early log phase. Cultures were exposed to 10 mM CaCl2 and/or 1 mM TC and grown microaerobically until mid-log phase (OD600 ∼0.2). Microscope slides were prepared with soft agarose pads (1%). Cell cultures (5 μl) were applied to slides and coverslips added before being visualized by fluorescence microscopy. Fluorescence microscopy was performed on a Zeiss LSM 710 confocal laser scanning microscope using a 63× oil immersion objective lens. ZEN 2012 software was used for the acquisition of data. The 488 line of a 30-mW argon ion laser was used for both photobleaching and the subsequent fluorescence excitation/recording. The radius of the laser in the focal plane was estimated to 0.2 μm. For photobleaching, a power of 1.4 mW was applied onto selected regions within one of the compartments of the cell for 40 ms. Images were taken before photobleaching (t = −2 s), immediately after photobleaching (t = 0 s), and for 30 s at an interval of 2 s. After photobleaching, the fluorescence within the compartment exposed was nearly fully depleted. FRAP recovery data were analyzed as described previously (37). Briefly, the recovery of fluorescence intensity was recorded within two equally sized circular regions, one in the middle of the bleached compartment A1(t) and the other in the middle of the unbleached compartment A2(t). The fluorescence recovery data were normalized to the total remaining fluorescence for any given time point t. The recovery rate was thus calculated as follows: 100 × {A1(t)/[A1(t) + A2(t)]}.
C14-labeled taurocholate uptake assays.
Overnight cultures of the wild type and ΔtolC mutants (31) were subcultured at 1:100 in LB medium with or without added CaCl2 aerobically until reaching mid-log phase (OD600 ∼0.2). The cells were concentrated 10-fold in uptake buffer (1 mM MgCl2, 10 mM Tris-Cl [pH 7.5], 137 mM NaCl) with or without CaCl2 and then incubated with 200 μM taurocholate and 20 μM [14C]taurocholate. Aliquots of cells were taken at the indicated time points, the samples were pelleted, and the counts per minute (cpm) of 14C in the cells were measured on a Beckman scintillation counter. TC accumulation is reported as the percentage of cell-bound cpm compared to the total cpm.
Supplementary Material
ACKNOWLEDGMENTS
We thank Srujana Yadavalli, Jasmine Zhao, and Andrea Jordan for technical support and Mark Goulian for helpful discussions.
This study was supported by NIH/NIAID R01 grants AI080654 and AI120489 to J.Z., NSF GRFP fellowship DGE-1321851 to A.J.H., and NSFC grants 31300082 and 31470244 to M.Y.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00707-16.
REFERENCES
- 1.Faruque SM, Albert MJ, Mekalanos JJ. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol Mol Biol Rev 62:1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun 75:5542–5549. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yildiz FH, Visick KL. 2009. Vibrio biofilms: so much the same yet so different. Trends Microbiol 17:109–118. doi: 10.1016/j.tim.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu J, Mekalanos JJ. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell 5:647–656. doi: 10.1016/S1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]
- 6.Hay AJ, Zhu J. 2015. Host intestinal signal-promoted biofilm dispersal induces Vibrio cholerae colonization. Infect Immun 83:317–323. doi: 10.1128/IAI.02617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Lopez A, Behnsen J, Nuccio SP, Raffatellu M. 2016. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol 16:135–148. doi: 10.1038/nri.2015.17. [DOI] [PubMed] [Google Scholar]
- 8.Johansson ME, Ambort D, Pelaseyed T, Schutte A, Gustafsson JK, Ermund A, Subramani DB, Holmen-Larsson JM, Thomsson KA, Bergstrom JH, van der Post S, Rodriguez-Pineiro AM, Sjovall H, Backstrom M, Hansson GC. 2011. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci 68:3635–3641. doi: 10.1007/s00018-011-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eastwood MA, Boyd GS. 1967. The distribution of bile salts along the small intestine of rats. Biochim Biophys Acta 137:393–396. doi: 10.1016/0005-2760(67)90116-6. [DOI] [PubMed] [Google Scholar]
- 10.DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krukonis ES, DiRita VJ. 2003. DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol Cell 12:157–165. doi: 10.1016/S1097-2765(03)00222-3. [DOI] [PubMed] [Google Scholar]
- 12.Kovacikova G, Lin W, Skorupski K. 2003. The virulence activator AphA links quorum sensing to pathogenesis and physiology in Vibrio cholerae by repressing the expression of a penicillin amidase gene on the small chromosome. J Bacteriol 185:4825–4836. doi: 10.1128/JB.185.16.4825-4836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacikova G, Lin W, Skorupski K. 2010. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J Bacteriol 192:4181–4191. doi: 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Wang H, Zhou Z, Naseer N, Xiang F, Kan B, Goulian M, Zhu J. 2016. Differential thiol-based switches jump-start Vibrio cholerae pathogenesis. Cell Rep 14:347–354. doi: 10.1016/j.celrep.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. 2011. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci U S A 108:810–815. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin W, Kovacikova G, Skorupski K. 2007. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64:953–967. doi: 10.1111/j.1365-2958.2007.05693.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun 77:4111–4120. doi: 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plecha SC, Withey JH. 2015. Mechanism for inhibition of Vibrio cholerae ToxT activity by the unsaturated fatty acid components of bile. J Bacteriol 197:1716–1725. doi: 10.1128/JB.02409-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun 75:1946–1953. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A 110:2348–2353. doi: 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofmann AF, Mysels KJ. 1992. Bile acid solubility and precipitation in vitro and in vivo: the role of conjugation, pH, and Ca2+ ions. J Lipid Res 33:617–626. [PubMed] [Google Scholar]
- 23.Berridge MJ, Lipp P, Bootman MD. 2000. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol 1:11–21. doi: 10.1038/35036191. [DOI] [PubMed] [Google Scholar]
- 24.Permyakov EA, Kretsinger RH. 2009. Cell signaling, beyond cytosolic calcium in eukaryotes. J Inorg Biochem 103:77–86. doi: 10.1016/j.jinorgbio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Bilecen K, Yildiz FH. 2009. Identification of a calcium-controlled negative regulatory system affecting Vibrio cholerae biofilm formation. Environ Microbiol 11:2015–2029. doi: 10.1111/j.1462-2920.2009.01923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominguez DC, Guragain M, Patrauchan M. 2015. Calcium binding proteins and calcium signaling in prokaryotes. Cell Calcium 57:151–165. doi: 10.1016/j.ceca.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Sarkisova S, Patrauchan MA, Berglund D, Nivens DE, Franklin MJ. 2005. Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J Bacteriol 187:4327–4337. doi: 10.1128/JB.187.13.4327-4337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore EW, Celic L, Ostrow JD. 1982. Interactions between ionized calcium and sodium taurocholate: bile salts are important buffers for prevention of calcium-containing gallstones. Gastroenterology 83:1079–1089. [PubMed] [Google Scholar]
- 29.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun 69:4681–4685. doi: 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan F, Liu Z, Jabeen N, Birdwell LD, Zhu J, Kan B. 2014. Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect Immun 82:1676–1682. doi: 10.1128/IAI.01377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donovan JM, Jackson AA. 1997. Transbilayer movement of fully ionized taurine-conjugated bile salts depends upon bile salt concentration, hydrophobicity, and membrane cholesterol content. Biochemistry 36:11444–11451. doi: 10.1021/bi9705927. [DOI] [PubMed] [Google Scholar]
- 34.Merritt ME, Donaldson JR. 2009. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J Med Microbiol 58:1533–1541. doi: 10.1099/jmm.0.014092-0. [DOI] [PubMed] [Google Scholar]
- 35.Kamp F, Hamilton JA, Kamp F, Westerhoff HV, Hamilton JA. 1993. Movement of fatty acids, fatty acid analogues, and bile acids across phospholipid bilayers. Biochemistry 32:11074–11086. doi: 10.1021/bi00092a017. [DOI] [PubMed] [Google Scholar]
- 36.van den Wildenberg SMJL, Bollen YJM, Peterman EJG. 2011. How to quantify protein diffusion in the bacterial membrane. Biopolymers 95:312–321. doi: 10.1002/bip.21585. [DOI] [PubMed] [Google Scholar]
- 37.Skoog K, Soderstrom B, Widengren J, von Heijne G, Daley DO. 2012. Sequential closure of the cytoplasm and then the periplasm during cell division in Escherichia coli. J Bacteriol 194:584–586. doi: 10.1128/JB.06091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nenninger A, Mastroianni G, Robson A, Lenn T, Xue Q, Leake MC, Mullineaux CW. 2014. Independent mobility of proteins and lipids in the plasma membrane of Escherichia coli. Mol Microbiol 92:1142–1153. doi: 10.1111/mmi.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray N, Nenninger A, Mullineaux CW, Robinson C. 2005. Location and mobility of twin arginine translocase subunits in the Escherichia coli plasma membrane. J Biol Chem 280:17961–17968. doi: 10.1074/jbc.M413521200. [DOI] [PubMed] [Google Scholar]
- 40.Hofer AM, Brown EM. 2003. Extracellular calcium sensing and signaling. Nat Rev Mol Cell Biol 4:530–538. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 41.Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol 47:1459–1473. doi: 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 42.Gu JJ, Hofmann AF, Tonnu HT, Schteingart CD, Mysels KJ. 1992. Solubility of calcium salts of unconjugated and conjugated natural bile acids. J Lipid Res 33:635–646. [PubMed] [Google Scholar]
- 43.Zimniak P, Little JM, Radominska A, Oelberg DG, Anwer MS, Lester R. 1991. Taurine-conjugated bile acids act as Ca2+ ionophores. Biochemistry 30:8598–8604. doi: 10.1021/bi00099a015. [DOI] [PubMed] [Google Scholar]
- 44.Hofmann AF. 1999. Bile acids: the good, the bad, and the ugly. News Physiol Sci 14:24–29. [DOI] [PubMed] [Google Scholar]
- 45.Douglas LM, Konopka JB. 2016. Plasma membrane organization promotes virulence of the human fungal pathogen Candida albicans. J Microbiol 54:178–191. doi: 10.1007/s12275-016-5621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haas BL, Matson JS, DiRita VJ, Biteen JS. 2015. Single-molecule tracking in live Vibrio cholerae reveals that ToxR recruits the membrane-bound virulence regulator TcpP to the toxT promoter. Mol Microbiol 96:4–13. doi: 10.1111/mmi.12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown EM, MacLeod RJ. 2001. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81:239–297. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz L, Sanchez B, Ruas-Madiedo P, de los Reyes-Gavilan CG, Margolles A. 2007. Cell envelope changes in Bifidobacterium animalis ssp lactis as a response to bile. FEMS Microbiol Lett 274:316–322. doi: 10.1111/j.1574-6968.2007.00854.x. [DOI] [PubMed] [Google Scholar]
- 49.Malik-Kale P, Parker CT, Konkel ME. 2008. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J Bacteriol 190:2286–2297. doi: 10.1128/JB.01736-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prouty AM, Gunn JS. 2000. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect Immun 68:6763–6769. doi: 10.1128/IAI.68.12.6763-6769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsiao A, Liu Z, Joelsson A, Zhu J. 2006. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci U S A 103:14542–14547. doi: 10.1073/pnas.0604650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 53.Hammer BK, Bassler BL. 2007. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc Natl Acad Sci U S A 104:11145–11149. doi: 10.1073/pnas.0703860104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Miyashiro T, Tsou A, Hsiao A, Goulian M, Zhu J. 2008. Mucosal penetration primes Vibrio cholerae for host colonization by repressing quorum sensing. Proc Natl Acad Sci U S A 105:9769–9774. doi: 10.1073/pnas.0802241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.