

Abstract
Alveolar type II (ATII) epithelial cells are the primary site of influenza virus replication in the distal lung. Development of acute respiratory distress syndrome in influenza-infected mice correlates with significant alterations in ATII cell function. However, the impact of infection on ATII cell surfactant lipid metabolism has not been explored. C57BL/6 mice were inoculated intranasally with influenza A/WSN/33 (H1N1) virus (10,000 plaque-forming units/mouse) or mock-infected with virus diluent. ATII cells were isolated by a standard lung digestion protocol at 2 and 6 days postinfection. Levels of 77 surfactant lipid-related compounds of known identity in each ATII cell sample were measured by ultra-high-performance liquid chromatography-mass spectrometry. In other mice, bronchoalveolar lavage fluid was collected to measure lipid and protein content using commercial assay kits. Relative to mock-infected animals, ATII cells from influenza-infected mice contained reduced levels of major surfactant phospholipids (phosphatidylcholine, phosphatidylglycerol, and phosphatidylethanolamine) but increased levels of minor phospholipids (phosphatidylserine, phosphatidylinositol, and sphingomyelin), cholesterol, and diacylglycerol. These changes were accompanied by reductions in cytidine 5′-diphosphocholine and 5′-diphosphoethanolamine (liponucleotide precursors for ATII cell phosphatidylcholine and phosphatidylethanolamine synthesis, respectively). ATII cell lamellar bodies were ultrastructurally abnormal after infection. Changes in ATII cell phospholipids were reflected in the composition of bronchoalveolar lavage fluid, which contained reduced amounts of phosphatidylcholine and phosphatidylglycerol but increased amounts of sphingomyelin, cholesterol, and protein. Influenza infection significantly alters ATII cell surfactant lipid metabolism, which may contribute to surfactant dysfunction and development of acute respiratory distress syndrome in influenza-infected mice.
Keywords: influenza, acute respiratory distress syndrome, alveolar type II cell, surfactant, phospholipid, liponucleotide, lipidomics, mouse
the rapid and unexpected emergence of new influenza A/WSN/33 (H1N1) strains in 2009 highlighted critical failings in current pandemic preparedness and influenza management strategies (22, 38). These limitations were further emphasized by the poor match between the 2014 influenza vaccine and circulating influenza strains (18). Hence, there is an urgent need to develop new treatments for potentially lethal influenza. One way to do this is to identify and target host cell factors that regulate viral replication and pathogenesis. However, our understanding of the impact of influenza on host cells remains incomplete, particularly at the metabolic level.
Alveolar type II (ATII) epithelial cells are essential to normal lung function. These cells synthesize and recycle surfactant lipids and proteins, regulate the depth of the alveolar lining fluid by active vectorial ion transport, and participate in the innate pulmonary immune response (33). Importantly, they are also the primary site of influenza A virus infection and replication in the distal lung (25). Nevertheless, the effects of influenza A virus infection on ATII cell metabolism have not been investigated.
We have shown that infection of mice with influenza A/WSN/33 (H1N1) virus results in onset of acute respiratory distress syndrome (ARDS) by 2 days postinoculation (dpi); ARDS is associated with significant impairment of ATII cell ion transport and reduced synthesis of surfactant proteins SP-A, SP-C, and SP-D (24, 50, 56). Infection also results in reduced static and dynamic lung compliance (2). Given these findings, we hypothesized that influenza infection will also result in significant dysregulation of ATII cell surfactant lipid metabolism. We used ultra-high-performance liquid chromatography-mass spectrometry (UHPLC/MS) to measure levels of 77 surfactant-related phospholipids, cholesterol, and phospholipid precursors in methanol extracts of ATII cells isolated from influenza-infected C57BL/6 mice. We found that, relative to mock-infected controls at the same time points, >85% of lipid species analyzed significantly changed at 2 and/or 6 dpi. Several major surfactant phospholipids significantly decreased, particularly when severe ARDS was present at 6 dpi (50). In contrast, most minor surfactant phospholipids increased after infection, as did cholesterol. The liponucleotides cytidine 5′-diphospho (CDP)-choline (CDP-choline) and CDP-phosphoethanolamine, which are essential precursors for de novo phospholipid synthesis, were undetectable in ATII cells from infected mice. This suggests that reduced synthesis of major phospholipids is a consequence of impaired liponucleotide generation. ATII cells from infected mice also contained fewer lamellar bodies, with abnormal morphology. Finally, we also found lower levels of major surfactant phospholipids and increased sphingomyelin (SM), cholesterol, and protein in bronchoalveolar lavage fluid (BALF) from influenza-infected mice.
Together, these data indicate that influenza A virus infection results in significant perturbation of surfactant phospholipid and cholesterol metabolism in ATII cells. The resulting alterations in intra-alveolar surfactant lipid composition may contribute appreciably to development of reduced lung compliance in influenza-infected mice, which is a hallmark of ARDS.
MATERIALS AND METHODS
Mice.
C57BL/6AnNCr mice (Mus musculus) were purchased from the National Cancer Institute (Frederick, MD). The mice were housed in sterile, static micro-isolator cages with solid flooring [to permit coprophagy (21)] and allowed free access to sterile, triple-filtered water and autoclaved Teklad 7012 mouse/rat diet (19.1% crude protein, 5.8% fat, and 4.6% crude fiber; Harlan, Haslett, MI). All procedures were approved by The Ohio State University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Infection of mice.
Eight- to 12-wk-old mice were inoculated intranasally with 10,000 plaque-forming units (pfu)/mouse of egg-grown, endotoxin- and mycoplasma-free influenza A/WSN/33 (H1N1) virus in 50 μl of virus diluent (PBS with 0.1% BSA) (1). This inoculum causes moderate ARDS by 2 dpi and severe ARDS at 6 dpi (50). Mock controls received an equal volume of virus diluent. To ensure that groups could be directly compared, mock and viral inoculations for each time point were performed at the same time and on the same day.
Murine ATII cell isolation.
As in our prior studies (24), ATII cells were isolated from euthanized mice at 2 and 6 dpi by a standard lung digestion protocol (10). All animals were euthanized at approximately the same time in the light−dark cycle. Purity of isolated ATII cell preparations was determined by visualization of lamellar bodies in modified Papanicolaou-stained cytospins. Cells were then repelleted and stored at −80°C.
Analysis of ATII cell metabolome.
Metabolomic analyses were conducted at Metabolon (Durham, NC). Briefly, ATII cell pellets were disrupted using a GenoGrinder (OPS Diagnostics, Lebanon, NJ) at 675 strokes/min for 2 min and then subjected to methanol extraction. Extracts were split into four aliquots and processed for analysis by UHPLC/MS in the positive, negative, or polar ion mode. Metabolites were identified by automated comparison of ion features (retention index and accurate mass match) to a reference library of authentic chemical standards followed by visual inspection for quality control. Each ion peak was quantified by a proprietary method. Data were normalized to sample lysate Bradford protein concentration for statistical analysis.
Fatty acid nomenclature.
All fatty acids are listed with their trivial name followed by their lipid number in parentheses. Lipid numbers take the form A:B, where A is the number of carbon atoms in the fatty acid and B is the number of double bonds in the fatty acid.
Transmission electron microscopy.
Whole lungs were perfusion-fixed with glutaraldehyde and prepared for transmission electron microscopy analysis by standard methods (5). Ultrastructure was visualized using a JEM-1400 transmission electron microscope (JEOL, Peabody, MA) linked to an Olympus SIS Veleta 2K camera (Olympus Soft Imaging Solutions).
Analysis of BALF lipids.
BALF total phospholipid was measured by ELISA (BioAssay Systems, Haywood, CA). Phosphatidylcholine (PC) and cholesterol were assayed using colorimetric assay kits (Cayman Chemical, Ann Arbor, MI). Phosphatidylglycerol (PG) and phosphatidylserine (PS) were measured with mouse-specific ELISA kits (MyBiosource, San Diego, CA). SM was quantified by fluorometric assay (Abcam, Cambridge, MA). All assays were performed in accordance with manufacturers' instructions.
Statistical analysis.
Welsh's two-factor t-test was performed on protein-normalized data in Array Studio (OmicSoft, Cary, NC) to identify metabolites that differed significantly between experimental groups. Any missing values were assumed to be below the limits of detection and were imputed with the compound minimum (minimum value imputation). An estimate of the false discovery rate (q value) was calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies, with q < 0.05 used as an indication of high confidence in a result. Differences between all mock- and influenza-infected samples at 2 and 6 dpi were determined by one-way ANOVA with a post hoc Tukey-Kramer multiple-comparison posttest using Instat software (GraphPad, San Diego, CA). P < 0.05 was considered significant.
RESULTS
ATII cell isolation.
C57BL/6 mice were infected intranasally with an acutely lethal dose (10,000 pfu/mouse in 50 μl of PBS and 0.1% FBS) of influenza A/WSN/33 (H1N1) virus or mock-infected with an equal volume of virus diluent. Infection was confirmed by loss of body weight: infected mice lost 8% of body weight by 2 dpi and 25% of body weight by 6 dpi (not shown). These changes were consistent with results of previous experiments. Mock-infected mice did not lose weight over the same time course. As in our earlier studies (23, 24), ATII cells were isolated to >95% purity by a standard lung digestion protocol (10). Characteristic Papanicolaou-positive lamellar bodies (refractile inclusions containing stored surfactant lipids) were visible in cytospins (11). Viability of all ATII cell preparations was >95% on the basis of Trypan blue exclusion.
Influenza A/WSN/33 virus infection significantly alters levels of large numbers of surfactant lipid metabolites in mouse ATII cells.
UHPLC/MS analysis was used to measure a total of 77 surfactant lipid-related compounds of known identity in methanol extracts from each ATII cell sample. Relative to mock-infected controls, infection with influenza A/WSN/33 virus for 2 or 6 days resulted in statistically significant increases or decreases in levels of 87% (67 of 77) of analyzed surfactant lipid species in ATII cells [n = 6 per group, except mock-infected mice at 6 dpi (n = 5)]. Of these, 62% were increased, including 6 at 2 dpi only, 12 at 6 dpi only, and 30 at both postinfection time points (Fig. 1A); 25% of surfactant lipid metabolites decreased after infection, including 1 at 2 dpi only, 13 at 6 dpi only, and 5 at both time points (Fig. 1B). A summary of changes in levels of all lipids measured by lipid class is provided in Table 1 (see Supplemental Fig. S1 in Supplemental Material for this article at the Journal website for a complete list of analyzed metabolites and a heat map of relative postinfection changes).
Fig. 1.
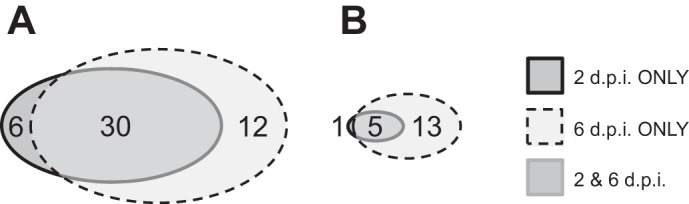
Influenza A/WSN/33 virus infection significantly alters levels of many surfactant lipid metabolites in mouse alveolar type II (ATII) cells. A: numbers of C57BL/6 mouse ATII cell metabolites significantly (P < 0.05) increased at 2 and/or 6 days after intranasal inoculation with influenza A/WSN/33 (H1N1) virus (10,000 plaque-forming units/mouse) relative to mock-infected controls at the same time points [2 and 6 days postinoculation (2 and 6 dpi)] (n = 48 total). B: numbers of ATII cell metabolites significantly decreased at 2 and/or 6 dpi relative to mock-infected controls at the same time points (n = 19 total). A total of 77 metabolites were analyzed. n = 6 per group, except mock-infected mice at 6 dpi (n = 5).
Table 1.
Summary of effects of in vivo influenza A/WSN/33 (H1N1) virus infection on murine ATII cell lipid content by lipid class
|
n‡ at 2 dpi |
n at 6 dpi |
n at 2 and 6 dpi |
n Total |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Class n† | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | |
| Phosphatidylcholine | 22 | 3 | 1 | 1 | 5 | 8 | 1 | 12 | 7 |
| Phosphatidylglycerol | 4 | 1 | 0 | 1 | 1 | 0 | 1 | 2 | 2 |
| Phosphatidylethanolamine | 11 | 1 | 0 | 1 | 3 | 3 | 2 | 5 | 5 |
| Phosphatidylserine | 5 | 1 | 0 | 2 | 0 | 1 | 0 | 4 | 0 |
| Phosphatidylinositol | 6 | 0 | 0 | 0 | 1 | 3 | 1 | 3 | 2 |
| Sphingolipid | 20 | 0 | 0 | 6 | 0 | 11 | 0 | 17 | 0 |
| Glycerolipid | 7 | 0 | 0 | 1 | 2 | 3 | 0 | 4 | 2 |
| Sterol | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| Inositol | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Total | 77 | 6 | 1 | 12 | 13 | 30 | 5 | 48 | 19 |
ATII, alveolar type II; dpi, days postinoculation.
Total number of metabolites analyzed for lipid class.
Number of metabolites in metabolite class significantly changed by influenza A virus infection (P < 0.05 vs. mock-infected at the same time point, by Welch's 2-sample t-test).
Influenza A virus infection decreases levels of many major surfactant phospholipids in ATII cells.
Pulmonary surfactant, which is synthesized by and stored in ATII cells, is a complex mixture consisting of 90% lipids and 10% proteins by weight. Approximately 80% of all surfactant lipids are phospholipids, and the phospholipid composition of surfactant is highly conserved in mammals (4). Infection with influenza A/WSN/33 virus for 2–6 days resulted in significant changes in 89% (33 of 37) of major ATII cell phospholipids analyzed. Importantly, this was the only lipid class in which a large number (33%) of metabolites decreased.
About 70% of surfactant phospholipid is PC, primarily in its disaturated form, 1,2-dipalmitoyl-PC [DPPC (16:0/16:0)] (4). DPPC and related PC species are also the major components of the outer leaflet of the plasma membrane. Influenza infection for 6 days significantly reduced ATII cell DPPC relative to mock-infected controls (Fig. 2A).
Fig. 2.

Influenza A virus infection decreases ATII cell levels of many major surfactant phospholipids. A–F: effect of mock infection or influenza A virus infection on ATII cell levels of 1,2-dipalmitoyl phosphatidylcholine [DPPC (16:0/16:0)], 1-palmitoyl,2-palmitoleoyl phosphatidylcholine [PPOPC (16:0/16:1)], 1-palmitoyl,2-linoleoyl phosphatidylcholine [PLPC (16:0/18:2)], 1,2-dipalmitoyl phosphatidylglycerol [DPPG (16:0/16:0)], 1-palmitoyl,2-linoleoyl phosphatidylethanolamine [PE (16:0/18:2)], and 1-palmitoyl,2-oleoyl phosphatidylglycerol [POPG (16:0/18:1)]. *P < 0.05, **P < 0.005, #P < 0.001 vs. mock-infected mice at the same time point. §P < 0.001 vs. influenza A virus-infected mice at 2 dpi. n = 6 per group, except mock-infected mice at 6 dpi (n = 5). Data in boxes represent 1st quartile, median, and 3rd quartile for each sample group; whiskers indicate highest and lowest sample values within the group.
Surfactant also contains other saturated and unsaturated palmitoylated lipids with a PC backbone, including significant amounts of 1-palmitoyl,2-palmitoleoyl-PC [PPOPC (16:0/16:1)], together with some 1-palmitoyl,2-myristoyl-PC [PMPC (16:0/14:0)], 1-palmitoyl,2-oleoyl-PC [POPC (16:0/18:1)], and 1-palmitoyl,2-linoleoyl-PC [PLPC (16:0/18:2)]. Infection resulted in reduced PPOPC (Fig. 2B) and PLPC (Fig. 2C) at 6 dpi. However, POPC modestly (but significantly) increased at 2 and 6 dpi (not shown). PMPC was not measured.
Cell membranes contain low levels of palmitoylated PG, but PG is highly enriched in surfactant and may be central to its surface tension-lowering properties (7). The other major phospholipid component of surfactant is phosphatidylethanolamine (PE), which is also enriched in mitochondrial membranes (53). At 6 dpi, ATII cells contained significantly lower levels of 1,2-dipalmitoyl-PG [DPPG (16.0:16.0)] and 1-palmitoyl,2-linoleoyl-PE (16:0/18:2) (Fig. 2, D and E). However, infection did not change 1-palmitoyl,2-oleoyl-PG [POPG (16:0/18:1)] at this time point (Fig. 2F).
ATII cells from influenza-infected mice contain increased amounts of several minor surfactant phospholipids and cholesterol.
In addition to DPPC, DPPG, and PE, pulmonary surfactant contains smaller amounts of PS, phosphatidylinositol (PI), and SM (7). PS, which is also enriched in the inner leaflet of the plasma membrane and in endosomal membranes, is generated from PC or PE by base exchange with serine. PI is synthesized from CDP-diacylglycerol (CDP-DAG) and myo-inositol (52, 53). The sphingoid base, sphingosine, can be synthesized de novo via condensation of palmitoyl-CoA and serine to 3-ketosphinganine, which is then reduced to sphinganine and acylated to ceramide. Ceramide can then serve as a substrate for sphingosine and SM synthesis.
Infection did not alter ATII cell serine content (not shown). However, 1-stearoyl,2-arachidonoyl-PS [PS (18:0/20:4)] levels were increased at 6 dpi (Fig. 3A). Myo-inositol tended to decrease at 6 dpi (not shown), but effects of infection on PI-based lipids were variable. Palmitoylated PI species such as 1-palmitoyl,2-linoleoyl-PI [PLPI (16:0/18:2)] decreased by 6 dpi (Fig. 3B), but stearoylated PI lipids such as 1-stearoyl,2-linoleoyl-PI [SLPI (18:0/18:2)] increased at both time points (Fig. 3C). Finally, infection had no effect on sphinganine levels (not shown) but increased both sphingosine (Fig. 3D) and SM (Fig. 3E) levels at 6 dpi.
Fig. 3.

ATII cells from influenza-infected mice contain increased amounts of several minor surfactant phospholipids and cholesterol. A–F: effect of mock infection or influenza A virus infection on ATII cell levels of 1-stearoyl,2-arachidonoyl phosphatidylserine [PS (18:0/20:4)], 1-palmitoyl,2-linoleoyl phosphatidylinositol [PLPI (16:0/18:2)], 1-stearoyl,2-linoleoyl phosphatidylinositol [SLPI (18:0/18:2)], sphingosine, sphingomyelin, and cholesterol. *P < 0.05, **P < 0.005, #P < 0.001 vs. mock-infected mice at the same time point. †P < 0.05, §P < 0.001 vs. influenza A virus-infected mice at 2 dpi. n = 6 per group, except mock-infected mice at 6 dpi (n = 5). Data in boxes represent 1st quartile, median, and 3rd quartile for each sample group; whiskers indicate highest and lowest sample values within the group.
Pulmonary surfactant and cell membranes also contain relatively large amounts of the neutral lipid cholesterol, which is taken up from the diet or synthesized de novo in the cytoplasm from 3-hydroxy-3-methylglutaryl-CoA. There was a trend toward increased cholesterol in ATII cells at 2 dpi, which became very significant at 6 dpi (Fig. 3F).
Availability of CDP-conjugated liponucleotide precursors for de novo DPPC and PE synthesis is reduced in ATII cells isolated from influenza-infected mice.
Approximately 45% of DPPC is derived from de novo synthesis by the well-known Kennedy pathway, which is illustrated in Fig. 4 to provide context to our findings (14). The remainder is generated by remodeling. De novo DPPC synthesis requires the liponucleotide CDP-choline and glycerol-3-phosphate. CTP-phosphocholine cytidylyltransferase-α (CCT-α) catalyzes synthesis of CDP-choline from choline phosphate and CTP. This step is rate-limiting for DPPC synthesis. Glycerol-3-phosphate can be generated by phosphorylation of glycerol or dehydrogenation of dihydroxyacetone phosphate, which is derived from glucose by glycolysis. Glycerol-3-phosphate is pamitoylated at the sn-1 position to form lysophosphatidic acid (16:0) by acyl-CoA:glucose-3-phosphate acyltransferase. Acyl-CoA:lysophosphatidic acid acyltransferase then catalyzes a second palmitoylation of lysophosphatidic acid (16:0) at the sn-2 position to generate phosphatidic acid (16:0/16:0). Dephosphorylation of phosphatidic acid by phosphatidic acid phosphatase produces 1,2-dipalmitoyl-DAG (16:0/16:0). Finally, choline phosphotransferase catalyzes the reaction of DAG with CDP-choline to yield DPPC (16:0/16:0).
Fig. 4.
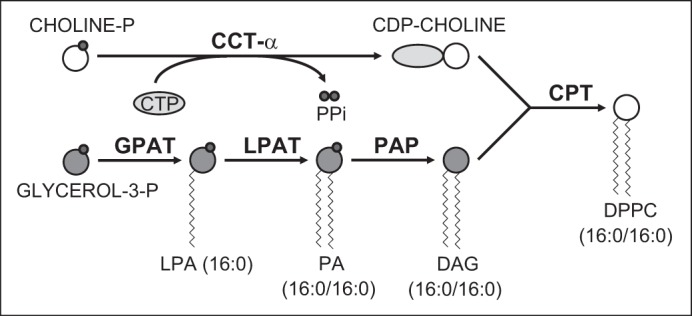
De novo synthesis of DPPC by the CDP-choline pathway. DPPC (16:0/16:0) is synthesized via the CDP-choline (Kennedy) pathway. In this pathway, CTP-phosphocholine cytidylyltransferase-α (CCT-α) makes the liponucleotide cytidine 5′-diphosphate-choline (CDP-choline) from choline phosphate (choline-P) and cytidine 5′-triphosphate (CTP). This reaction, which is rate-limiting for DPPC synthesis, also generates inorganic pyrophosphate (PPi). 1,2-Diacylglycerol [DAG (16:0/16:0)] is synthesized from glycerol-3-phosphate (glycerol-3-P) by sequential acylation. Glycerol-3-P is first palmitoylated at the sn-1 position to form lysophosphatidic acid [LPA (16:0)] by acyl-CoA:glucose-3-P acyltransferase (GPAT). Acyl-CoA:LPA acyltransferase (LPAT) then catalyzes a second palmitoylation of LPA at the sn-2 position to generate phosphatidic acid [PA (16:0/16:0)]. Both reactions also yield CoA (not shown). Dephosphorylation of PA by PA phosphatase (PAP) generates DAG (16:0/16:0). The final reaction of CDP-choline with DAG (16:0/16:0) to yield DPPC (16:0/16:0), glycerol (not shown), and cytidine 5′-monophosphate (CMP; not shown) is catalyzed by choline phosphotransferase (CPT). Enzymes are shown in bold font. Gray circles, phosphate moiety.
ATII cell glycerol was significantly elevated at 6 dpi (not shown), but glycerol-3-phosphate decreased (Fig. 5A). 1-Oleoyl,3-linoleoyl-DAG (18:1/18:2) increased at 2 and 6 dpi (Fig. 5B). However, 1-palmitoyl,3-linoleoyl-DAG (16:0/18:2) did not change (not shown). 1,2-Dipalmitoyl-DAG (16:0/16:0) was not measured. Levels of the CDP-choline precursors choline (not shown) and choline phosphate (Fig. 5C) did not change after infection. However, CDP-choline was undetectable at 2 and 6 dpi (Fig. 5D).
Fig. 5.

Availability of CDP-conjugated liponucleotide precursors for de novo DPPC and PE synthesis is reduced in ATII cells isolated from influenza-infected mice. A–F: effect of mock infection or influenza A virus infection on ATII cell levels of glycerol-3-phosphate (glycerol-3-P), 1-oleoyl,3-linoleoyl diacylglycerol [DAG (18:1/18:2)], choline phosphate (choline-P), cytidine 5′-diphosphocholine (CDP-choline), phosphoethanolamine, and cytidine 5′-diphosphoethanolamine (CDP-ethanolamine). *P < 0.05, **P < 0.005, #P < 0.001 vs. mock-infected mice at the same time point. ‡P < 0.005, §P < 0.001 vs. influenza A virus-infected mice at 2 dpi. n = 6 per group, except mock-infected mice at 6 dpi (n = 5). Data in boxes represent 1st quartile, median, and 3rd quartile for each sample group; whiskers indicate highest and lowest sample values within the group.
De novo PE synthesis also occurs via the Kennedy pathway (53), but in this case CDP-ethanolamine serves as the liponucleotide precursor. In an analogous fashion to CCT-α, the rate-limiting enzyme CTP:phosphoethanolamine cytidyltransferase catalyzes the formation of CDP-ethanolamine from phosphoethanolamine and CTP. The subsequent reaction of CDP-ethanolamine with DAG generates PE. PE can also be produced by decarboxylation of PS in mitochondria. The CDP-ethanolamine precursor phosphoethanolamine was significantly increased at 6 dpi (Fig. 5E). However, like CDP-choline, CDP-ethanolamine was undetectable at 2 and 6 dpi (Fig. 5F). Hence, reductions in ATII cell DPPC and PE following infection were accompanied by a decrease in liponucleotide precursors of de novo biosynthesis of both PC and PE.
Influenza virus infection results in abnormal ATII cell lamellar body morphology.
Surfactant lipids and proteins are stored within ATII cells in characteristic lamellar bodies. Transmission electron microscopy showed that lamellar bodies were fewer in number and smaller in ATII cells from influenza-infected mice than mock-infected controls (Fig. 6). Lamellar bodies in cells from infected mice also tended to be more electron-dense and had disordered lamellae. Alveoli also contained significant amounts of proteinaceous fluid at 6 dpi.
Fig. 6.

Influenza infection results in abnormal ATII cell lamellar body morphology. A: effect of mock infection for 6 days on ATII cell lamellar body ultrastructure. B: effect of influenza A virus infection on ATII cell lamellar body ultrastructure at 6 dpi. Magnification ×10,000; scale bar = 5 μm. No adjustments of tone or scale were made to captured images.
Changes in the ATII cell surfactant lipidome are accompanied by changes in BALF surfactant lipid composition.
Infection resulted in significant decreases in BALF PC (Fig. 7A) and PG (Fig. 7B) at 6 dpi. In contrast, BALF PS levels did not change (Fig. 7C), and both SM (Fig. 7D) and cholesterol (Fig. 7E) increased at 6 dpi. Finally, as in previous studies (56), BALF protein was very significantly elevated at 6 dpi (Fig. 7F).
Fig. 7.

Changes in the ATII cell surfactant lipidome are accompanied by changes in bronchoalveolar lavage fluid (BALF) lipid composition. A–F: effect of mock infection or influenza A virus infection on BALF phosphatidylcholine (PC), phosphatidylglycerol (PG), phosphatidylserine (PS), sphingomyelin (SM), cholesterol, and protein. #P < 0.001 vs. mock-infected mice at the same time point. §P < 0.001 vs. influenza A virus-infected mice at 2 dpi. n = 5 per group for mock-infected mice at 2 and 6 dpi; n = 6 per group for infected mice at 2 and 6 dpi. Data in boxes represent 1st quartile, median, and 3rd quartile for each sample group. Whiskers indicate highest and lowest sample values within the group.
DISCUSSION
Influenza accounts for ∼200,000 hospitalizations and >30,000 excess deaths per year (40, 49). Around 20% of patients with severe influenza develop ARDS, which is associated with poor prognosis (31). Once ARDS has developed, the only treatment option is nonspecific supportive management in the intensive care unit. Therefore, there is a great need for new influenza treatments that can prevent, retard, or manage progression of severe influenza to ARDS. Because influenza viruses are adept at developing resistance to antiviral drugs, these treatments would ideally be directed against pathophysiologically relevant host determinants of influenza severity. However, this necessitates a better understanding of the impact of infection on host cell function.
Considerable work has been done to investigate the effects of influenza on the transcriptome, primarily at the whole lung level. The primary functional consequence of changes in the host cell transcriptome following infection is altered cellular metabolism (41). However, very little is known about the impact of influenza infection on host lipid metabolism, particularly at the level of viral target cells in the lung. Studies in the 1970s showed that influenza A virus infection resulted in decreased surfactant activity and DPPC in mouse lung tissue (32, 45), but no further detailed analysis of the impact of infection on other surfactant lipids was performed. In the current study we show for the first time that infection results in significant global derangement of ATII cell surfactant lipid metabolism. More than 85% of the lipid metabolites measured in ATII cells changed after infection, and 60% (48 of 77) of them, including many minor surfactant phospholipids and cholesterol, increased. More importantly, however, infection also resulted in a progressive and significant reduction in most major surfactant phospholipids, particularly DPPC, DPPG, and PE. To the extent measured, these postinfection changes were reflected in BALF. Effects on ATII cell and BALF surfactant phospholipids and cholesterol were most pronounced late in infection when ARDS severity is increased. This implies a direct correlation between severity of influenza-induced ARDS and the extent of changes in the ATII cell lipidome. However, we found previously that lung viral titers do not differ significantly between 2 and 6 dpi, which indicates that viral effects are indirect or the influenza virus acts primarily as an initiating stimulus for lung injury.
Previous studies have shown that influenza infection alters lipid metabolism in A549 cells in vitro (36, 48). However, A549 cells do not recapitulate many primary respiratory epithelial cell functions, so the relevance of these studies is dubious (15). With use of an in vitro primary ATII cell infection model, it might be possible to determine whether alterations in the lipidome are a direct consequence of the virus. However, this would be technically challenging, since lipidomic analysis requires large numbers of cells. Moreover, ATII cells tend to differentiate ex vivo, which might significantly alter their lipidome, even in the absence of infection, and make data interpretation difficult. Other investigators have performed lipidomic analysis on whole lung tissue from influenza-infected mice (34–36, 47), but the surfactant lipidome was not a focus of any of these studies. Moreover, whole lung lipidomics is relatively noninformative, as it is highly likely that different resident lung cell types have different baseline lipid profiles and respond differently to infection and inflammation. It is also very probable that whole lung lipidomes change over the course of infection as a result of infiltration by inflammatory leukocytes, all of which will have different basal lipid metabolism. Additionally, lipid profiles in different subsets of infiltrating leukocytes will themselves change in different ways over time. Hence, the signal-to-noise ratio for whole lung lipidomics is very poor, which means that biologically significant changes in lipid metabolism may be underestimated, overestimated, or even missed entirely. This may be particularly true later in infection when inflammation and leukocyte infiltration are more severe. We therefore focused on ex vivo analysis of a single cell type that is highly relevant to both normal lung function and influenza infection. We believe that this approach allowed us to overcome some of the limitations of earlier studies and to generate a more relevant data set.
The significant reduction in ATII cell DPPC, DPPG, and PE at 6 dpi implies that these phospholipids are being secreted faster than they can be synthesized or that their synthesis is reduced. We showed previously that BALF from influenza-infected mice contains increased levels of ATP and adenosine (1, 3), both of which are potent surfactant secretagogues (6). However, in this study we found that BALF PC and PG were also reduced. Moreover, ATII cells from infected mice contained elevated levels of glycerol, DAG, and phosphoethanolamine, but levels of choline and choline phosphate were unchanged. Nevertheless, CDP-choline and CDP-ethanolamine were undetectable after infection. This suggests that, rather than increasing phospholipid secretion, infection results in reduced liponucleotide synthesis and, consequently, an imbalance between availability of DAG and liponucleotide substrates for phospholipid synthesis. Given that CDP-choline and CDP-ethanolamine were undetectable before DPPC and PE declined, we hypothesize that inhibition of liponucleotide synthesis early in infection is the proximate cause of later decreases in ATII cell PC and PE. We and others have shown that ATII cells can proliferate and/or differentiate into alveolar type I cells in response to influenza-induced lung injury. It is therefore possible that the cessation of liponucleotide synthesis is a component of these processes. However, the specific mechanism underlying reduced liponucleotide synthesis by ATII cells in influenza is yet to be determined.
It is believed that the tight intermolecular packing of DPPC is largely responsible for the surface tension-lowering properties of alveolar surfactant (54). The hydrophobic surfactant proteins SP-B and SP-C contribute to this function. Because of its conical shape, PE modulates membrane curvature and may play an important role in both membrane fusion and alveolar surfactant architecture. Cholesterol is also important for regulation of lipid bilayer fluidity. Together, PG, PE, and cholesterol facilitate adsorption, spreading, and fluidity of the surfactant film. Overall, therefore, the changes in the ATII cell surfactant lipidome are likely to have a very significant detrimental impact on the surface tension-lowering properties of surfactant in infected mice. In the current study we found a very significant increase in BALF protein at 6 dpi. We previously showed that influenza A virus infection also resulted in decreased ATII cell SP-C synthesis (24). Both effects are likely to further impair surfactant function. However, we have not yet directly measured the functional consequences of influenza infection for BALF surface tension.
A host cell-derived lipid envelope is an essential component of the influenza virion (37). However, the envelope lipid composition does not directly reflect that of the host cell: viral envelopes contain more sphingolipids, cholesterol, PE, and PS, but much less PC (17, 26). This configuration may facilitate fusion to target cells by altering viral membrane curvature. Envelope lipids also differ between virus strains (26). In addition to its detrimental effects on surfactant function, altered ATII cell lipid metabolism following infection may therefore significantly impact viral replication. Normal surfactant has been shown to inhibit hemagglutinin proteolysis by airway tryptases, which is necessary for infection (8, 29). Similarly, POPG blocks viral attachment to the host cell and, therefore, has strong antiviral activity against influenza (39). An intact SM synthesis pathway is necessary for intracellular transport of viral glycoproteins, and cholesterol in recycling endosomes is necessary for virus assembly and packaging (27, 46). Sphingolipid-cholesterol rafts are also required for transport of hemagglutinin to the apical cell surface prior to budozone formation (27). Hence, the changes in the ATII cell surfactant lipidome (particularly increased SM and cholesterol) might facilitate viral entry, replication, and infectivity of progeny virions.
Targeted metabolomics has been performed on exhaled breath, BALF, and plasma samples from patients with sepsis and ARDS, although, to our knowledge, this has not been done on samples from patients with influenza-induced ARDS (13, 19, 20, 42). However, there appears to be wide variation in metabolite profiles between patients, and individual studies have identified completely different groups of metabolites associated with death in sepsis (41). This may be a reflection of the highly variable nature of the patient populations sampled. A significant advantage of the mouse model is that effects of influenza infection on specific cell types or biological fluids of interest can be evaluated ex vivo in a genetically homogeneous sample population where experimental subjects and controls are matched for age, sex, health status, diet, microbiota, and environment. The influenza strain and dose inoculated, disease severity, and sampling time postinoculation can also be tightly controlled, and matched controls can be generated. This lack of variability further improves the signal-to-noise ratio for detecting differences between infected and mock-infected samples. Moreover, in the mouse model, sampling is not restricted to bodily fluids. Finally, changes in the lipidome of ATII cells isolated from an infected mouse will reflect the intrinsic effects of the virus on infected cells and the extrinsic effects of infection on the inflammatory response and cellular metabolism in other organ systems. This is not the case for cultured cells.
Although untargeted lipidomics with internal calibration standards allows for quantification of individual metabolites with high confidence, our study does have some limitations. First, the assay system we used is proprietary and not truly quantitative, although data are normalized to protein and are in concordance with quantitative findings from BALF assay. In follow-up studies we plan to perform quantitative targeted lipidomics to confirm the changes described here. Second, our data provide only a snapshot of the consequences of influenza infection for the host cell and provide no information regarding the dynamic flux of lipid metabolites. Hence, it is not possible to determine whether a reduction in the level of a particular metabolite is due to decreased synthesis or increased utilization. Detailed ATII cell-specific metabolic flux studies could address this question, but such studies are likely to be very challenging in an in vivo model. Similarly, because many metabolic reactions occur by mass action, lipidomic data do not provide a complete picture of the mechanisms underlying altered lipid metabolism in ATII cells from infected mice. To do so, they must ultimately be combined with data on the effects of influenza infection on expression and catalytic activity of enzymes involved in lipid metabolism. Moreover, because phospholipid synthesis and degradation are intimately linked to metabolism of other lipids, carbohydrates, amino acids, and nucleotides, our lipidomic data need to be viewed within the context of the entire ATII cell metabolome in order to be fully understood. We are currently investigating the impact of infection on other metabolites, but such studies are beyond the scope of the current report. Finally, because ATII cells are involved in surfactant lipid and protein synthesis, release, and reuptake, it is possible that changes in surfactant lipid profiles in ATII cells and BALF are due to altered exo- or endocytosis. This may be a primary effect of the virus on the infected cell or secondary to the viral inhibition of surfactant protein synthesis, which we recently described (24).
A further methodologic concern is that the nature and duration of the ATII cell isolation procedure may alter ATII cell metabolism from its status in vivo. Unfortunately, lipidomics cannot be performed on cells from fixed tissues, so there is no other way to generate samples from infected lungs for analysis. However, metabolite levels remained highly consistent between mock groups at both time points, yet, relative to both mock controls and each other, many differed significantly between the two infected groups. This suggests that changes in metabolite levels in ATII cells from infected mice are not an artifact. Similarly, the postinfection increase in the majority of other lipids suggests that reductions in DPPC, DPPG, and PE were not simply due to increased ATII cell death.
Although ex vivo ATII cell studies have not been performed in ARDS models, investigators have consistently reported changes in phospholipid content of BALF from human ARDS patients that are comparable to those in ATII cells and BALF from influenza-infected mice with ARDS. These include decreases in total BALF PC (13, 19, 43), DPPC (13, 42–45), and PG (19, 42, 43, 45), together with an increase in SM (19, 42, 45). Our findings are consistent with these earlier studies, although we did not detect the increase in BALF PS reported by Stinson et al. (45). Nevertheless, our data suggest that 1) consequences of lung injury for ATII cell lipid synthesis and release are similar regardless of its proximal cause and 2) the mouse influenza model may be a useful system for testing novel surfactant therapeutics relevant to all forms of ARDS. Recent trials of surfactant replacement therapy in ARDS patients were inconclusive or showed no benefit (12, 55). Nevertheless, intratracheal administration of natural or artificial surfactant improved outcomes in H1N1 influenza-infected mice (16, 51) and in a pediatric patient infected with pandemic H1N1 influenza (9). However, this approach proved ineffective in a second patient (30). Based on our data, we propose that it may be more effective to target the surfactant phospholipid synthesis pathway directly.
To our knowledge, this is the first study showing that influenza infection alters ATII cell phospholipid metabolism. It is also the first study to demonstrate that surfactant deficiency in influenza-induced ARDS may be due to impaired synthesis of liponucleotide phospholipid precursors, rather than simply ATII cell death. Our findings may provide an explanation for the failure of surfactant replacement therapy in ARDS (12, 55).
GRANTS
This work was supported by the C. Glenn Barber Fund and National Heart, Lung, and Blood Institute Grant R01 HL-102469.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.S.W., L.M.D., L.E.R., L.M.J., and E.P.C. performed the experiments; P.S.W., L.M.D., L.E.R., L.M.J., and I.C.D. analyzed the data; P.S.W., L.M.D., L.E.R., E.P.C., and I.C.D. interpreted the results of the experiments; P.S.W., L.M.D., L.E.R., L.M.J., E.P.C., and I.C.D. approved the final version of the manuscript; I.C.D. prepared the figures; I.C.D. drafted the manuscript; I.C.D. edited and revised the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
Current address of P. S. Woods: Section of Pulmonary/Critical Care, University of Chicago, Chicago IL.
REFERENCES
- 1.Aeffner F, Bratasz A, Flaño E, Powell KA, Davis IC. Post-infection A77-1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol 47: 543–551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aeffner F, Traylor ZP, Yu EN, Davis IC. Double-stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am J Physiol Lung Cell Mol Physiol 301: L99–L109, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol 88: 10214–10227, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agassandian M, Mallampalli RK. Surfactant phospholipid metabolism. Biochim Biophys Acta 1831: 612–625, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alli AA, Brewer EM, Montgomery DS, Ghant MS, Eaton DC, Brown LA, Helms MN. Chronic ethanol exposure alters the lung proteome and leads to mitochondrial dysfunction in alveolar type 2 cells. Am J Physiol Lung Cell Mol Physiol 306: L1026–L1035, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 293: L259–L271, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bernardino de la Serna J, Hansen S, Berzina Z, Simonsen AC, Hannibal-Bach HK, Knudsen J, Ejsing CS, Bagatolli LA. Compositional and structural characterization of monolayers and bilayers composed of native pulmonary surfactant from wild type mice. Biochim Biophys Acta 1828: 2450–2459, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Böttcher-Friebertshäuser E, Klenk HD, Garten W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis 69: 87–100, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busani S, Girardis M, Biagioni E, Pasetto A, Sambri V. Surfactant therapy and intravenous zanamivir in severe respiratory failure due to persistent influenza A/H1N1 2009 virus infection. Am J Respir Crit Care Med 182: 1334, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14: 309–315, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Dobbs LG. Isolation and culture of alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 258: L134–L147, 1990. [DOI] [PubMed] [Google Scholar]
- 12.Dushianthan A, Cusack R, Goss V, Postle A, Grocott M. Exogenous surfactant therapy for acute lung injury/acute respiratory distress syndrome—where do we go from here? Crit Care 16: 238, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dushianthan A, Goss V, Cusack R, Grocott M, Postle A. Altered molecular specificity of surfactant phosphatidylcholine synthesis in patients with acute respiratory distress syndrome. Respir Res 15: 128, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fagone P, Jackowski S. Phosphatidylcholine and the CDP-choline cycle. Biochim Biophys Acta 1831: 523–532, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forbes B, Ehrhardt C. Human respiratory epithelial cell culture for drug delivery applications. Eur J Pharm Biopharm 60: 193–205, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Fukushi M, Yamashita M, Miyoshi-Akiyama T, Kubo S, Yamamoto K, Kudo K. Laninamivir octanoate and artificial surfactant combination therapy significantly increases survival of mice infected with lethal influenza H1N1 virus. PLos One 7: e42419, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerl MJ, Sampaio JL, Urban S, Kalvodova L, Verbavatz JM, Binnington B, Lindemann D, Lingwood CA, Shevchenko A, Schroeder C, Simons K. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J Cell Biol 196: 213–221, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grohskopf LA, Olsen SJ, Sokolow LZ, Bresee JS, Cox NJ, Broder KR, Karron RA, Walter EB. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)-United States, 2014–2015 influenza season. MMWR Morb Mortal Wkly Rep 63: 691–697, 2014. [PMC free article] [PubMed] [Google Scholar]
- 19.Günther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med 153: 176–184, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Hallman M, Spragg R, Harrell JH, Moser KM, Gluck L. Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J Clin Invest 70: 673–683, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammond PD, Stutzenberger FJ, Butler RN, Read LC, Davidson GP. Factors affecting the validity of the 13C-urea breath test for in vivo determination of Helicobacter pylori infection status in a mouse model. Helicobacter 4: 260–265, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Hessel L. Pandemic influenza vaccines: meeting the supply, distribution and deployment challenges. Influenza Other Respir Viruses 3: 165–170, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hickman-Davis JM, Nicholas-Bevensee C, Davis IC, Ma HP, Davis GC, Bosworth CA, Matalon S. Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am J Respir Crit Care Med 173: 334–344, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofer CC, Woods PS, Davis IC. Infection of mice with influenza A/WSN/33 (H1N1) virus alters alveolar type II cell phenotype. Am J Physiol Lung Cell Mol Physiol 308: L628–L638, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80: 7469–7480, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanova PT, Myers DS, Milne SB, McClaren JL, Thomas PG, Brown HA. Lipid composition of the viral envelope of three strains of influenza virus—not all viruses are created equal. ACS Infect Dis 1: 435–442, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawaguchi A, Hirohama M, Harada Y, Osari S, Nagata K. Influenza virus induces cholesterol-enriched endocytic recycling compartments for budozone formation via cell cycle-independent centrosome maturation. PLoS Pathog 11: e1005284, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol 140: 1357–1367, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kido H, Sakai K, Kishino Y, Tashiro M. Pulmonary surfactant is a potential endogenous inhibitor of proteolytic activation of Sendai virus and influenza A virus. FEBS Lett 322: 115–119, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Ku AS, Chan LT. The first case of H5N1 avian influenza infection in a human with complications of adult respiratory distress syndrome and Reye's syndrome. J Paediatr Child Health 35: 207–209, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Li G, Yilmaz M, Kojicic M, Fernández-Pérez E, Wahab R, Huskins WC, Afessa B, Truwit JD, Gajic O. Outcome of critically ill patients with influenza virus infection. J Clin Virol 46: 275–278, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loosli CG, Stinson SF, Ryan DP, Hertweck MS, Hardy JD, Serebrin R. The destruction of type 2 pneumocytes by airborne influenza PR8-A virus: its effect on surfactant and lecithin content of the pneumonic lesions of mice. Chest 67: 7S–14S, 1975. [DOI] [PubMed] [Google Scholar]
- 33.Mason RJ. Biology of alveolar type II cells. Respirology 11 Suppl S12–S15, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Milner JJ, Rebeles J, Dhungana S, Stewart DA, Sumner SC, Meyers MH, Mancuso P, Beck MA. Obesity increases mortality and modulates the lung metabolome during pandemic H1N1 influenza virus infection in mice. J Immunol 194: 4846–4859, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milner JJ, Wang J, Sheridan PA, Ebbels T, Beck MA, Saric J. 1H NMR-based profiling reveals differential immune-metabolic networks during influenza virus infection in obese mice. PLos One 9: e97238, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, Watanebe T, Sakabe S, Daidoji T, Nakamura S, Kadowaki A, Ohto T, Nakanishi H, Taguchi R, Nakaya T, Murakami M, Yoneda Y, Arai H, Kawaoka Y, Penninger J, Arita M, Imai Y. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 153: 112–125, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Nayak DP, Balogun RA, Yamada H, Zhou ZH, Barman S. Influenza virus morphogenesis and budding. Virus Res 143: 147–161, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicoll A, Brown C, Karcher F, Penttinen P, Hegermann-Lindencrone M, Villanueva S, Ciotti M, Jean-Gilles L, Rehmet S, Nguyen-Van-Tam JS. Developing pandemic preparedness in Europe in the 21st century: experience, evolution and next steps. Bull World Health Organ 90: 311–317, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Numata M, Kandasamy P, Nagashima Y, Posey J, Hartshorn K, Woodland D, Voelker DR. Phosphatidylglycerol suppresses influenza A virus infection. Am J Respir Cell Mol Biol 46: 479–487, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reichert TA, Simonsen L, Sharma A, Pardo SA, Fedson DS, Miller MA. Influenza and the winter increase in mortality in the United States, 1959–1999. Am J Epidemiol 160: 492–502, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Rogers AJ, Matthay MA. Applying metabolomics to uncover novel biology in ARDS. Am J Physiol Lung Cell Mol Physiol 306: L957–L961, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt R, Meier U, Yabut-Perez M, Walmrath D, Grimminger F, Seeger W, Günther A. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med 163: 95–100, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt R, Markart P, Ruppert C, Wygrecka M, Kuchenbuch T, Walmrath D, Seeger W, Guenther A. Time-dependent changes in pulmonary surfactant function and composition in acute respiratory distress syndrome due to pneumonia or aspiration. Respir Res 8: 55, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simonato M, Baritussio A, Ori C, Vedovelli L, Rossi S, Massara L, Rizzi S, Carnielli VP, Cogo PE. Disaturated-phosphatidylcholine and surfactant protein-B turnover in human acute lung injury and in control patients. Respir Res 12: 1–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stinson SF, Ryan DP, Hertweck S, Hardy JD, Hwang-Kow SY, Loosli CG. Epithelial and surfactant changes in influenzal pulmonary lesions. Arch Pathol Lab Med 100: 147–153, 1976. [PubMed] [Google Scholar]
- 46.Tafesse FG, Sanyal S, Ashour J, Guimaraes CP, Hermansson M, Somerharju P, Ploegh HL. Intact sphingomyelin biosynthetic pathway is essential for intracellular transport of influenza virus glycoproteins. Proc Natl Acad Sci USA 110: 6406–6411, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tam V, Quehenberger O, Oshansky C, Suen R, Armando A, Treuting P, Thomas P, Dennis E, Aderem A. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 154: 213–227, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanner LB, Chng C, Guan XL, Lei Z, Rozen SG, Wenk MR. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J Lipid Res 55: 1357–1365, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289: 179–186, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Traylor ZP, Aeffner F, Davis IC. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses 7: 472–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Daal GJ, Bos JA, Eijking EP, Gommers D, Hannappel E, Lachmann B. Surfactant replacement therapy improves pulmonary mechanics in end-stage influenza A pneumonia in mice. Am Rev Respir Dis 145: 859–863, 1992. [DOI] [PubMed] [Google Scholar]
- 52.Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic 16: 1–18, 2015. [DOI] [PubMed] [Google Scholar]
- 53.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831: 543–554, 2013. [DOI] [PubMed] [Google Scholar]
- 54.Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol 10: 371–393, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willson DF, Truwit JD, Conaway MR, Traul CS, Egan EE. The adult calfactant in acute respiratory distress syndrome trial. Chest 148: 356–364, 2015. [DOI] [PubMed] [Google Scholar]
- 56.Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, Durbin JE, Davis IC. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med 178: 969–976, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.