

Abstract
Nucleosomes have structural and regulatory functions in all eukaryotic DNA-templated processes. The position of nucleosomes on DNA and the stability of the underlying histone–DNA interactions affect the access of regulatory proteins to DNA. Both stability and position are regulated through DNA sequence, histone post-translational modifications, histone variants, chromatin remodelers, and transcription factors. Here, we explored the functional implications of nucleosome properties on gene expression and development in Caenorhabditis elegans embryos. We performed a time-course of micrococcal nuclease (MNase) digestion and measured the relative sensitivity or resistance of nucleosomes throughout the genome. Fragile nucleosomes were defined by nucleosomal DNA fragments that were recovered preferentially in early MNase-digestion time points. Nucleosome fragility was strongly and positively correlated with the AT content of the underlying DNA sequence. There was no correlation between promoter nucleosome fragility and the levels of histone modifications or histone variants. Genes with fragile nucleosomes in their promoters tended to be lowly expressed and expressed in a context-specific way, operating in neuronal response, the immune system, and stress response. In addition to DNA-encoded nucleosome fragility, we also found fragile nucleosomes at locations where we expected to find destabilized nucleosomes, for example, at transcription factor binding sites where nucleosomes compete with DNA-binding factors. Our data suggest that in C. elegans promoters, nucleosome fragility is in large part DNA-encoded and that it poises genes for future context-specific activation in response to environmental stress and developmental cues.
The fundamental unit of eukaryotic chromatin is the nucleosome, which consists of 147 bp of DNA wrapped around an octamer of histone proteins (Luger et al. 1997). Nucleosomes have important structural and regulatory functions in organizing the genome and restricting access of regulatory factors to the DNA sequence (Henikoff 2008). As such, the interactions between nucleosomes and DNA strongly influence the regulation of gene expression by determining DNA accessibility for transcription factors (TFs) and RNA polymerase. In addition to regulated nucleosome assembly and disassembly through the action of histone chaperones and chromatin remodelers, nucleosome stability is influenced by histone modifications, histone variants, DNA features encoded in cis, and competition with DNA-binding factors in trans (Chereji and Morozov 2015). A complete picture of the mechanisms governing nucleosome stability is fundamental to understanding how gene expression is dynamically regulated.
Nucleosome stability has been studied in vitro using sensitivity to enzymatic digestion or salt concentration (Bloom and Anderson 1978; Burton et al. 1978; Li et al. 1993; Polach and Widom 1995; Wu and Travers 2004; Jin and Felsenfeld 2007). Genome-wide adaptations of these methods have been used to identify nucleosome position and stability in vivo. Studies in yeast, Drosophila, plants, and mammals have used varying concentrations of the enzyme micrococcal nuclease (MNase) to identify nucleosomes with differential sensitivity to MNase digestion in vivo (Weiner et al. 2010; Henikoff et al. 2011; Xi et al. 2011; Lombraña et al. 2013; Vera et al. 2014; Chereji et al. 2015; Kubik et al. 2015). Nucleosomes sensitive to low concentrations of MNase have been labeled as “fragile” and have been associated with TF binding sites (TFBSs) (Vera et al. 2014), active origins of replication (Lombraña et al. 2013), gene promoters (Xi et al. 2011), and genomic sequences with high AT content (Chereji et al. 2015). Thus, both DNA-encoded sequence features and trans-factors influence nucleosome fragility. However, the functional implication of nucleosome fragility remains unclear. For example, one study reported fragile nucleosomes at the promoters of repressed stress-response genes during normal growth (Xi et al. 2011), while another found fragile nucleosomes at the promoters of highly transcribed genes in yeast (Kubik et al. 2015). We performed a time-course of MNase digestion in Caenorhabditis elegans mixed-stage embryos to study the relationship between fragility and gene activity in a developing multicellular organism.
Results
A digestion time-course identifies nucleosomes with differential MNase sensitivity
We postulated that functionally distinct nucleosomes in C. elegans could be distinguished by the length of time it took them to be liberated from bulk chromatin by MNase digestion. Previous studies using this approach defined nucleosomes released early in the time-course as “fragile” and those released later in the time-course as “resistant” (Xi et al. 2011). To identify nucleosomes of differential sensitivity genome-wide, we isolated mixed-stage embryos from C. elegans, treated them with formaldehyde to cross-link the chromatin, isolated nuclei, and digested the chromatin with MNase (Fig. 1A). After 2, 4, 8, 15, and 30 min of digestion, we removed a chromatin aliquot and performed paired-end Illumina sequencing on the mononucleosomal fragments liberated at each time point (Fig. 1B). We performed two replicate experiments on native chromatin and two replicates on formaldehyde-fixed chromatin samples. Results from the native and fixed chromatin were very similar (Supplemental Fig. 1). We therefore focused our downstream analysis on fixed chromatin for maximum compatibility with previously generated data sets. Although the genome-wide occupancy profiles of mononucleosomal fragments were globally similar across the timepoints (Fig. 1B; Supplemental Fig. 2), there were a number of substantial differences in the nucleosome maps among the timepoints (Fig. 1C; Supplemental Fig. 2F).
Figure 1.

An MNase digestion time-course on C. elegans embryos. (A) Mixed-stage embryos were collected from gravid hermaphrodites by bleach treatment. Dissociated nuclei from mixed-stage embryos were incubated with MNase for 2, 4, 8, 15, or 30 min. (B) Paired-end reads from each timepoint were mapped to the C. elegans genome, normalized, and Gaussian smoothed for display. High signals represent regions of the genome protected from MNase digestion. Region plotted: Chr IV position 12,074,951–12,084,347. (C) Calculation of fragility and resistance scores. (Fragility) For each nucleosome, the average occupancy of the intermediate timepoints is subtracted from the 2-min timepoint. (Resistance) For each nucleosome, the average occupancy of the intermediate timepoints is subtracted from the 30-min timepoint. Intermediate timepoints are 4, 8, and 15 min. Region plotted is Chr IV position 12,076,980–12,078,364. (D) Distribution of fragility and resistance scores at all nucleosomes. The top 10% of each class (shaded in green and orange, respectively) were considered “fragile” or “resistant.”
To systematically study nucleosomes of differential sensitivity to MNase, we assigned each nucleosome both a fragility score and a resistance score as follows (Supplemental Fig. 2G). For each timepoint, we first called nucleosome positions and then assigned each nucleosome an occupancy score (for details, see Methods). The fragility score for a nucleosome is defined by subtracting the average occupancy score of the intermediate timepoints (4, 8, and 15 min) from the occupancy score of the 2-min timepoint. Conversely, a resistance score is computed by subtracting the average occupancy score of the intermediate timepoints from that of the 30-min timepoint (Fig. 1C). Thus, fragility and resistance scores were generally reciprocal to each other at a given nucleosome, but not necessarily so. We defined the top 10% of nucleosomes with the highest fragility or resistance scores as “fragile” or “resistant” nucleosomes, respectively (Fig. 1D).
Trans-factors increase nucleosome fragility
We sought to address whether nucleosome fragility was a consequence of competition with DNA-binding proteins and other trans-factors. Trans-acting factors disrupt nucleosomes by competing with histones for binding to the DNA sequence (Simpson 1990; Adams and Workman 1995). We first examined regions of the genome where we expected to find nucleosomes destabilized by competition with other DNA-binding factors, for example, at TFBSs. We collected a set of 35,062 TFBSs bound at any stage of C. elegans development, as identified by TF ChIP-seq from the modENCODE Consortium (Araya et al. 2014). The DNA immediately surrounding TFBSs in the C. elegans genome on average show strong affinity to histones in vitro (Locke et al. 2013). A nucleosome occupancy model based solely on DNA sequence also predicted C. elegans TFBSs to be nucleosome bound (Fig. 2A; Kaplan et al. 2009). In vivo, however, these sites show a local decrease in nucleosome occupancy, consistent with the footprint of TF binding. Moreover, TFBSs had high fragility scores on average (Fig. 2B). These data are in agreement with previous reports from yeast to humans that TFs compete with nucleosomes for access to DNA (Wang et al. 2012; Ozonov and van Nimwegen 2013; Barozzi et al. 2014). To further investigate the relationship between TF binding and fragility, we broke TFBSs into groups depending on the number of TFs bound at a site. Although the majority of TFBSs identified in C. elegans are bound by a single factor, some sites are bound by many TFs (Araya et al. 2014; Boyle et al. 2014; Chen et al. 2014). Fragility scores increased with the number of TFs bound at a single TFBS (Fig. 2C).
Figure 2.

Competition with transcription factors influences nucleosome fragility. (A) Average reconstituted nucleosome occupancy (Locke et al. 2013) and computational nucleosome occupancy model scores (Kaplan et al. 2009) at 35,062 regions bound at any stage by any number of transcription factors. (B) Average fragility, resistance, and intermediate nucleosome occupancy scores are plotted around the same set of intervals from A. (C) Boxplot of average fragility or resistance scores at groups of sites bound by different numbers of transcription factors. Nfactors = number of transcription factors bound. Nsites = number of regions in each category. (D) Cartoon characterization of how embryo-specific and L4-specific HOT regions were identified. (E) Model to distinguish whether trans (top) or cis (bottom) effects result in nucleosome fragility at a given nucleosome in the embryo. Hypothetical fragility scores are represented. (F) Fragility, resistance, and nucleosome occupancy scores measured in the embryo at 119 embryo-specific HOT regions. (G) Fragility, resistance, and intermediate nucleosome occupancy scores at 88 L4-specific HOT regions.
We found that TFBSs had high fragility scores despite their intrinsic preference for nucleosome formation in vitro (Fig. 2A). One possible explanation is that TFs destabilize nucleosomes at their binding sites, causing the fragility at TFBSs. To investigate this possibility, we identified a set of TFBSs specifically bound at different developmental stages (Fig. 2D). We hypothesized that if active competition with TFs increases nucleosome fragility, then TFBSs bound only in the embryo should be fragile in embryos, whereas TFBSs bound only in the L4 larval stage should not be fragile in embryos (Fig. 2E, top). Alternatively, if DNA sequence influences nucleosome fragility, then the embryo-specific and L4-specific TFBSs should be equally fragile in embryos (Fig. 2E, bottom). Due to their high fragility scores and dynamic nature, we focused our analysis on HOT regions, TFBSs where significant enrichment (false-discovery rate <5%) in multiple TFBSs are observed (Araya et al. 2014). We found that embryo-specific HOT regions had high nucleosome fragility and low nucleosome occupancy in our data set, which was obtained in embryos (Fig. 2F; Supplemental Fig. 3). In contrast, L4-specific HOT regions showed lower fragility and higher nucleosome occupancy in the embryonic samples (Fig. 2G). These results support the hypothesis that active competition with TFs in vivo contributes to nucleosome fragility despite their intrinsically nucleosome favoring properties in vitro.
Nucleosome fragility increases throughout heat-shock genes upon induction
The preceding analysis found a correlation between TF binding and nucleosome fragility. We next sought to test the relationship between fragile nucleosomes and trans-factors more explicitly. At extremely highly transcribed genes, such as heat-shock–responsive genes after induction, it has been proposed that RNA polymerase II (Pol II) molecules occupy the entire gene body (Schwabish and Struhl 2004; Merz et al. 2008; Cole et al. 2014). We hypothesized that fragility would increase at gene bodies after inducing high levels of transcription, as a result of nucleosome competition with transcribing Pol II. To test whether we could induce nucleosome fragility, we designed a heat-shock experiment in conjunction with an MNase-seq time-course (Fig. 3A).
Figure 3.

Heat-shock increases nucleosome fragility at the promoter and gene body of up-regulated genes. (A) Experimental overview. Mixed-stage embryos were either incubated at room temperature (RT) or heat-shocked at 34°C (HS) for 20 min. Subsequently, embryos were fixed and used for an MNase-seq time-course or stored in TRIzol and used for RNA-seq. (B) mRNA-seq identifies differentially expressed genes after HS at 34°C. Significantly differentially expressed genes (padj < 0.1) shown in red. (C) RNA, fragility, resistance, and nucleosome occupancy scores with and without HS are plotted in the region surrounding F33E5.4 and F33E5.5, two divergently transcribed hsp-70 orthologs. Region plotted is Chr II position 11,749,925–11,770,394. (D) Nucleosome fragility and nucleosome occupancy at 14 significantly differentially expressed genes with and without HS. (E) Nucleosome fragility, resistance, and occupancy scores at all 20,785 coding genes with and without HS.
Heat-shock in C. elegans activates HSF-1 and HSF-2, two homologs of the mammalian HSF1 TF, which bind heat-shock elements (HSEs) in the promoters of heat-shock–responsive genes to up-regulate their expression (Åkerfelt et al. 2010). By using RNA-seq, we identified 14 genes that are rapidly up-regulated after a brief (20-min) heat-shock at 34°C (Fig. 3B; Supplemental Fig. 4A). We then analyzed how fragility scores changed at those genes after heat-shock (Supplemental Fig. 4B,C). Though nucleosome occupancy decreased at heat-shock–responsive genes, we found nucleosome fragility dramatically increased both 5′ and 3′ of heat-shock genes, as well as in the gene body itself (Fig. 3C,D). Notably, promoter and +1 nucleosome fragility increased on average genome-wide, although gene-body fragility was specific to the set of heat-shock–induced genes (Fig. 3E).
High transcription rates have been suggested to remove the entire histone octamer, superseding FACT-mediated H2A-H2B recycling (Kireeva et al. 2002; Kulaeva et al. 2010). But nucleosomes can also be removed from gene bodies independently of transcription. Previous studies of the Hsp70 locus in Drosophila have shown that heat-shock induces rapid and transcription-independent loss of gene-body nucleosomes (Petesch and Lis 2008). Finally, it is possible that the 14°C temperature increase itself perturbs nucleosomes. Chereji et al. (2015) observed a related effect: Nucleosomes in Drosophila S2 cells cultured at 18°C are more stable than when cultured at 27°C. Future experiments may clarify the exact mechanism by which gene body nucleosomes become fragile after heat-shock.
Nucleosome fragility or resistance is associated with stereotypic nucleosome locations within genes
We found high fragility scores at genomic locations where we expected to find destabilized nucleosomes, like TFBSs and the gene bodies of newly induced genes. We next investigated the genome-wide distribution of fragile nucleosomes (nucleosomes with the highest 10% of fragility scores) (Fig. 1D) in detail (Fig. 4). Fragile nucleosomes were enriched 5′ and 3′ of genes, specifically at the promoter −2, −1, and +1 nucleosomes and at the terminal nucleosome (TN) and TN + 1 nucleosomes (Fig. 4B; Supplemental Fig. 1H). Resistant nucleosomes (nucleosomes with the highest 10% of resistance scores) (Fig. 1D) were enriched in gene bodies (Fig. 4C; Supplemental Fig. 1H).
Figure 4.

Fragility is enriched 5′ and 3′ of genes and is anti-correlated with gene expression. (A) Log2 DESeq2-normalized number of reads measured by mRNA-seq at 20,785 genes, ordered by their relative expression. (B) Heatmap of fragility scores (green) at genes ordered as in A. Genes were aligned at the center of the first nucleosome downstream from the transcript start site, known as the +1 or 5′ boundary nucleosome (yellow line). (C) Same as in B, except resistance scores are plotted in orange. (D) Same as B, except HTZ-1 input-normalized ChIP-seq signals (Ho et al. 2014) are plotted. (E) Same as B, except the average GC content (as a percentage of 100%) in 5-bp windows is plotted. (F–H) Fragility (F), resistance (G), and nucleosome occupancy (H) scores around the 5′ and 3′ boundary nucleosomes averaged over expression quintiles (highest expressed 20% in dark orange, dark green, or black; lowest expressed 20% in lightest orange, lightest green, or lightest gray). (Quintile 1) 0–4.5 normalized counts; (quintile 2) 4.5–65; (quintile 3) 65–619; (quintile 4) 619–2209; and (quintile 5) more than 2209.
Nucleosome fragility and resistance is not correlated with nucleosome occupancy
To investigate whether nucleosome fragility or resistance scores were a consequence of nucleosome positioning or occupancy, we asked whether fragility or resistance scores were correlated with nucleosome occupancy or the standard deviation of the nucleosome center (“fuzziness”). Nucleosome fragility scores were not correlated with nucleosome occupancy in the intermediate MNase timepoint (R = −0.02) or with nucleosome occupancy as measured by an independent histone H3 ChIP (R = −0.14) (Supplemental Results; Supplemental Fig. 5). In addition, neither nucleosome fragility nor nucleosome resistance scores were correlated with the fuzziness of the nucleosome at the intermediate timepoint (fragility vs. fuzziness: R = 0.03; resistance vs. fuzziness: R = −0.06), suggesting that susceptibility to MNase digestion is an independent feature of nucleosomes and is not a direct consequence of nucleosome occupancy or positioning (Supplemental Fig. 5).
Nucleosome fragility near genes is anti-correlated with expression
Gene expression levels on average were anti-correlated with nucleosome fragility at both promoters and gene bodies (R = −0.17) (Fig. 4A,B,F; Supplemental Fig. 6B) and positively correlated with nucleosome resistance (R = 0.11) (Fig. 4A,B,G; Supplemental Fig. 6B). In contrast to our earlier observation at the heat-shock genes, we found no correlation between expression and nucleosome fragility at the gene bodies of the most highly transcribed genes (cf. Fig. 3D and Supplemental Fig. 6C). It is possible that only newly induced genes display gene-body fragility or that extremely high levels of transcription are required to induce fragility in gene bodies.
Although nucleosome fragility scores were generally high at the 5′ and 3′ ends of all genes, including at the majority of TFBSs (Fig. 2), fragile nucleosomes occurred preferentially at the promoters of lowly expressed genes (Fig. 4B,F; Supplemental Fig. 6A). Examination of the existing ChIP data did not yield clues to the mechanism underlying the preferential nucleosome fragility at low-expressing gene promoters. No single TF profiled in the embryo significantly overlapped the distribution of fragile nucleosomes (Supplemental Fig. 6A). Further, although previous reports have suggested that the histone variant H2A.Z may act to promote nucleosome instability (Jin and Felsenfeld 2007; Jin et al. 2009; Xi et al. 2011), we did not observe a significant overlap between previously identified H2A.Z-containing nucleosomes (Ho et al. 2014) and fragile nucleosomes (Fig. 4D; Supplemental Fig. 6B,D). Our data, placed in to the context of the existing literature, suggest that two separate mechanisms account for nucleosome fragility, depending on the genomic context. In places where nucleosomes are directly in competition with TFs (Fig. 2) or in the bodies of exceptionally highly expressed or newly induced genes (Fig. 3), fragility arises through competition with TFs or other DNA-binding proteins. In contrast, fragility at the 5′ and 3′ end of genes at locations with few TF binding events appears to be determined by another mechanism, which we explored next.
Nucleosome fragility is correlated to cis-encoded DNA features
We hypothesized that cis features may be responsible for the fragility of nucleosomes at the promoters of lowly expressed genes. We examined the DNA sequences occupied by fragile and resistant nucleosomes and compared these to sequences occupied by all nucleosomes in the genome (Fig. 5). Compared to the set of all nucleosomes, DNA sequences occupied by fragile nucleosomes had lower GC sequence content on average (Fig. 5A). Lower GC content generally disfavors nucleosome formation. We then asked whether these sequences were likely to form nucleosomes based on a previously reported in vitro reconstitution assay (Locke et al. 2013). We found that DNA sequences occupied by fragile nucleosomes in the embryo were generally less likely to be nucleosomal in vitro (Fig. 5C). We also observed that sequences occupied by fragile nucleosomes were less conserved across nematodes than DNA sequences occupied by the set of all nucleosomes (Fig. 5E).
Figure 5.
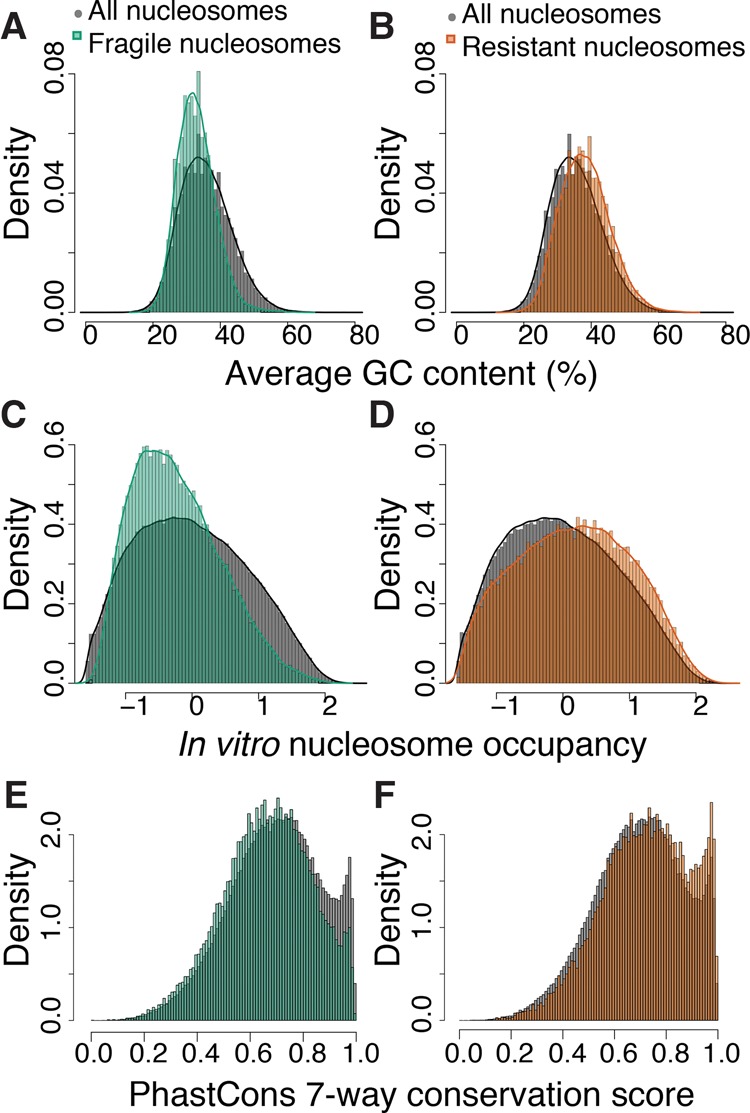
Fragile nucleosomes contain AT-rich, nucleosome disfavoring, and poorly conserved DNA. (A) Histogram of average GC content at fragile (green) or all nucleosomes (gray). (B) Same as A for resistant (orange) or all nucleosomes (gray). (C) Histogram of in vitro nucleosome occupancy scores at fragile or all nucleosomes. (D) Same as C for resistant nucleosomes or all nucleosomes. (E) Histogram of PhastCons seven-way conservation score at fragile or all nucleosomes. (F) Same as E for resistant or all nucleosomes.
Poly(dA:dT) tracts disrupt nucleosome formation and tend to increase transcription of downstream genes (Raveh-Sadka et al. 2012), while TATA box motifs in yeast are associated with bendable promoters sensitive to chromatin remodelers (Albert et al. 2007; Tirosh et al. 2007). In C. elegans, the number of T-block motifs (three to five consecutive thymine nucleotides, often spaced at 10-bp periodicity) have been positively correlated with expression: Genes with more than five T-blocks have fivefold higher expression than genes with fewer than four T-blocks (Grishkevich et al. 2011), presumably through a reduction in promoter nucleosome occupancy. T-blocks were not enriched at fragile or resistant nucleosomes, whereas TATA box motifs were enriched at fragile nucleosomes (Supplemental Fig. 7A). Taken together, our data indicate that fragile nucleosomes in gene promoters are correlated with high AT content and TATA box motifs. It seems that most promoters are fragile at least in part due to high AT content (see residual fragility in Fig. 2G), but that this cis effect of DNA sequence becomes apparent only at sites where the observation is not confounded by TF binding.
Nucleosome fragility is not associated with a specific epigenetic state
Through comparison with previously generated data sets, we asked whether any histone post-translational modifications, histone variants, or chromatin states were positively associated with nucleosome fragility or resistance (Ooi et al. 2010; Ho et al. 2014). No specific chromatin modification or combination of modifications were associated with nucleosome fragility. Only “low signal” chromatin states and chromatin extracted with 80 mM salt (another method proposed to identify unstable nucleosomes) were associated with fragile nucleosomes (Supplemental Fig. 7; Ooi et al. 2010; Ho et al. 2014). Although longer linkers were weakly correlated with increased fragility levels (Supplemental Fig. 7E), the GC content of the nucleosome was the strongest predictor of overall nucleosome fragility score (Supplemental Fig. 7F–J).
Fragile nucleosomes are associated with genes expressed in context-specific situations
To infer potential functional implications of nucleosome fragility in the developing embryo, we next asked which genes were significantly associated with fragile nucleosomes. We identified one set of genes that contained fragile nucleosomes, and another set of genes that contained resistant nucleosomes, and then looked for enriched Gene Ontology (GO) terms in each of the gene sets. Genes with fragile nucleosomes were enriched for GO terms related to neuronal response, immune response, and stress response genes (“sensory perception of chemical stimulus,” “defense response,” “pharynx development,” “immune system process”) (Fig. 6A). Because of their anti-correlation with expression and TF binding, this class of fragile nucleosomes in the embryo is unlikely to be fragile due to the action of trans-factors. Rather, these fragile nucleosomes were associated with lowly transcribed genes that are expressed in a context-specific fashion during stress response or development. In contrast, genes with resistant nucleosomes were enriched for general embryogenesis and cell cycle related terms (“mitotic cell cycle,” “RNA processing,” “regulation of developmental process,” “organic substance transport”) (Fig. 6B).
Figure 6.

Fragile nucleosomes are enriched at genes that will be expressed in the future and in specific contexts. Top 10 Gene Ontology biological process functional annotation terms associated with genes with fragile (A) and resistant (B) nucleosomes. (C) Bar plot representation of expression levels and coefficient of variation (CV) for R07H5.2 and F26F2.10. R07H5.2 has a low CV and is an example of a stably expressed gene (yellow); F26F2.10 has a high CV and is an example of a developmentally regulated gene (violet). (D) Average plot of fragility and nucleosome occupancy scores at 1000 stably expressed genes (left) or developmentally regulated genes (right) as determined by their coefficient of variation across seven different life stages: early embryo, late embryo, larval stages L1, L2, L3, L4, and young adult.
To confirm the association between fragile nucleosomes and future context-specific expression with an independent method, we used the modENCODE transcriptome data from seven different life stages to define a set of “developmentally regulated” genes and a set of “stably expressed” genes (Fig. 6C; Spencer et al. 2011; Gerstein et al. 2014; Pérez-Lluch et al. 2015). We hypothesized that if promoter nucleosome fragility is related to context-specific expression as our GO analysis suggested, then we should find higher fragility signals near developmentally regulated genes. When we plotted the average fragility scores around these genes, we indeed saw higher nucleosome occupancy and fragility signals at developmentally regulated genes compared with the set of stably expressed genes (Fig. 6D). While both sets of genes have fragile promoters, our data indicate that fragility is enriched at genes that tend to be expressed specifically during development, stress, or environmental stimulus response. Together, we suggest that these sequences may reflect a specialized promoter architecture that is primarily determined by high AT content, which acts to allow future disruption of nucleosome stability and thereby the rapid induction of gene expression in a context-specific fashion (Fig. 7).
Figure 7.

We propose a model whereby nucleosome fragility is determined by two distinct mechanisms: one that operates in cis at all genes and one that operates in trans at a subset of genes. (Left) Competition in trans with transcription factors and polymerase machinery destabilizes nucleosomes at the promoters of stably expressed genes. (Right) Condition-specific and developmentally regulated genes contain promoters with high levels of nucleosome fragility, determined primarily in cis by high AT content. (Green line) High AT content is sequence-encoded at all promoters but is highest at condition-specific genes; (orange cylinders) resistant nucleosomes found in the gene body of highly and stably expressed genes; (green cylinders) fragile nucleosomes compete (single arrow) with transcription factors and RNA Pol II at stably expressed genes. Fragile nucleosomes at condition-specific genes “treadmill” on the DNA (three arrows) due to destabilizing DNA elements like TATA-box motifs and high AT content.
Discussion
We performed an MNase digestion time-course, a simple modification to the traditional MNase digestion assay, in C. elegans embryos. Our experiment measured which individual nucleosomes were most quickly released from their polynucleosome context after exposure to MNase. Sensitivity to MNase digestion, and thereby fragility or resistance as defined in this study, could be determined by a number of factors. These include (1) a DNA sequence that is preferentially cut by MNase, (2) longer linker regions, (3) low DNA-histone affinity, or (4) competition with TFs. Two lines of evidence suggest that nucleosome fragility reflects nucleosome instability. First, we found that nucleosomes can be made fragile by competition with TFs and Pol II. Second, we observed that nucleosome fragility was associated with nucleosome-disfavoring DNA sequences, including high AT content and TATA-box motifs. All of these factors have been shown to cause nucleosome instability in previous studies (Widom 2002; Ozonov and van Nimwegen 2013).
We performed our experiments in nuclei derived from whole embryos, which reflect a mixture of cell types. This creates challenges for data interpretation. For example, from this heterogeneous mixture of cells, we observed that promoter nucleosome fragility decreases with increasing gene expression. In our data, ubiquitously expressed genes are more likely to fall into the “highly expressed” category, while a gene expressed in a cell-type–specific fashion is more likely to be part of the “lowly expressed” category due to its lower expression on average in the mixture of many cell types. Therefore, for genes expressed in every cell (or most cells), our data are simpler to interpret relative to nucleosome behavior.
Further, we may be less able to detect fragile nucleosomes that occur in a small fraction of the cell population. This may include nucleosomes at the promoters of genes expressed in a cell-type–specific manner, which are likely to differ in their fragility between expressing and nonexpressing cells. For example, promoter nucleosomes of myosin genes may be fragile in muscle cells and nonfragile in neuronal or intestinal cell types. As we observed in the heat-shock experiment, response to a stimulus can affect nucleosome fragility. Thus, fragile nucleosomes at these cell-type–specific genes are potentially underrepresented in our data. Future experiments using embryo dissociation and FACS enrichment for a certain cell or tissue types may be able to test this hypothesis.
MNase-resistant nucleosomes tend to be in gene bodies and correlated with expression
We found a class of nucleosomes that required relatively long durations of MNase digestion to be removed from chromatin. Traditional expectations might be that unstable nucleosomes would be found in the body of transcribed genes, and stable nucleosomes in silent, heterochromatic genomic regions. However, recent reports illustrate that nucleosomes in the gene body of transcribed genes are consistently well-positioned and highly occupied due to a number of factors, including the activity of Pol II and the histone chaperone FACT (facilitates chromatin transcription) (Jiang and Pugh 2009; Bai and Morozov 2010). Indeed, we found resistant nucleosomes enriched in the gene body of actively transcribed housekeeping genes. Histone modifications such as H3K36me3, which we found positively correlated with resistant nucleosomes, are also thought to contribute to nucleosome stability and maintenance of transcription fidelity (Lieb and Clarke 2005; Lickwar et al. 2009). Together, our measurements agree with an emerging picture of highly regulated nucleosome stability throughout the genome, which is likely critical for regulation of DNA templated events like transcription, splicing, and DNA replication (Tilgner et al. 2009; Eaton et al. 2010; Bintu et al. 2011; Kwak et al. 2013).
MNase-sensitive fragile nucleosomes are 5′ enriched and anti-correlated with expression
Differential MNase digestion and salt fractionation have been previously used to probe nucleosome–DNA stability. Results from yeast (Weiner et al. 2010; Xi et al. 2011; Kubik et al. 2015), plants (Vera et al. 2014), mouse (Lombraña et al. 2013; Deng et al. 2015; Iwafuchi-Doi et al. 2016; Mieczkowski et al. 2016), worm (Ooi et al. 2010), and fly (Henikoff et al. 2009; Chereji et al. 2015) have identified highly labile nucleosomes in 5′ and 3′ “nucleosome-free” regions. In yeast, Xi et al. (2011) observed that fragile nucleosomes were associated with H2A.Z-containing promoter nucleosomes, believed to be involved in stress response (Li et al. 2005; Zhang et al. 2005). In vertebrates, individual nucleosomes containing both H3.3 and H2A.Z histone variants are unstable (Jin and Felsenfeld 2007; Jin et al. 2009). We did not observe a correlation between H2A.Z incorporation and nucleosome fragility in C. elegans as measured by our assay. Rather, H2A.Z distribution is strongly biased toward active genes (Whittle et al. 2008; Liu et al. 2011). This distinction could be due to a divergence in H2A.Z properties between yeast and C. elegans (Zlatanova and Thakar 2008). It is also possible that the H2A.Z-containing nucleosomes used for analysis in this study (measured by Ho et al. 2014) were comprised of the particularly stable, homotypic type of H2A.Z nucleosomes (Ishibashi et al. 2009).
Previous reports disagree about the relationship between nucleosome fragility and expression. Studies in yeast, C. elegans, Drosophila, and maize have used salt profiling and different MNase concentrations to identify a positive correlation between promoter fragility and expression (Henikoff et al. 2009; Ooi et al. 2010; Vera et al. 2014; Kubik et al. 2015). However, Xi et al. (2011) used the same approach and observed the opposite effect. One possible explanation for the discrepancy is that some techniques may recover fragments from nucleosome-depleted loci, like the nucleosome-free regions in the promoters of active genes. A recent study performed a careful comparison of hypersensitive (fragile) nucleosomes and histone occupancy and concluded that fragile nucleosomes can come from one of two classes: (1) from nucleosome-depleted open-chromatin regions that are easily cleaved by light MNase, or (2) from truly nucleosome-occupied regions with destabilized nucleosomes (Iwafuchi-Doi et al. 2016). It is possible that the fragile nucleosomes we observe at highly transcribed genes are of the first class: nucleosome-depleted regions that are rapidly digested during low MNase conditions. In contrast, the highly fragile nucleosomes observed at the promoters of lowly transcribed genes—where we see the highest fragility signals—are more likely to be highly occupied nucleosomes that also have high fragility scores. This interpretation may help to clarify the apparently contradictory reports of nucleosome fragility enriched at either highly transcribed genes or at lowly transcribed genes. Further, slight variations in MNase digestion conditions may bias the capture of these two different classes of fragile nucleosomes.
In addition to sensitivity to MNase, other methods have been proposed to identify unstable or fragile nucleosomes. Washing chromatin with different ionic-strength buffers can be used to isolate nucleosomes of fundamentally different stabilities: Unstable nucleosomes are liberated at low salt concentrations, whereas more stable nucleosomes require higher salt concentrations to be disrupted. Ooi et al. (2010) used differential salt profiling followed by tiling microarray to investigate where 80 mM salt-extracted nucleosomes are located on chromatin isolated from C. elegans embryos. Both fragile nucleosomes and 80 mM salt extracted nucleosomes are enriched 5′ and 3′ of genes (Supplemental Fig. S7K–N). However, 80 mM salt nucleosomes are more highly recovered from highly expressed genes. In contrast, fragile nucleosomes observed in this study show the opposite enrichment, with higher fragile nucleosome recovery at lowly expressed genes.
Xi and colleagues identified nucleosome fragility at nearly one-third of all promoters of protein-coding genes, and found fragile nucleosomes to be enriched at the promoters of genes involved in stress response (Xi et al. 2011). When we assessed the types of functional annotations that were enriched at promoters with fragile nucleosomes in C. elegans embryos, we identified GO terms related to context-specific expression: sensory perception of chemical stimulus, defense response, immune system process. Based on our findings in conjunction with those of Xi et al. (2011), we propose that nucleosome fragility may serve to poise genes for rapid activation in response to developmental or external stimuli. This is consistent with previous work investigating the transcriptional activation of mammalian primary response genes, where unstable nucleosomes are used to achieve rapid induction independent of chromatin remodeling complexes (Ramirez-Carrozzi et al. 2009).
We found high fragility scores at the −2, −1, and +1 nucleosomes of developmentally regulated genes in comparison to stably expressed housekeeping genes. Previous work found that the promoters of developmentally regulated genes lack the histone post-translational modifications associated with active genes, like H3K4me3 (Pérez-Lluch et al. 2015). Likewise, Iwafuchi-Doi noticed few histone modifications associated with sensitive nucleosomes at tissue-specific and stage-specific enhancers (Iwafuchi-Doi et al. 2016). Similarly, we were unable to find an association between fragile nucleosomes and any histone post-translational modifications examined by the modENCODE group. Given the increased fragility of these nucleosomes, it is possible that (1) these nucleosomes at developmentally regulated genes were lost from standard chromatin preparation protocols and are thus underrepresented in the histone modification ChIPs, or (2) developmentally regulated genes use promoter nucleosome fragility as a mechanism for gene regulation. Iwafuchi-Doi proposes that the activity of pioneer TFs like FOXA1/A2 is responsible for destabilizing nucleosomes or displacing the stabilizing linker histone H1 (Iwafuchi-Doi et al. 2016). We observed no correlation between TF binding and fragile nucleosomes at cell-type–specific genes. This could be for a number of reasons, including (1) the TF ChIP was also performed on a heterogenous mixture of cells, making it difficult to recover signal from cell-type–specific binding events, or (2) the cell-type–specific/pioneering TF was not a member of the modENCODE ChIP collection.
Our results are reminiscent of previous reports from yeast, which propose that promoter structures can generally be classified as containing depleted proximal nucleosomes (DPNs) or occupied proximal nucleosomes (OPNs) (Tirosh and Barkai 2008). In yeast, DPN genes have low transcriptional plasticity (defined as the capacity to modulate transcription levels upon changing conditions), have well positioned nucleosomes, and are enriched for TF binding sites and H2A.Z. In contrast, OPN genes have high transcriptional plasticity, higher evolutionary divergence, and higher nucleosome turnover and are sensitive to chromatin regulation (Lickwar et al. 2012). The yeast DPN genes may correspond to the set of stably expressed genes we defined in C. elegans, which have DPNs. The yeast OPN genes may correspond to the set of developmentally regulated genes we defined in C. elegans, which have high promoter fragility and highly OPNs. To our knowledge, OPN- and DPN-type promoters have not been previously described or defined in C. elegans. Our results are consistent with a model in which nucleosome fragility is encoded at the promoters of OPN-type genes, potentiating the high transcriptional plasticity observed at these sites. The presence of these promoter structures in yeast, human, and now C. elegans suggests a well-conserved strategy that uses nucleosome architecture to regulate the dynamics of gene expression.
Methods
Worm strains and growth in liquid culture
Wild-type N2 worms were obtained from the Caenorhabditis Genome Center and maintained at 20°C in liquid culture as previously described (Ercan et al. 2011). Mixed-stage embryos were isolated from gravid adults by bleach hypochlorite treatment.
MNase digestion time-course
MNase digestion was performed as previously described (Ercan et al. 2011), with slight alterations. For each MNaseTC experiment, MNase was added, and at each timepoint (0, 2, 4, 8, 15, or 30 min after enzyme addition), a fraction of the reaction was removed, purified, and used for sequencing.
Heat-shock
Mixed-stage embryos were isolated and split into two pools. One pool was incubated at 34°C for 20 min, while the other pool nutated at room temperature. After 20 min, an aliquot from each pool was saved for RNA-seq, while the remaining embryos were fixed for 30 min at room temperature.
RNA isolation
Embryos were dropped into TRIzol (Life Technologies) and flash frozen in liquid nitrogen after incubation for 20 min at room temperature or 34°C heat-shock. Total RNA was isolated using a TRIzol/chloroform extraction followed by RNeasy mini (Qiagen) preparation.
Illumina library preparation
Individual libraries were prepared with unique barcodes for each timepoint from the time-course. MNaseTC libraries were prepared from 100 ng of gel-extracted DNA using the Illumina TruSeq DNA library preparation kit v2 (FC-121-2001) according to the manufacturer's instructions. RNA-seq libraries were prepared from 2 µg of total RNA using the Illumina TruSeq RNA library preparation kit v2 (RS-122-2001) according to the manufacturer's instructions.
Illumina sequencing and post-processing
Paired-end sequencing was performed by the Princeton University Sequencing Core Facility according to Illumina protocols. Paired end reads were mapped to the UCSC Oct. 2010 (WS220/ce10) genome release using Bowtie (v1.1.2) (Langmead et al. 2009).
Nucleosome analysis
Reads with insert sizes between 100 and 250 bp were kept for downstream analysis.
Replicates were first processed individually and then pooled after confirming a high degree of correlation between replicates. Nucleosome analysis was performed as described previously (Kaplan et al. 2010; Gossett and Lieb 2012). Additional details can be found in the Supplemental Methods.
Nucleosome fragility and resistance scores
To identify regions of the genome that were liberated earlier or later than average, we subtracted the occupancy of the pooled sample from either the 2-min (2m – pool = fragility score) or the 30-min (30m – pool = resistance score) samples. To highlight regions significantly enriched with this signal, we considered the 10% of nucleosomes with the highest fragility or resistance scores as fragile or resistant nucleosomes.
GO analysis
Gene lists were uploaded to the FatiGO web server (babelomics.bioinfo.cipf.es) and compared against the background set of all C. elegans genes (Al-Shahrour et al. 2004). P-values were calculated using the Fisher's exact test and corrected for multiple testing using the FDR procedure of Benjamini and Hochberg (1995). Corrected P-values and GO terms were then input in to REVIGO to reduce and visualize significantly enriched GO clusters (Supek et al. 2011).
Stable and developmentally regulated genes
Prenormalized transcriptome sequencing data were downloaded from: https://www.encodeproject.org/comparative/transcriptome/ (Spencer et al. 2011; Gerstein et al. 2014). For each gene, we calculated the coefficient of variation (CV): cv = σ/μ. We took the 1000 genes with the highest CVs as the set of developmentally regulated genes, and the set of 1000 genes with the lowest CVs as the set of stably expressed genes.
RNA-seq analysis
RNA-seq reads were mapped to the C. elegans WS220 gene annotation model using TopHat2 (v0.7) (Trapnell et al. 2012). The resulting alignment files were quantified using HT-Seq (v0.4.1) and the RefSeq gene annotations for WS220 (Anders et al. 2015). Total read counts per gene were normalized for differential expression using DESeq2 (v1.0.19) in R (v3.0.1) (Love et al. 2014).
Additional data sets
A brief description of the additional publicly available data sets used in this study and their accession numbers can be found in Supplemental Table S1.
Data access
All sequencing data from this study (i.e., RNA-seq and MNase-seq) have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP072274. Additionally, processed and raw data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE79567.
Supplementary Material
Acknowledgments
We thank Coleen Murphy and members of the Lieb laboratory for feedback and comments on the manuscript. This project was supported by the National Institutes of Health (NIH) National Human Genome Research Institute (NHGRI) grant U01 HG004270. T.E.J. was supported by NHGRI predoctoral training grant T32HG003284-10S1. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Author contributions: Experiments were designed by T.E.J. and J.D.L. Experiments and bioinformatics analyses were performed by T.E.J. with guidance and feedback from J.D.L. Manuscript was prepared by T.E.J. and edited by J.D.L.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.208173.116.
References
- Adams CC, Workman JL. 1995. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol 15: 1405–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åkerfelt M, Morimoto RI, Sistonen L. 2010. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F, Diaz-Uriarte R, Dopazo J. 2004. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 20: 578–580. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D, et al. 2014. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature 512: 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Morozov AV. 2010. Gene regulation by nucleosome positioning. Trends Genet 26: 476–483. [DOI] [PubMed] [Google Scholar]
- Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, Natoli G. 2014. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell 54: 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 57: 289–300. [Google Scholar]
- Bintu L, Kopaczynska M, Hodges C, Lubkowska L, Kashlev M, Bustamante C. 2011. The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat Struct Mol Biol 18: 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom KS, Anderson JN. 1978. Fractionation of hen oviduct chromatin into transcriptionally active and inactive regions after selective micrococcal nuclease digestion. Cell 15: 141–150. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Araya CL, Brdlik C, Cayting P, Cheng C, Cheng Y, Gardner K, Hillier LW, Janette J, Jiang L, et al. 2014. Comparative analysis of regulatory information and circuits across distant species. Nature 512: 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Butler MJ, Hyde JE, Phillips D, Skidmore CJ, Walker IO. 1978. The interaction of core histones with DNA: equilibrium binding studies. Nucleic Acids Res 5: 3643–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RAJ, Stempor P, Down TA, Zeiser E, Feuer SK, Ahringer J. 2014. Extreme HOT regions are CpG-dense promoters in C. elegans and humans. Genome Res 24: 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Morozov AV. 2015. Functional roles of nucleosome stability and dynamics. Brief Funct Genomics 14: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Kan T-W, Grudniewska MK, Romashchenko AV, Berezikov E, Zhimulev IF, Guryev V, Morozov AV, Moshkin YM. 2015. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res 44: 1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Ocampo J, Iben JR, Chereji RV, Clark DJ. 2014. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res 42: 12512–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T, Zhu ZI, Zhang S, Postnikov Y, Huang D, Horsch M, Furusawa T, Beckers J, Rozman J, Klingenspor M, et al. 2015. Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers. Genome Res 25: 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. 2010. Conserved nucleosome positioning defines replication origins. Genes Dev 24: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan S, Lubling Y, Segal E, Lieb JD. 2011. High nucleosome occupancy is encoded at X-linked gene promoters in C. elegans. Genome Res 21: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, Brown JB, Davis CA, Hillier L, Sisu C, Li JJ, et al. 2014. Comparative analysis of the transcriptome across distant species. Nature 512: 445–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett AJ, Lieb JD. 2012. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet 8: e1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishkevich V, Hashimshony T, Yanai I. 2011. Core promoter T-blocks correlate with gene expression levels in C. elegans. Genome Res 21: 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S. 2008. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet 9: 15–26. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. 2009. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res 19: 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. 2011. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci 108: 18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JWK, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn K-A, Minoda A, Tolstorukov MY, Appert A, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512: 449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausió J. 2009. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry 48: 5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Donahue G, Kakumanu A, Watts JA, Mahony S, Pugh BF, Lee D, Kaestner KH, Zaret KS. 2016. The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol Cell 62: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Felsenfeld G. 2007. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev 21: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. 2009. H3.3/H2A.Z double variant–containing nucleosomes mark “nucleosome-free regions” of active promoters and other regulatory regions. Nat Genet 41: 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Hughes TR, Lieb JD, Widom J, Segal E. 2010. Contribution of histone sequence preferences to nucleosome organization: proposed definitions and methodology. Genome Biol 11: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. 2002. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell 9: 541–552. [DOI] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone J-L, Rougemont J, Shore D. 2015. Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol Cell 60: 422–434. [DOI] [PubMed] [Google Scholar]
- Kulaeva OI, Hsieh F-K, Studitsky VM. 2010. RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc Natl Acad Sci 107: 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Nagaraja S, Delcuve GP, Hendzel MJ, Davie JR. 1993. Effects of histone acetylation, ubiquitination and variants on nucleosome stability. Biochem J 296: 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Pattenden SG, Lee D, Gutierrez J, Chen J, Seidel C, Gerton J, Workman JL. 2005. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histonemodifications and chromatin remodeling. Proc Natl Acad Sci 102: 18385–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Rao B, Shabalin AA, Nobel AB, Strahl BD, Lieb JD. 2009. The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS One 4: e4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Mueller F, Hanlon SE, McNally JG, Lieb JD. 2012. Genome-wide protein–DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb JD, Clarke ND. 2005. Control of transcription through intragenic patterns of nucleosome composition. Cell 123: 1187–1190. [DOI] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. 2011. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res 21: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke G, Haberman D, Johnson SM, Morozov AV. 2013. Global remodeling of nucleosome positions in C. elegans. BMC Genomics 14: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombraña R, Almeida R, Revuelta I, Madeira S, Herranz G, Saiz N, Bastolla U, Gómez M. 2013. High-resolution analysis of DNA synthesis start sites and nucleosome architecture at efficient mammalian replication origins. EMBO J 32: 2631–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Merz K, Hondele M, Goetze H, Gmelch K, Stoeckl U, Griesenbeck J. 2008. Actively transcribed rRNA genes in S. cerevisiae are organized in a specialized chromatin associated with the high-mobility group protein Hmo1 and are largely devoid of histone molecules. Genes Dev 22: 1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, et al. 2016. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SL, Henikoff JG, Henikoff S. 2010. A native chromatin purification system for epigenomic profiling in Caenorhabditis elegans. Nucleic Acids Res 38: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonov EA, van Nimwegen E. 2013. Nucleosome free regions in yeast promoters result from competitive binding of transcription factors that interact with chromatin modifiers. PLoS Comput Biol 9: e1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Lluch S, Blanco E, Tilgner H, Curado J, Ruiz-Romero M, Corominas M, Guigó R. 2015. Absence of canonical marks of active chromatin in developmentally regulated genes. Nat Genet 47: 1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT. 2008. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 134: 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J. 1995. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254: 130–149. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. 2009. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138: 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E. 2012. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44: 743–750. [DOI] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K. 2004. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24: 10111–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RT. 1990. Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature 343: 387–389. [DOI] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, et al. 2011. A spatial and temporal map of C. elegans gene expression. Genome Res 21: 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcárcel J, Guigó R. 2009. Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol 16: 996–1001. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. 2008. Two strategies for gene regulation by promoter nucleosomes. Genome Res 18: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Berman J, Barkai N. 2007. The pattern and evolution of yeast promoter bendability. Trends Genet 23: 318–321. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera DL, Madzima TF, Labonne JD, Alam MP, Hoffman GG, Girimurugan SB, Zhang J, McGinnis KM, Dennis JH, Bass HW. 2014. Differential nuclease sensitivity profiling of chromatin reveals biochemical footprints coupled to gene expression and functional DNA elements in maize. Plant Cell 26: 3883–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. 2012. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22: 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. 2010. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20: 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CM, McClinic KN, Ercan S, Zhang X, Green RD, Kelly WG, Lieb JD. 2008. The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genet 4: e1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom J. 2002. Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys 34: 269–324. [DOI] [PubMed] [Google Scholar]
- Wu C, Travers A. 2004. A “one-pot” assay for the accessibility of DNA in a nucleosome core particle. Nucleic Acids Res 32: e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Yao J, Chen R, Li W, He X. 2011. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 21: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Roberts DN, Cairns BR. 2005. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, Thakar A. 2008. H2A.Z: view from the top. Structure 16: 166–179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.