

Abstract
Folding of G-protein coupled receptors (GPCRs) according to the two-stage model (Popot, J. L., and Engelman, D. M. (1990) Biochemistry 29, 4031–4037) is postulated to proceed in 2 steps: partitioning of the polypeptide into the membrane followed by diffusion until native contacts are formed. Herein we investigate conformational preferences of fragments of the yeast Ste2p receptor using NMR. Constructs comprising the first, the first two, and the first three transmembrane (TM) segments, as well as a construct comprising TM1–TM2 covalently linked to TM7 were examined. We observed that the isolated TM1 does not form a stable helix nor does it integrate well into the micelle. TM1 is significantly stabilized upon interaction with TM2, forming a helical hairpin reported previously (Neumoin, A., Cohen, L. S., Arshava, B., Tantry, S., Becker, J. M., Zerbe, O., and Naider, F. (2009) Biophys. J. 96, 3187–3196), and in this case the protein integrates into the hydrophobic interior of the micelle. TM123 displays a strong tendency to oligomerize, but hydrogen exchange data reveal that the center of TM3 is solvent exposed. In all GPCRs so-far structurally characterized TM7 forms many contacts with TM1 and TM2. In our study TM127 integrates well into the hydrophobic environment, but TM7 does not stably pack against the remaining helices. Topology mapping in microsomal membranes also indicates that TM1 does not integrate in a membrane-spanning fashion, but that TM12, TM123, and TM127 adopt predominantly native-like topologies. The data from our study would be consistent with the retention of individual helices of incompletely synthesized GPCRs in the vicinity of the translocon until the complete receptor is released into the membrane interior.
Keywords: G protein-coupled receptor (GPCR), membrane protein, nuclear magnetic resonance (NMR), protein folding, structural biology, Ste2p, Topology
Introduction
G-protein coupled receptors (GPCRs)3 are a large family of integral membrane proteins that transmit signals into cells upon activation by a set of highly chemically heterogeneous inducers (1). GPCRs form a seven-transmembrane (TM) helical bundle wherein the individual helices are connected by three extracellular and intracellular loops. The helix bundle is attached to an extracellular N-terminal domain of highly variable size and structure, and an intracellular and mostly flexible C terminus that often contains an eighth helix (2–4). Binding of an extracellular agonist stabilizes the conformation of the receptor that activates the now more accessible heterotrimeric G-protein (5). The first high-resolution structure of a GPCR, that of bovine rhodopsin, was published in 2000 (3), followed in 2007 by the first X-ray structure of a recombinantly produced GPCR (6). Subsequently, many structures of GPCRs in ground and activated states have been published (2, 4). Importantly, coordinates of an agonist-bound GPCR coupled to a G-protein have been released (7), and the structure of a GPCR-arrestin complex was solved (8).
Although our knowledge of the structure of GPCRs in various states and their mode of activation and desensitization is rapidly increasing, detailed information on their folding pathways is still lacking. The popular refined two-stage model from Popot and Engelman (9–11) postulates that secondary structures form when the peptide chain partitions into the membrane-water interface. However, proteins destined for membrane insertion are generally subjected to the concerted action of translating ribosomes in the cytoplasm and translocon complexes located in the endoplasmic reticulum (ER) of eukaryotes or in the plasma membrane of bacteria (12). To traffic proteins to membranes in most cells the signal-recognition particle targets the nascent chains emerging from the ribosome tunnel to the translocon complex (13). After folding within the ribosome-translocon complex (14–16), individual helices will then insert into and diffuse laterally in the membrane until native contacts are formed and the bundle fully assembles. In this co-translational insertion/folding process, segments of sufficient hydrophobicity are laterally gated from the translocon into the membrane interior.
A remarkable agreement has been observed between the purely biophysical Wimley-White scale (17) and the biological translocon scale from the von Heijne group (18). This agreement suggests that formation of the TM helices and their insertion into the membrane are largely governed by thermodynamic factors that are related to the amino acid sequence of the GPCRs (12, 18–20). Folding of polytopic membrane proteins is the result of a series of events that include helix insertion into the hydrophobic core and sequestering of loop sequences into cytosolic or extracellular space (11, 21). The timing of the chain insertion and the localization of TM helices would be expected to be a consequence of the amino acid sequence and the interaction of the growing polypeptide chain with the membrane. Recently, however, the first evidence was obtained that helices might change their location during synthesis of later portions of the polypeptide chain (22–24) emphasizing the aspect of context for proper folding.
An apparent conceptual problem with the sequential, hydrophobicity-based, folding model is that TM helices of some membrane proteins are only marginally hydrophobic. Helix-bundle membrane proteins display a large number of inter-helical contacts that are often formed between polar or even charged residues (25–28). We noticed that some TM segments of GPCRs that are under study in our lab do not display favorable energies for full membrane insertion (29, 30), an observation not unlike that reported for individual TMs of bacteriorhodopsin (31). It is therefore highly questionable whether in the absence of other interacting helices these “hydrophilic” TM segments would still fully insert into the membrane. To answer whether insertion occurs, topology-mapping methods that use terminally fused reporter moieties have been developed by the von Heijne group (32). Although they very successfully allow the rapid attainment of a quantitative picture of the location of the TM termini, they unfortunately do not provide any detailed structural information on the TM segments or on protein-membrane interactions.
To address some of these issues we have developed a systematic approach to investigate conformational preferences of N-terminal fragments of the Ste2p receptor, a yeast GPCR, using solution NMR methods. Considering that proteins are synthesized starting with the N terminus of the polypeptide chain, TM1 is expected to be the first segment that is inserted into the hydrophobic core, followed by TM2 and so on (21). We therefore report on studies of polypeptides corresponding to the overlapping fragments TM1, TM1–TM2 (TM12), and TM1–TM2–TM3 (TM123) (supplemental Fig. S1). TM1, TM2, and TM7 form a distinct subcore in the fully assembled helix bundle, and, based on analysis of published GPCR structures, often more contacts exist between TM7 and TM1 and TM2 than between TM3 and TM2 (4). Therefore, we have also examined a chimeric three TM helix construct, TM1–TM2–TM7 (TM127). In this construct the N-terminal end of TM7 is linked to TM2 using portions of IL1 (intracellular loop 1) and EL3 (extracellular loop 3, supplemental Fig. S1). NMR is being used to determine conformational preferences of these Ste2p fragments and their overall topology in detergent micelles. As the size of these polypeptides has increased from 60–80 (TM12) to 160–180 (3-TM constructs) residues, the NMR assignments became more challenging. We, therefore, have probed whether chemical shift assignments, which are more easily obtained for smaller fragments, can be transferred to these larger fragments. To ensure that no significant artifacts are introduced by conducting NMR studies in micelles, which do not represent true bilayers, we have also investigated TM127 incorporated into nanodiscs with different lipid compositions. Finally, we have monitored insertion of all these constructs into ER-derived membranes using an established insertion-glycosylation assay to obtain data on their integration into true biological membranes. Our data indicate that TM insertion is indeed context-dependent.
Results
Protein Expression and Purification
The Ste2p fragments TM1 (Gly31-Thr78), TM12 (Gly31-Thr110), TM123 (Gly31-Arg161), and TM127 (Gly31-Thr114, Thr274-Leu340) were expressed as C-terminal fusions to the hydrophobic TrpΔLE sequence (33). In all fragments cysteine and methionine residues were replaced by amino acids with similar properties (Ser for Cys; Ile, Leu, and Val for Met residues) to retain feasibility of chemical cleavage and preserve functionality of the receptor as reported before (34, 35). Several Escherichia coli expression strains (BL21(DE3), BL21-AI, BL21(DE3) pLysS, BL21(DE3) STAR pLysS, BL21 Rosetta pLysS, C41, and C43) were screened for expression (see “Experimental Procedures”). Inclusion bodies were solubilized in 70% TFA and, following chemical cleavage of the fusion proteins with CNBr, polypeptides of interest were purified using RP-HPLC. This resulted in expression yields of about 15 and 5 mg of purified protein per liter of 15N,13C-labeled and 15N,13C,2H-labeled M9 culture, respectively, for TM1 (using the strain BL21-AI), 11 and 5 mg/liters for TM123 (BL21-AI) (36), and 15 and 5 mg/liters for TM127 (BL21(DE3)STARpLysS). Expression details of TM12 have been reported previously (37). To obtain additional long-range distance restraints for TM127, the single cysteine mutants S47C, S75C, and S104C containing the full N terminus (Ser2-Thr114, Thr274-Leu340), and an N-terminal His10 tag separated by a cloning insertion coding for a C3 site (His10-Leu-Glu-Val-Leu-Phe-Gln-Gly-Pro-Ser2 … ) were expressed. After RP-HPLC purification, 40 mg of the mutant proteins per liter of 15N-M9 medium culture were obtained. These sulfhydryl-containing mutants were successfully coupled to MTSL maleimide.
NMR Studies
Resonance Assignments
For NMR measurements, samples were measured in 150 mm 1-palmitoyl-2-hydroxy-sn-glycero-3-(phospho-rac-(1-glycerol)) (LPPG)/dodecyl-phosphocholine (DPC) (4:1 mol/mol) micelles as the membrane mimetic, using 40 mm potassium phosphate buffer at pH 6.4. These conditions were similar to those previously used in our investigation of TM12 (37). The NMR samples of TM1, TM123, and TM127 all exhibit homogeneous line widths and good signal dispersion considering their highly α-helical nature (see Fig. 1 and supplemental Figs. S3–S6). Almost complete backbone assignment and partial side chain assignment could be achieved for every fragment (see below) using three-dimensional triple resonance as well as HCCH and 13C-resolved NOESY spectra. For TM1, 97% of the backbone and 66% of the side chain assignments were achieved. The assignments for TM12 as well as details of its structure calculation have been published previously (37).
FIGURE 1.

The N-terminal half of the entire Ste2p receptor seven-transmembrane bundle, schematically depicted at the top, is dissected into various fragments (TM1, TM12, TM123, and TM127) of increasing length. Typical 15N,1H-HSQC spectra in LPPG/DPC micelles are depicted at the bottom.
Samples containing TM123 exhibited a strong tendency to form soluble aggregates. The rate of aggregate formation depended on the detergent and the deuteration scheme; the TM123 sample with reverse ILV methyl labeling (completely perdeuterated protein with only the methyl groups of Ile, Leu, and Val residues protonated) aggregated completely within hours in deuterated detergents. Non-deuterated samples of the same polypeptide aggregated after several days. Aggregation was judged by the NMR line width and confirmed by size exclusion chromatography-multiangle light scattering (SEC-MALS) experiments (supplemental Fig. S2). Despite these challenges about 95% of the backbone resonances of TM123 could be assigned, the only missing residues being the two N-terminal amino acids, as well as two prolines (Pro79 and Pro117) and two residues at the beginning of TM2 (Asn84 and Gln85). Due to the high redundancy of certain amino acids and the resulting spectral overlap, as well as broad lines in the proton-carbon correlation spectra (HCCH-TOCSY and 13C-resolved NOESY) only 55% of side chains could be assigned.
In contrast to TM123, TM127 did not aggregate in the micellar environment (supplemental Fig. S2) and it was possible to assign 93% of the backbone resonances and significantly more of the side chain resonances (63%) than for TM123. The two N-terminal amino acids and a region in the center of TM7 containing the Leu-Pro-Leu (residues 289–291) sequence were missing resulting in the slightly lower fraction of assigned backbone resonances despite the higher quality of the TM127 spectra compared with those of TM123. We suspect that conformational exchange in TM7 broadens these latter signals beyond detection. Selective reverse methyl labeling of Ile, Leu, and Val allowed us to assign most of the methyl groups but many of the remaining side chain atoms could not be uniquely assigned. As is often the case with membrane proteins, only a small number of these assignments yielded unambiguous NOE restraints.
TM12 and TM127 or TM123 constitute two pairs of polypeptides in which nearly 80 residues are identical (those from TM12). We used these pairs to determine whether assignments on shorter fragments of GPCRs could be used to facilitate the assignments of the longer fragments. To transfer assignments, we compared corresponding strips in the three-dimensional 15N-resolved NOESY spectra of TM12 (37) with those of TM123 and TM127 for residues from TM1 and TM2. The rationale behind this approach is that the amide protons of a given residue will be close to protons from its own side chain or from neighboring residues, but will likely not be close to side chain protons from other helices that form tertiary contacts. In fact, it is very rare to observe inter-chain NOEs for HN interactions with side chain protons of even closely packed helices because these are almost always more than 5 Å apart. Starting with cross-peaks in the TM123 or TM127, 15N,1H correlation spectra that were in the vicinity of an assigned peak in the TM12 correct assignment was derived from a peak pattern match of strips in the three-dimensional 15N-resolved NOESY spectrum. When using 15N,1H-HSQC and three-dimensional 15N-NOESY spectra were recorded at 900 MHz, 70 (93%) of the 75 strips of TM12 could be matched correctly for TM127. Similarly, 68 (90%) of the 75 strips of TM12 were successfully matched for the TM123 fragment. Details on this assignment procedure will be reported elsewhere.4
Conformational Preferences
Usually, structure calculations of helical membrane proteins suffer from an insufficient number of long-range restraints, partially due to the fact that complete side chain assignments are difficult to obtain. More importantly, suboptimal packing of helices in the not fully formed helix bundle, the fact that detergents are not a perfect mimic for biological membranes, and the inherent flexibility of membrane proteins result in exchange broadening that tends to damp out the weak but structurally important long-range NOEs. To compensate for the low number of NOE restraints, residual dipolar couplings (RDCs), paramagnetic relaxation enhancements (PREs), and chemical shift-derived restraints were used. We have also probed access to a water-soluble spin label to reveal which residues are solvent exposed. All these data were used to obtain restraints for the final structure calculation and to orient the fragments in the micelles. To judge how well primary NMR data were represented by the ensemble, back-prediction of the raw data from conformers of the NMR ensemble was carried out.
Based on TALOS-N (38), backbone chemical shift data were used to predict the propensity of regions of Ste2p to form helices (Fig. 2). In general, all putative helices seem to be in the regions that are predicted by hydropathy algorithms. However, in the single TM fragment TM1 is clearly destabilized in the center of the helix around a GXXXG (GVRSG, residues 56–60) motif. In contrast this same region is significantly rigidified upon packing against TM2 in TM12. Accordingly the largest chemical shift differences between TM1 and TM2 for the overlapping segment are observed for the GVRSG residues (supplemental Fig. S8). The rigidification of the GVRSG region in TM1 is also observed in TM123 and TM127. Despite this rigidification there is a small but reproducible destabilization of the TM1 helices in the GVRSG domain also in the longer constructs. Based on the TALOS predictions the N-terminal end of the TM2 helix is destabilized in TM12, TM123, and TM127. In the latter polypeptide some assignments are missing, possibly indicating the presence of conformational exchange. A short helix in the extracellular segment from Phe38 to Val45 of the N terminus is also visible in all fragments. This helix is likely surface-associated and was also observed in an N-terminal fragment of the NPY4 GPCR (39). A region in the loop between TM2 and the third helix in both TM123 and TM127 displays some disposition to form a helical secondary structure. In TM127, the secondary structure in the center of TM7 is not well defined, largely due to the missing assignments for residues Leu-Pro-Leu (residues 289–291). The fact that no peaks were found for Leu289 and Leu291 indicates that conformational transitions occur close to the central Pro residue. Similarly, the center of the third helix in TM123 is destabilized around several polar residues.
FIGURE 2.

Secondary structure predictions obtained from TALOS-N for TM1, TM12, TM123, and TM127. The propensity of residues for forming α-helices as predicted by TALOS-N is shown. Putative helical regions are shaded in gray.
The structure of TM1 was computed based on distance restraints derived from 15N- and 13C-resolved NOESY spectra. It reveals two well defined short helices comprising residues Thr48-Phe55 and Ala61-Ile71. However, the relative orientation of the two helices is ill-defined (Fig. 3, left). We noted above that the chemical shift data suggest that the helical regions in TM1 are interrupted by the flexible central GVRSG pentapeptide. TM1 in TM12 is significantly stabilized due to packing against TM2, resulting in formation of a helical hairpin (Fig. 3, right).
FIGURE 3.

NMR structures of TM1 (left) and TM12 (right) in LPPG/DPC micelles. Residues 48–55 and 61–71 of TM1 have been superimposed either individually, as well as part of a continuous chain encompassing residues 48–71. In case of TM12 residues the 20 lowest-energy structures were superimposed for residues 49–103.
Due to its oligomerization tendency, TM123 could not be investigated beyond the location of its secondary structure and its mode of membrane integration (see below). The structure of TM127 could not be calculated in the usual way using NOE-derived upper distance limits due to incomplete side chain assignments and the poor quality of the 13C-resolved NOESY spectra. For TM127 only a few characteristic long-range NOEs that indicated interhelical contacts, mainly between residues from the surface associated N-terminal helix and the C-terminal end of TM2, were observed. To augment these, we sought to obtain long-range distance restraints for TM127 from PREs. Briefly, chemical moieties harboring an unpaired electron, e.g. the doxyl group in S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL), can result in distance-dependent line broadening of signals from remote protons in a range up to 20 Å. To obtain information on the global topology of TM127, we constructed three different single cysteine mutants: S47C at the N terminus of TM1, S75C in the short loop between TM1 and TM2, and S104C at the beginning of the relatively long loop between TM2 and TM7, and coupled these to MTSL. Using EPR methods we could demonstrate quantitative coupling and full functionality of the spin label for the constructs S75C and S104C, whereas S47C was partially deactivated.
The PREs from the S75C and S104C mutants are summarized in Fig. 4. The S75C mutant, labeled in the loop between TM1 and TM2, exhibited decreased intensity (high signal attenuation) throughout the entire TM2 helix and the second half of the TM1 helix. Attenuations are propagated further in TM2 compared with TM1 because the N-terminal end of TM2 is slightly destabilized as already observed in the TALOS predictions (Fig. 2). Signal attenuations were also observed at the end of TM7 and the C terminus, revealing spatial proximity between the first intracellular loop (IL1) and the C-terminal end of TM7. The spin-labeled S104C mutant also displays signal attenuations that are most severe at the C terminus of TM2 and the N terminus of TM7, respectively, and medium attenuations for residues of the surface-associated short N-terminal helix. These latter PREs indicate weak interactions between the second loop and the N terminus.
FIGURE 4.

Attenuations from spin labels. Residual intensities are depicted for site-directed spin labels S75C (left) and S104C (right) in TM127. Red bars indicate residues with no observable intensities.
Structure calculations were performed using the short and medium-range NOE restraints, the few observed long-range NOE restraints, and the PRE restraints. Additionally, consistency with NH RDC data were checked (see below). The calculations indicated that TM127 is a highly dynamic system, not a rigid helix bundle. Whereas the helices appear to be firmly integrated into the micelle (see below), inter-helical contacts seem to be transient. A homology model of the Ste2p structure (40) was used to simulate the PREs expected for the S75C spin-labeled TM127. Strong signal attenuations for residues from the C-terminal end of TM7 are expected (Fig. 5A). However, the PRE measurements on this mutant show only about 50% signal attenuations in the C-terminal part of TM127. Other back calculations of the PRE data, based on several of our NMR conformers (Fig. 5B), result in a better agreement between the simulated and experimental results. We conclude that the PRE data would be consistent with multiple conformations at the C terminus of TM7 (Fig. 5B, left and right panels) that result from movements about the flexible hinge formed by the Leu-Pro-Leu tripeptide in the center of TM helix 7. Because it was not possible to identify a distinct set of conformers that satisfy the experimental data we consider it prudent to focus on a qualitative rather than a quantitative description of the structures of these GPCR fragments. As discussed further below we expect that such structures may provide insights into conformations that are assumed by fragments of Ste2p in various stages of the folding of the incomplete receptor.
FIGURE 5.

PRE results calculated from various structural models for TM127. Experimental (black) and back-calculated (red) residual peak intensity of amide cross-peaks from the 15N,1H-TROSY spectra of the S75C MTSL-labeled TM127 sample. For the back-calculations, either the homology model from Eilers (40) (A) or two principal conformers derived from the structure calculations using NOE and PRE restraints (B, see text) were used. Values for TM helices are indicated by dark gray and for the surface-associated N-terminal helix by light gray-shaded boxes. The corresponding structures are depicted on top with TM helices 1, 2, and 7 in gold, red, and blue, respectively.
To aid our structural information and in particular to help in the alignment of the TM helices with respect to each other, we attempted to measure RDCs. The latter allow the determination of the orientations of vectors connecting two nuclei (e.g. the N and H of an amide moiety) relative to a molecular axis system. This results in global structural information, thereby helping to define the orientation (but not translation) of two helices relative to each other. To overcome the problem that many solutions exist for a given RDC value, a number of different types of RDCs can be measured. Unfortunately in our experience, in practice only NH RDCs can be measured on large membrane proteins in detergent micelles. A TROSY-based ARTSY experiment (41) for measuring NH RDCs of TM127 using the dinucleotide 2′-deoxyguanylyl-(3′,5′)-2′-deoxyguanosine (dGpG) as the alignment agent resulted in RDC values between −24 and +14 Hz (supplemental Fig. S9). As mentioned above, the impact of single sets of RDCs in structure calculations is small, therefore we will interpret the RDC data only qualitatively. The RDC values for residues in TM1, TM2, and in the first half of TM7 are large, and hence cannot be extensively averaged. This observation is consistent with the situation that TM1, TM2, and the N-terminal half of TM7 form a folded nucleus that does not undergo large conformational transitions and the C terminus of TM7 is not packed against this nucleus and hence more flexible. This supports the PRE results.
Probing the Integration of TM123 and TM127 into Micelles Using Water-soluble Spin Labels
To map the topology of membrane-integrated TM123 and TM127 polypeptides, the soluble, inert paramagnetic probe Gd-(DTPA-BMA) (gadolinium diethylenetriamine-pentaacetic acid bis-methylamine) was added to the NMR sample. With this probe, in general, residues outside the micelle or closer to the micelle surface experience stronger signal attenuations compared with residues that are buried in the micelle interior. At 15 mm Gd-(DTPA-BMA), water-exposed residues usually exhibit less than 20% residual signal intensity, whereas residues that are buried in the micelle still display intensities of up to 90%.
The residues in the C terminus of TM123 and TM127 almost all exhibit less than 20% residual intensity in the presence of the water-soluble spin label. These serve as the internal control in the experiment. In contrast, most residues in the predicted TM regions of these polypeptides have residual intensities >50% and many have intensities between 70 and 80% (Fig. 6). Thus, overall, the TM regions of both constructs, TM123 and TM127, seem to be well integrated into the LPPG/DPC micelle, whereas both termini as well as portions of the loop regions are located outside of the micelle or near its surface (Fig. 6). Signals from the short N-terminal helix (residues Phe38-Val45) as well as part of the second loop (EL1) display moderately weaker attenuations, indicating that they still interact with the micelle surface. Signals from TM2 are slightly more attenuated than those of TM1, both in TM123 and TM127. TM7 in TM127 displays very limited signal attenuation at the N terminus, but stronger attenuations toward the C terminus. In addition, we probed for integration of TM127 into the micelles by monitoring the occurrence and magnitude of NOEs to detergent proteins, and the exchange peaks with water (supplemental Fig. S10). These data support the topology as established from the Gd-spin label data, although the latter, in addition, reveal water exposure of residues from the second loop. The Gd-(DTPA-BMA) data indicate that TM123 generally seems to be less well folded compared with TM127. For example, the center of TM3 is partially solvent exposed, in agreement with the occurrence of polar residues in that region of the TM3 sequence. The poor packing of the helices in TM123 may explain the tendency of TM123 to aggregate.
FIGURE 6.

Water accessibility. Relative intensities of TM123 (top) and TM127 (bottom) HNCO peaks after addition of 15 mm Gd-(DTPA-BMA). Blue bars indicate residual intensities below 30%. Locations of the TM helices are shaded in gray.
We have additionally investigated the membrane-insertion topology of TM1 and TM12. Although the data for the extracellular portions look very similar to each other and to TM123 or TM127, less solvent protection is seen for TM1 in the isolated TM1 peptide relative to the protection observed for the same region in TM12, TM123, and TM127 (supplemental Fig. S11B), supporting the view that the first transmembrane helix is not stably integrated into the micelle in TM1. Furthermore, we compared Gd-(DTPA-BMA)-dependent attenuations of TM12 with and without the His6 tag (supplemental Fig. S11A), and found the data to be largely identical, ruling out the possibility that differences between TM123 and TM127 are due to the presence of the N-terminal His tag in TM127.
To conclude, our data demonstrate that an isolated TM1 polypeptide does not form a stable helix that fully integrates into the hydrophobic environment of a micelle. However, TM1 becomes significantly stabilized upon packing against TM2. To test whether these peptides dimerize or oligomerize in micelles we ran SEC analyses. Unfortunately, it was impossible to separate empty and TM1- or TM12-loaded micelles on the SEC column, and hence we were unable to perform an analysis similar to that done for TM123 and TM127. In addition, chemical cross-linking experiments carried out on TM1 and TM12 failed to detect any cross-linked products (data not shown). The data suggest that neither TM1 nor TM12 form a measurable amount of dimer in micelles. It is possible, however, that the bifunctional cross-linking agent is sterically unable to react with two chains. Therefore, based on these experiments we cannot unequivocally rule out stabilization of TM12 due to dimer formation. Adding a third TM does not result in a stable 3-TM folding core for TM123 or TM127. Rather, the presence of polar residues in the center of TM3 or the Pro residue in the center of TM7 results in large conformational transitions of the C-terminal half of TM3 or TM7, respectively. In the case of TM123 the center of TM3 is partially exposed to water during these transitions.
Incorporation of TM127 into Nanodiscs
To determine whether the above results were influenced by the fact that the polypeptides were integrated into micelles rather than bilayers, we have additionally attempted to incorporate TM127 into nanodiscs with different lipid compositions. The nanodisc system (42, 43), represents a true bilayer composed of lipids surrounded by a so-called membrane scaffold protein (MSP). We have utilized nanodiscs, in which the belt protein was truncated by one helix to decrease the size of the total assembly (the so-called MSP1D1ΔH5) (42). Lipids with phosphatidylglycerol (PG) head groups were used to mimic the negatively charged plasma membrane of Saccharomyces cerevisiae (44).
To investigate TM127 in membranes of different fatty acid chain lengths and degrees of unsaturation, several lipids were used for the nanodisc preparation (supplemental Fig. S12). Size exclusion chromatography in combination with multiangle light scattering revealed that in DMPG nanodiscs TM127 was incorporated in a monomeric form (supplemental Fig. S13). In contrast, nanodisc assemblies with other lipids did not integrate monomeric TM127 into intact nanodiscs, and hence will not be described in more detail. Assignment of the 15N,1H-TROSY spectrum of TM127 in DMPG nanodiscs revealed strong peaks for residues of loops and termini. In general, peaks in both the 15N,1H (supplemental Fig. S14A) and the 13C,1H correlation maps (supplemental Fig. S15A) for TM127 in nanodiscs were much broader in comparison to those in the micelle, even though the protein was perdeuterated (the sample we used was the ILV-labeled TM127). In sharp contrast, amide peaks from both loops and helices in bacteriorhodopsin (BR), a bacterial 7-TM protein, were of similar intensity and line width when incorporated into nanodiscs (supplemental Fig. S14B), although this protein is clearly larger. Similarly, ILV methyl groups in BR display comparatively sharp peaks and superior signal dispersion, whereas the peaks are fairly clustered and much broader in TM127 (supplemental Fig. S15). All these observations are consistent with significant conformational broadening of TM127 in the DMPG nanodiscs, indicating that the different conformers are interconverting more slowly in the intermediate regime. We attribute this to the fact that the nanodisc environment exerts stronger topological restraints on TM127 folding because the composition and orientation of lipids in this system is more fixed than that of the detergent molecules in micelles, and less likely to adapt to the protein.
Integration and Folding of Ste2p-derived Constructs into Biological Membranes
To investigate the topology of the N-terminal fragments of Ste2p in an in vivo-like environment, we used a co-translational insertion/glycosylation assay in the presence of microsomal membranes (45). In this assay, the locations of the N- and C-terminal ends relative to the ER membrane (46) are identified using N-linked glycosylation as the topological reporter (47). This modification is performed by the oligosaccharyl transferase (OST) complex, which is adjacent to the translocon (48). OST scans nascent polypeptides for consensus acceptor sequences after the polypeptide emerges from the translocon pore to add sugar residues co-translationally (49). Glycosylation of a polypeptide region translated in vitro in the presence of microsomal membranes reveals that this region of the nascent protein is exposed to the OST active site on the luminal side of the ER membrane (50). Such glycosylation is easily detected by an increase in molecular mass of about 2.5 kDa for each glycosylation site relative to the mass of the protein expressed in the absence of microsomes. Using this approach, the membrane topology of Ste2p was tested by in vitro translation of a series of Ste2p truncations (Fig. 7A) containing native and, in the appropriate cases, an optimized N-linked glycosylation C-terminal reporter tag (51).
FIGURE 7.
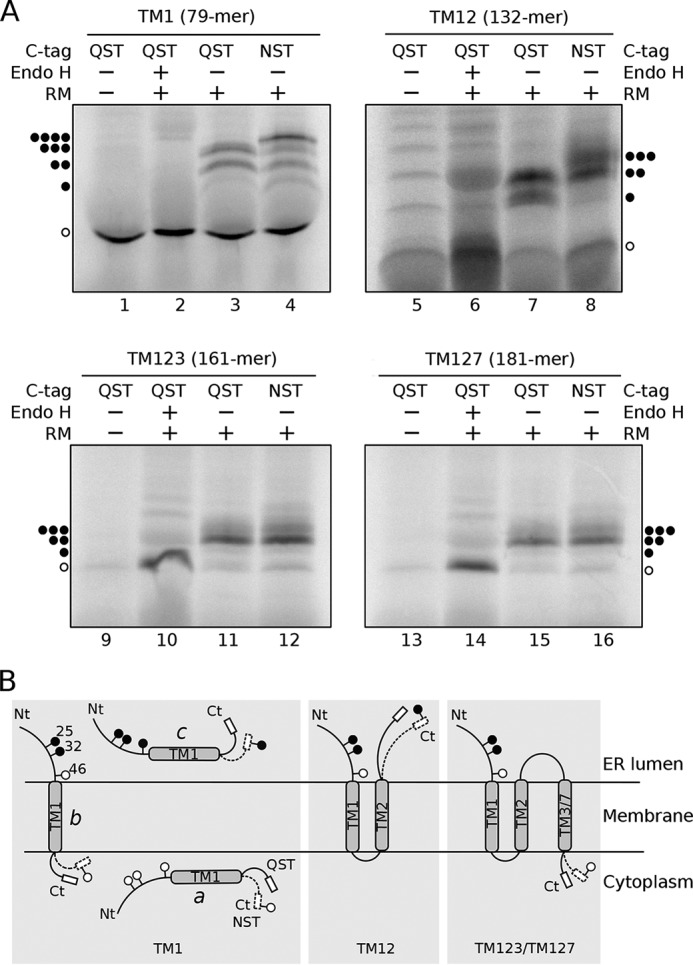
Membrane topology of TM1, TM12, TM123, and TM127 constructs. A, constructs TM1, TM12, TM123, and TM127 bearing a glycosylation acceptor site in the C-terminal (NST) or a mock site (QST) were in vitro transcribed and translated in the presence (+) or absence (−) of RM. Subsequently, samples were treated with endoglycosidase H (Endo H+) or mock treated (Endo H−). To determine the electrophoretic mobility samples were then run on a SDS-PAGE. The amino acid length of each construct is indicated in parentheses. The location of non-glycosylated protein is revealed with a single white dot, whereas one, two, three, or four black dots mark the presence of mono-, double-, triple-, and quadruple-glycosylated proteins, respectively. B, models of the membrane topology of truncated Step2-derived constructs. The QST and NST C-terminal tags are represented with solid and dotted lines, respectively. Glycosylation patterns are depicted for TM1 in the cytosolic (a) or periplasmic (c) locations as well as for transmembrane insertion (b).
The Ste2p sequence harbors 3 native glycosylation acceptor sites at the N terminus of the protein (Fig. 7B and supplemental Fig. S1) (positions 25, 32, and 46). These three acceptor sites are located preceding the first TM segment (TM1); subsequently, they can be used in all truncated Ste2p variants to monitor the location of the extramembranous N-terminal end. To test the location of the C-terminal end, we added an acceptor site (Asn-Ser-Thr, NST, Fig. 7) or a non-glycosylable (mock) sequence (Gln-Ser-Thr, QST, Fig. 7) at the C terminus of each truncated variant (52). When the truncated Ste2p carrying solely TM1 (residues 1–79), plus the non-glycosylable QST tag, was translated in vitro in the absence of membranes (rough microsomes, RM) we obtained a single polypeptide band corresponding to the non-glycosylated form (Fig. 7A, lane 1). In contrast, when this construct was translated in the presence of RM we observed higher molecular weight populations (Fig. 7A, lane 3). The nature of these higher molecular weight polypeptide species was analyzed by endoglycosidase H (Endo H) treatment, an enzyme that cleaves Asn-linked mannose oligosaccharides regardless of their localization. Treatment with Endo H eliminated all higher molecular mass bands (Fig. 7A, lane 2), confirming the sugar source of their retarded electrophoretic mobility.
Translation of TM1 carrying the mock C-terminal tag in the presence of membranes (lane 3) yielded mostly non-glycosylated polypeptides, suggesting that a high percentage is neither incorporated into membrane nor translocated into the ER lumen (Fig. 7B, scheme a). The doubly glycosylated population represents polypeptide molecules inserted into the membrane with an N-terminal luminal orientation (Fig. 7B, scheme b). One of the native glycosylation acceptor sites is located at position 46 (Asn46), only 6 residues prior to the N-terminal end of the TM1 helix (Ala52). Because Asn46 is closer to the membrane than the 14–15 residue minimal distance from the luminal boundary of a TM segment required for efficient glycosylation (53), it cannot be glycosylated by the membrane-bound OST (50). Therefore, insertion of TM1 into the membrane prevents the glycosylation of the acceptor site at Asn46 resulting in the addition of only two carbohydrate moieties at the N terminus of TM1. Finally, the presence of a band corresponding to triply glycosylated polypeptide molecules for the in vitro translation of the mock C-terminal labeled TM1 (lane 3) indicates that for some molecules all three N-terminal acceptor sites are modified, which can only happen if the truncated nascent polypeptides are fully translocated into the ER lumen (Fig. 7B, scheme c). These results were further corroborated by adding a glycosylation tag at the C terminus (Fig. 7B, NST C-tag, dotted line). Translation assays of this construct additionally revealed bands for the tetra-glycosylated forms (Fig. 7A, lane 4), confirming the existence of a population of species fully translocated into the ER lumen. Interestingly, even in the presence of the C-terminal glycosylation tag the presence of singly glycosylated molecules is negligible, excluding the existence of a significant population of proteins with an opposite topology (TM1 N-terminal cytosolic/C-terminal luminal).
Next, we tested the insertion of Ste2p truncations containing TM1 and TM2 (residues 1–132; TM12). When a construct, carrying the mock QST C-terminal tag was translated (Fig. 7A, lanes 5–7), essentially only glycosylated species were observed in the presence of microsomes (Fig. 7A, lane 7); in fact, a protein band corresponding to doubly glycosylated forms became predominant, whereas triply glycosylated forms were not observed. These results suggest that TM12 is being inserted into the membrane as a helical hairpin (26, 37), with both N and C termini oriented toward the ER lumen (Fig. 7B, central panel). When a glycosylable NST C-terminal tag was added (Fig. 7A, lane 8) the presence of triply glycosylated species supports the luminal orientation of the C terminus (Fig. 7B, central panel dotted line).
Subsequently, we tested TM123 (Fig. 7A, lanes 9–12) and compared the results with those of TM127 (Fig. 7A, lanes 13–16). Translation of both Ste2p-derived constructs rendered similar results, suggesting that swapping TM3 by TM7 does not significantly alter the membrane protein topology. Most of the glycosylated peptides (Fig. 7, lanes 11, 12, 15, and 16) are diglycosylated. The lack of non- and triply glycosylated protein forms when the constructs were translated in the presence of microsomes (Fig. 7, lanes 11 and 15) indicates that most of the proteins are properly inserted into the ER-derived membranes. In these cases, the addition of an NST C-terminal tag did not increase the apparent molecular weight of the chimeras, revealing that both TM3 and TM7 are being recognized by the translocon as truly TM segments (Fig. 7A, lanes 12 and 16) and efficiently inserted into the membrane with the NST C-terminal tag oriented toward the cytoplasm where it cannot be glycosylated (Fig. 7B, right panel).
Membrane Insertion of the Isolated TM1 and TM2 Segments
To test further the propensity of TM1 to insert into the ER membrane, we used an experimental system based on the E. coli inner membrane protein leader peptidase (Lep). Lep consists of two TM segments (H1 and H2) connected by a cytoplasmic loop (P1) and a large C-terminal domain (P2). Lep inserts into ER-derived microsomal membranes with both N and C termini facing the ER lumen (Fig. 8A, left). The region evaluated (TM-tested) is engineered into the luminal P2 domain and is flanked by two N-linked glycosylation acceptor sites (G1 and G2) (45). In this system, double glycosylation of the Lep derivatives indicates translocation of the tested segment across the membrane (Fig. 8A, right), whereas a single glycosylation denotes membrane integration (Fig. 8A, center) (52, 54).
FIGURE 8.

Insertion of STE2p TM1 into microsomal membranes. A, schematic representation of the leader peptidase (Lep) constructs used to report insertion into the ER membrane. The TM under study is inserted into the P2 domain of Lep, flanked by two artificial glycosylation acceptor sites (G1 and G2). The Lep chimera will be singly glycosylated when the TM being tested is inserted into the lipid bilayer and doubly glycosylated if it is translocated into the lumen of the microsomes. B, in vitro translation of Lep constructs in the presence (+) or absence (−) of RM. Control TM sequences were used to verify sequence translocation (lanes 1 and 2) and membrane integration (lanes 3 and 4) (52). Lep chimeras containing STE2p wild type TM1 sequence (lanes 5 and 6), or TM1 bearing R58L (lane 7), R58A (lane 8), R58E (lane 9), and S59L (lane 10) mutations are included. Bands of non-glycosylated protein forms are indicated by a white dot; singly and doubly glycosylated proteins are indicated by one and two black dots, respectively. C, comparison of in vitro translation of TM1 and TM2 constructs in the presence of rough microsomes together with insertion and translocation controls as described in B. D, the TM-tested sequence in each construct is shown accompanied by the corresponding insertion percentage (average of 3 independent experiments). Bold letters are used to highlight single residue mutations. The last two columns of the table display predicted (pred) and experimental (exp) ΔG values, respectively, in kcal/mol. Predictions were obtained with the ΔG prediction server on the basis of the amino acid sequence of the TM helix (18, 45).
The translation of chimeric constructs harboring the TM1 sequence of Ste2p in the presence of ER membranes produced primarily doubly glycosylated forms (Fig. 8B, lane 6). This result indicates a rather inefficient insertion (≈40%) of this Ste2p region when studied of its native context (Fig. 8). In contrast, the isolated TM2 sequence is much more efficiently inserted into the microsomal membrane (≈80%) (Fig. 8, C and D).
This observation is consistent with the results of our membrane topology study (Fig. 7). An in silico analysis of the TM1 sequence with the ΔG prediction algorithm revealed Arg58, followed by Ser59, as residues with a higher energy penalty for membrane insertion (supplemental Table S3). Substitution of Arg58 by hydrophobic residues such as Leu (R58L) or Ala (R58A) lowered the predicted ΔGapp (Fig. 8D) and increased the experimental percentage of insertion from 36 to 85 and 83%, respectively (Fig. 8B, lanes 7 and 8). Similarly, the replacement of Ser59 by Leu (S59L) raised the insertion percentage to 72% (Fig. 8B, lane 10). In contrast, replacing Arg58 by a negatively charged residue (Glu) lowered the insertion efficiency to 30% (Fig. 8, lane 9). Our results demonstrated that Ste2p TM1 is not efficiently recognized by the translocon as an independent TM segment, most likely, due to its low hydrophobicity (positive predicted ΔGapp value, Fig. 8D). TM2 is significantly more hydrophobic (see Fig. 8 and supplemental Table S3) and hence inserts more efficiently (Fig. 8, C and D). Once the second TM segment is synthesized during the biogenesis of the protein, the overall hydrophobicity of the TM12 polypeptide is higher and therefore both TM segments are properly inserted into the membrane and assume a native hairpin topology (Fig. 7, TM12 construct).
Discussion
Recent years have witnessed a dramatic increase in the amount of structural information on GPCRs. Detailed information on the ground and excited states, as well as on the mechanism of activation of GPCRs has been derived from crystallographic studies and MD simulations. However, a detailed picture of how these proteins actually fold is not yet available, partly due to the fact that crystallography cannot describe conformational preferences of folding intermediates. Our approach to understand the folding of a model GPCR, Ste2p, is based on the study of a series of overlapping N-terminal fragments of increasing size. Using solution NMR in micelles to determine the structure of these fragments and in vitro biochemical assays to determine the insertion and topology of the fragments into biological membranes we expect to learn about the conformational preferences of GPCR segments as the elongating polypeptide chain emerges from the ribosome during early stages of protein biosynthesis.
Conformational preferences of fragments of GPCRs have been investigated before. Yeagle and co-workers (55, 56) determined the structures of individual helices and loops of rhodopsin, and used the structure of these fragments to restrain a model of the entire receptor. Arseniev and co-workers (57–59) determined conformational preferences of the individual and combined first two TM regions of BR in organic solvents and SDS micelles. The Khorana group (60), in their seminal work, showed that BR could be cleaved enzymatically into two fragments, which assemble into a proton-translocating complex in the presence of retinal. Similarly, Dumont and co-workers (61) demonstrated that two complementary Ste2p fragments, which represented this GPCR split at every cytosolic or extracellular loop, could form a functional receptor when co-expressed. Although major contributions of loops for folding have been claimed in the case of mammalian rhodopsin (62), the above mentioned studies by the Khorana and Dumont groups (60, 61) and work on other polytopic membrane proteins (e.g. Lac permease) attribute a pivotal role to the formation of inter-helical contacts.
TM helices represent the building blocks of the cores of integral membrane proteins. Elegant studies on model helices such as glycophorin A indicate that the driving forces to form helical bundles are contained solely in the helical regions (63, 64). Connecting loops bring the TM helices of GPCRs into spatial proximity, increase their effective local concentration, and thereby facilitate formation of inter-helical contacts. The central question this study addresses is whether mutual interactions of multiple helices in the nascent GPCR increase the stability of the secondary structure of these helices and promote their proper integration into the hydrophobic core. Our working hypothesis was that the presence of polar residues in central positions of individual TMs would be in a thermodynamically unfavorable state in the lipid core and must, therefore, be stabilized through interhelical interactions. To investigate this question, fragments were required that contain TM helices that form contacts in the fully reconstituted receptor. Our analysis of GPCR structures solved by X-ray crystallography and of a homology model of Ste2p revealed such contacts between TM1, TM2, and TM3 at the N terminus. In addition, in all GPCR structures TM7 forms contacts with TM1 and TM2. Therefore, we chose also to investigate the conformation of the TM127 construct as well, realizing that the connecting loop is non-native.
Our NMR data reveal that the isolated first helix from the Ste2p receptor does not stably integrate into detergent micelles. This observation is supported by the glycosylation assays that employ biological microsomes purified from eukaryotic cells. The latter assays, on carefully designed TM1 mutants, also confirmed the assumption that polar or charged residues are responsible for the non-efficient membrane integration. The longer constructs described in this study were designed to test whether addition of one or more helices, which in the entire receptor form direct contacts with TM1, would help to stabilize this transmembrane domain and improve its integration into bilayers. Indeed, addition of TM2 to TM1 stabilizes the structure of TM1, potentiates its insertion into bilayers, and results in formation of a relatively stable helical hairpin. Unfortunately, addition of TM3 to the TM12 construct results in a protein (TM123) that tends to strongly oligomerize, and which therefore could not be investigated in detail. Interestingly, early CD studies on isolated Ste2p TM3-derived peptide both in SDS micelles and DMPC bilayers concluded that this peptide has a tendency to aggregate into β-sheet-like structures (65). As stated above, TM7 also forms many contacts with TM1 and TM2 (Fig. 1), and is strongly helical as an isolated peptide in micelles and bilayers (65). The NMR analysis of TM127 indicated that, despite the fact that it remained perfectly monomeric, TM7 did not stably pack against TM1 and/or TM2. The glycosylation assays, however, indicated that most of TM127 does display the correct membrane insertion/topology, and this was also the case for TM123. We conclude that 3-TM constructs can form metastable bundles that can fold with the correct topology in a true membrane environment. However, additional contacts with other TMs appear to be necessary to form stable native-like folds and to prevent aggregation.
What factors are critical in determining membrane protein folding and insertion? The simple two-stage model for the folding of helical membrane proteins postulated by Popot and Engelman (10, 11) proposes that secondary structure forms once the entire TM segment partitions into the membrane. A rather surprising recent discovery is that the biological scale for partitioning amino acids between the translocon and the ER membrane (45) is very similar to the biophysical data for partitioning between water and lipid bilayers (17). Perhaps based in part on this finding Popot and Engelman (10, 11) entitled their recent commentary on membrane protein folding “Membranes do not tell proteins how to fold,” emphasizing that membrane protein folding is primarily encoded by the amino acid sequence and largely independent of the specifics of the environment as long as it is sufficiently hydrophobic (66). Accordingly, membrane protein folding in their view is also not determined by specific properties of the translocon.
Helical TM-bundle proteins often contain polar or even charged residues at internal positions. Some of the individual helices are only marginally stable even in a membrane environment. In the folded TM-bundle the polar or charged residues form contacts with complementary groups in other TM helices. These contacts contribute to the assembly of the TM-bundle, and in particular to the specificity of inter-helical contacts (25, 28, 67, 68). Insertion of single TM helices into the hydrophobic core exposes these polar moieties to the lipids, an unfavorable interaction that is expected to expel the corresponding part from the membrane interior and transfer it to the interface. Krishnamani and Lanyi (69) performed MD simulations of individual BR helices in SDS micelles. Their data indicated that partitioning of the helices into the hydrophobic core largely depended on the hydrophobicity of the individual helices, and the presence of charged (but not polar) residues prevented full insertion of the TM portions into the hydrophobic core of the membrane and resulted in significant destabilization of secondary structure. We observed in this study that the first helix is significantly destabilized around the central polar GVRSG motif. Packing of TM2 against TM1 apparently stabilizes secondary structure in TM1 as demonstrated by the fact that TM12 forms a much more stable α-helical hairpin (Figs. 3 and 7). We expected that adding another TM helix would even stabilize the fragment further. In fact, we observed formation of a comparably stable hydrophobic core in TM127, but not in TM123, as evident, for example, from penetration of water into central parts of TM3.
Considering the possibility that individual helices may not be sufficiently stable in a fully membrane-inserted mode and do require inter-acting partners to remain inserted, timing of the release of TM segments into the membrane is important. Proteins are synthesized starting at the N terminus, and the co-translational folding model postulates that folding occurs after the nascent chain is released via the translocon into the membrane compartment (70, 71). Our NMR data supported by the microsomal topology studies, however, indicate that TM1 does not exist in one well defined conformation, and is not efficiently membrane-integrated on its own, whereas TM2 more efficiently inserts into the membrane. Addition of TM2 therefore significantly stabilizes TM1 in TM12, TM123, and TM127. These results and the biochemical topology studies would be consistent with integration of the growing polypeptide into the bilayer only after several TM helices have been biosynthesized.
Structural analysis of the TM127 construct concludes that TM7 does not stably pack against TM1 and TM2. In particular, hinge motions about Leu-Pro-Leu in TM7 likely exist as inferred from the fact that amide signals for the two Leu residues are broadened beyond detection, and that both straight and kinked TM7 conformers are obtained in the structure calculations. Moreover, the close to parallel alignment of the TM helices that is observed in the crystal structures of full GPCRs is not observed in the detergent micelle. TM127 rather seems to form a rapidly interconverting set of conformers, in which TM7 packs in multiple ways against the hydrophobic core of TM1 and TM2, forming a loosely packed 3-TM helix bundle in micelles (Fig. 5), and likely also in nanodiscs as inferred from the observed exchange-broadening of TM residues. Based on the paramagnetic broadening experiments with the Gd-based reagent (Fig. 6), in micelles we can exclude any bundle in which the individual three TM127 helices present lipid-associated but well separated surface-associated entities. This is despite the presence of various polar or even charged residues within the TM helices. Although we cannot present a structure of TM123 at present, it seems to be structurally more inhomogeneous than TM127, existing in various oligomeric states, as TM3 is not well integrated into the hydrophobic core.
Recently we performed an exhaustive topological study on folding of N and C terminally truncated forms of the Y4 receptor, a human GPCR (29). The data from this study indicated that dual topologies (C terminus in and out) are more likely to occur for short fragments (e.g. TM1 or TM12), whereas more unique and correct topologies were encountered for proteins that comprise most of the TM helices (e.g. TM1–6). Again, we suspected that the presence of uncompensated polar or charged residues at central positions within the TM helices is responsible for that behavior.
What happens to helices that do not readily insert into the membrane because polar or charged residues prevent insertion? In the case of a sequential exit from the translocon, our data suggest that such helices (e.g. TM1 from the Ste2p receptor) will localize, at least to some extent, in the interface, and, due to their hydrophobicity, will always remain associated with the membrane in some way. In principle, the isolated TM1 may also exist in a V-shaped arrangement in which two membrane-inserted short hydrophobic helices are connected by a more polar linker that is located in the interface. Although such a scenario cannot be ruled out, the reduced secondary chemical shifts and the lack of long-range NOEs between these helices indicate that the overall topology of TM1 is rather flexible. Similarly, our data indicate that also central parts of TM3 have solvent access indicating that TM3 is not fully inserted into the membrane. Would such interfacial helices later be pulled back into the membrane interior when their interacting partners become available? Although we cannot exclude this scenario per se, the fact that refolding of GPCRs, in general, is very difficult and that they denature irreversibly indicates that TM domains that initially misfold would likely not spontaneously fold correctly into the hydrophobic core when interacting helices become available. Alternatively, nascent helices may accumulate within or near the translocon, where the bundle is pre-assembled and then fully released into the membrane. Photocross-linking experiments demonstrated that TM helices formed either specific or nonspecific contacts with residues from the translocon (72). Interestingly, these experiments also revealed that TM helix bundles leave the translocon and enter the lipid bilayer in a concerted manner (73–76). High and co-workers (77) using chemical cross-linking methodology demonstrated that large portions of opsin containing multiple TM domains remain bound or associated to the translocon. The rate of release of these portions depended more on their amino acid sequence than on their position in the protein. In this concerted model the nascent chain remains within the translocon or attached to other translocon-associated components until the protein has been fully synthesized (for a more general discussion of the issue see also the review by Skach (71)).
In the experiments described in this work we have probed the conformational preferences of overlapping N-terminal polypeptides of increasing length of a yeast GPCR. Thereby we were able to obtain biophysical evidence about the putative behavior of these protein fragments when they are released into the membrane hydrophobic core from the translocon. Based on the fact that TM1 does not form a stable helix and does not integrate well into micelles or microsomal membranes and that TM123 and TM127 do not form stable three-helix bundles it is reasonable to conclude that large portions of the nascent receptor remain either within the translocon or associated with nearby proteins until compensatory tertiary intramolecular contacts between polar or charged residues are available. Otherwise, some of these TM helices would not remain fully inserted in the membrane core, instead they would accumulate at the interface and might be prone to form aggregates as found for TM123. The exact folding pathway of a GPCR will likely depend on its the amino acid sequence, and hence may vary from receptor to receptor.
Finally we believe that the more detailed structural picture of membrane protein folding that can be derived from the NMR data, as compared with using conventional folding/unfolding studies of entire GPCRs or when using only topology mapping experiments (see below), provides important additional insights into GPCR folding. Given the rather complex pathway a membrane polypeptide must take from ribosomal synthesis, to membrane insertion and folding, a combination of biochemical and biophysical methods is essential to fully decode these latter processes.
Experimental Procedures
Chemicals and Solutions
15NH4Cl, [13C-d7]d-glucose, and D2O were purchased from Spectra Stable Isotopes (Andover, MA). LPPG and DPC were bought from Avanti Polar Lipids (Alabaster, AL). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), and 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) were purchased from Anatrace Inc. Bio-BeadsTM SM-2 Resin was bought from Bio-Rad. All other chemicals were obtained from Sigma (Buchs, Switzerland).
The pGEM1 plasmid, rabbit reticulocyte lysate, and the TnT-coupled transcription/translation system were purchased from Promega (Madison, WI). The ER rough microsomes from dog pancreas and the SP6 RNA polymerase were purchased from tRNA Probes (College Station, TX). The [35S]Met/Cys labeling mix was purchased from PerkinElmer Life Sciences (Waltham, MA). The restriction enzymes and endoglycosidase H were purchased from New England Biolabs (Ipswich, MA). The DNA plasmid, RNA clean up, and PCR purification kits were from Qiagen (Hilden, Germany). The PCR mutagenesis kit, QuikChange was from Stratagene (La Jolla, CA). All the oligonucleotides were purchased from Thermo (Ulm, Germany).
Expression and Purification
Cloning procedures are described under supplementary materials. Ste2p constructs were expressed with ΔTrpLE as a N-terminal fusion as previously described (34). In most constructs the N terminus of Ste2p was truncated at residue 30 (78). Small scale growths in BL21-AI cells (for TM1 and TM123) and BL21 Star(DE3)pLysS (for TM127) were used to optimize expression. Inoculation was carried out at an A600 of 0.5–1.0, and cultures were subsequently cooled to 20–22 °C and harvested after 16–22 h. Expression was carried out in M9 minimal medium with uniform 15N/13C/2H, 15N/13C, or selective reverse methyl labeling (supplementary materials). Additionally, the TM1 (BL21-AI) and TM127 (BL21(DE3)) constructs were expressed directly containing the full N terminus (and N-terminal His6 tag in case of TM127) at 37 and 22 °C, respectively. Preparation of inclusion bodies, cyanogen bromide cleavage of the ΔTrpLE sequence, and subsequent RP-HPLC purification were carried out as described previously (36). Direct expression products were purified analogously without the chemical cleavage step. For further details of expression and purification, see supplementary materials.
Coupling of the MTSL spin label to single cysteine mutants was achieved by first dissolving 0.4 mm protein in 40 mm Tris/HCl buffer at pH 7.5 containing 6 m guanidinium hydrochloride, followed by addition of 2 mm dithiothreitol (DTT) and gentle shaking for 4 h at room temperature to assure complete reduction of cysteines. After the solution was purged with nitrogen for several minutes to remove oxygen, a 15- to 20-fold excess of the MTSL spin label dissolved in DMSO was added, and the solution was incubated at room temperature overnight. The reaction mixture was injected directly onto a nickel-nitrilotriacetic acid column and washed with about 100 column volumes of the reaction buffer to remove unreacted spin label and residual DTT. The reaction product was eluted with 500 mm imidazole after another wash step with 10 mm imidazole, and collected fractions were dialyzed against water and lyophilized. For NMR sample preparation the protein was dissolved in hexafluoroisopropanol (HFIP)/H2O (4:1, v/w), and half of the solution was deactivated with 10 mm ascorbic acid and afterward neutralized with sodium hydroxide. Subsequent sample preparation was carried out as described below.
The expression and purification of the nanodisc membrane scaffold protein MSP1D1ΔH5 was carried out based on a published protocol (79). The MSP/lipid stoichiometry was optimized for each lipid empirically using size exclusion chromatography in combination with multiangle light scattering (SEC-MALS). Nanodiscs carrying TM127 were assembled based on an established protocol (80). Briefly, TM127 and the particular lipid solubilized in SDS as well as the MSP were mixed with a 5-fold excess (relative to the membrane protein concentration) of empty nanodiscs. Bio-Beads were added to remove the SDS thereby inducing the self-assembly. After the formation of nanodiscs was complete, Bio-Beads were removed by filtration, and nanodiscs containing TM127 were isolated using a nickel-nitrilotriacetic acid column. After buffer exchange, TM127 nanodiscs were concentrated and characterized by SEC-MALS (for more details, see supplemental materials).
NMR Sample Preparation
NMR samples contained 40 mm K3PO4 buffer (pH 6.4) and 120 mm LPPG, 30 mm DPC, and were produced using a protocol slightly modified from the one described by Killian et al. (81). The protein was first dissolved in small amounts of HFIP/water (8:1), whereas detergents were dissolved in a phosphate buffer equivalent to their final sample concentrations. After mixing the two solutions, water was added stepwise to dilute the organic solvent and to allow micelles to form. After two lyophilization steps, the sample was taken up in 250 μl of H2O/D2O (9:1). In the case of TM123, several cycles of dissolving the sample in HFIP and H2O and subsequent lyophilization were necessary to achieve acceptable protein integration into the micelle.
Gd-(DTPA-BMA) was added from a ×100 stock solution to prepare samples that were used to probe micelle integration. Gd-(DTPA-BMA) concentrations of 5, 10, and 15 mm were measured and compared with a blank of 1 mm to reduce effects resulting from T1 relaxation.
RDC samples were prepared as follows. The dinucleotide 2′-deoxyguanylyl-(3′,5′)-2′-deoxy-guanosine (dGpG) was added as a powder to a freshly prepared NMR sample containing [15N/13C/2H]His-N-TM127 and LPPG/DPC (4:1) in 40 mm K3PO4 buffer (pH 6.4). KCl was then added from a concentrated stock solution in the same buffer and complete dissolution required several heating-cooling (45−4 °C) cycles. The volumes and weights were adjusted so that the final concentrations of dGpG, [15N/13C/2H]His-N-TM127, LPPG/DPC, and KCl were 30 mg/ml, 0.4 mm, 150 mm, and 100 mm, respectively. The KCl was necessary to ensure G-tetrade formation. The formation of a stable liquid crystal phase was monitored by observing the residual 2H2O quadrupolar coupling on the solvent signal.
NMR Spectroscopy
All samples for assignment purposes were measured on a Bruker AV700 spectrometer equipped with a triple-resonance cryoprobe at 317 K. NMR samples contained 0.2–0.4 mm protein in 40 mm phosphate buffer (pH 6.4), 120 mm LPPG, and 30 mm DPC as described previously (37). Proton chemical shifts were referenced to the water line at 4.47 ppm at 317 K, from which the nitrogen and carbon scales were derived indirectly by using the conversion factors of 0.10132900 (15N) and 0.25144954 (13C).
Backbone assignments were obtained from standard TROSY-type triple resonance experiments. Briefly, an HNCO data set was used to pick peaks in the 15N,1H-TROSY spectrum to recognize peak overlap, and to adjust peak positions in cases of peak overlap. Backbone and partial side chain assignments were then obtained from HNCACB and HN(CO)CACB experiments (or HNCA and HN(CO)CA) spectra. Side chain resonance assignment was accomplished using hCCH-TOCSY/COSY (82, 83) in combination with 13C,1H-HSQC and aliphatic/aromatic 13C-resolved NOESY experiments. Spectra for side chain assignments required the use of d36-LPPG and d38-DPC to eliminate the strong residual signals from detergent. All chemical shifts were finally correlated to peak positions in the 15N,1H- and 13C,1H-HSQC spectra.
For the automatic shift adaptation of TM1-TM2 in TM127, signals of the three-dimensional 15N-NOESY spectra were picked according the location of the parent amide peaks in the corresponding 15N,1H-HSQC spectra. The resulting strips were then matched in the following way. For each assigned “old” strip (from TM12) a number of potential “new” strips (from TM127) are pre-selected. This selection was based on the 1H(F3) and 15N(F2) shifts of the TM12 strip, defining a spectral window, within which all potential new strips were searched. This window was larger for residues at the beginning and the tail of the sequence. The set of new strips are then compared with the old strip by calculating chemical shift differences, and the best match is the one displaying the smallest differences. The procedure will be described in more detail elsewhere.
To evaluate PREs, the ratio of peak intensities in 15N,1H-HSQC spectra measured on freshly prepared samples was computed relative to samples in which the MTSL label was deactivated by ascorbic acid. Peaks that were not sufficiently separated and therefore could not be integrated reliably were omitted from further analysis.
Structure Calculations
Distance restraints were obtained from 15N-resolved NOESY spectra recorded on 15N/1H- and 15N/2H-labeled Ste2p TM127 samples with mixing times of 70 and 200 ms, respectively, and from 13C-resolved NOESY spectra with 100-ms mixing time. In addition, dihedral angle restraints obtained using the TALOS-N program (84), which uses chemical shifts of 1Hα, 13Cα, 13Cβ, 13C′, and backbone 15N nuclei, were added. Unassigned integrated peak lists from UNIO′10 were transferred to CYANA, which assigned the peak list in seven iterative cycles using the built-in macro “noeassign” (85). The final CYANA calculation was performed with 100 randomized starting structures, and the 20 CYANA conformers with the lowest target function values were selected to represent the NMR ensemble.
Three classes of PREs were distinguished. Strong PREs, with less than 10% of the original signal intensity, yielded 26 PRE restraints with an upper distance limit of 14.0 Å. Medium PREs, with residual peak intensities of 10–80%, yielded 49 distance restraints with a lower limit of 14 Å and an upper limit of 20 Å, and unaffected residues, with residual intensities larger than 80%, 48 distance restraints with a lower limit of 20 Å. PRE distance restraints required cross-checking for compliance with the short- to medium-range NOE restraint network, as well as the torsion angle restraints. 63 RDC restraints complemented the PRE-derived distance restraints.
To obtain a representative statistical analysis of the different conformations of TM127 we performed calculations starting from 5000 structures and clustering the 500 structures with the lowest energies based on their backbone root mean square deviations (<3 Å) in the helical regions. This resulted in 48 clusters that contained up to 95 structures. The four most prominent clusters with at least 30 structures have been analyzed by back-calculation of the raw PRE and RDC data and subsequently compared with the experimental values, as well as to the back-calculated values for the structure based on a homology model (for more details, see supplemental materials) (40). Important parameters of the structure calculations are summarized in supplemental Tables S1 and S2.
Glycosylation Assays
Truncated Ste2p constructs were obtained by using reverse primers at defined positions either with QST or NST C-terminal tags followed by tandem stop codons. mRNAs were transcribed from the SP6 promoter with SP6 RNA polymerase (tRNA probes) according to the manufacturer's instructions. The mRNA products were purified with a Qiagen RNeasy clean up kit and verified on a 1% agarose gel. In vitro translation of in vitro transcribed mRNA was performed in the presence of reticulocyte lysate, [35S]Met/Cys, and dog pancreas microsomes, as described previously (54). After translation, membranes were analyzed by SDS-PAGE. Finally, the gels were visualized on a Fuji FLA3000 phosphorimager with ImageGauge software. For Endo H treatment, the translation mixture was diluted in 4 volumes of 70 mm sodium citrate (pH 5.6) and centrifuged (100,000 × g for 20 min at 4 °C). The pellet was then resuspended in 50 μl of sodium citrate buffer with 0.5% SDS and 1% β-mercaptoethanol, boiled for 5 min, and incubated for 1 h at 37 °C with 0.1 milliunit of Endo H (52). The samples were analyzed by SDS-PAGE. Full-length Lep constructs were transcribed and translated in the TnT Quick system (Promega). Briefly, 1 μg of DNA template, 1 μl of [35S]Met/Cys (5 μCi), and 0.4 μl of microsomes (tRNA Probes) were added to 10 μl of TnT mixture at the start of the reaction, and samples were incubated for 90 min at 30 °C. Translation products were analyzed as previously described for the truncated molecules (86).
Quantification of the fractions of singly glycosylated (f1g) and doubly glycosylated (f2g) proteins allows calculation of the apparent equilibrium constant, Kapp, for the membrane insertion of a given TM sequence, Kapp = f1g/f2g. The Kapp value can be converted into the apparent free energy difference between the non-inserted state and the inserted state with the formula ΔGapp = −RT lnKapp, where R is the gas constant (r = 1.986 kcal K−1 mol−1), and T is the absolute temperature (T = 303 K).
Author Contributions
M. P. expressed proteins and analyzed NMR data on TM123 and TM127. S. J. developed tools for automatic resonance adaptations. K. E. F. and L. S. C. expressed proteins TM1, TM12, and TM123, and K. E. F. analyzed NMR data of TM1. L. M.-G. conducted the topology study. P. G. and D. G. performed structure calculations on TM127. P. G., I. M., F. N., and O. Z. supervised research, analyzed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Gunnar Jeschke for EPR measurements and Gerhard Wagner and Franz Hagn for providing the plasmid for the MSP.
This work was supported in part by Swiss National Science Foundation Grant 31003A_124469, Spanish Ministry of Economy and Competitiveness Grant BFU2016-79487, and Generalitat Valenciana Grant PROMETEOII/2014/061. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S15 and Tables S1–S3.
M. Poms, S. Jurt, P. Güntert, and O. Zerbe, unpublished data.
- GPCR
- G-protein coupled receptor
- TM
- transmembrane
- ER
- endoplasmic reticulum
- RP-HPLC
- reverse phase-HPLC
- LPPG
- 1-palmitoyl-2-hydroxy-sn-glycero-3-(phospho-rac-(1-glycerol))
- DPC
- dodecyl-phosphocholine
- SEC-MALS
- size exclusion chromatography-multiangle light scattering
- RDC
- residual dipolar coupling
- PRE
- paramagnetic relaxation enhancement
- MTSL
- S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate
- Gd-(DTPA-BMA)
- gadolinium diethylenetriamine-pentaacetic acid bis-methyl-amine
- MSP
- membrane scaffold protein
- PG
- phosphatidylglycerol
- BR
- bacteriorhodopsin
- OST
- oligosaccharyl transferase
- Endo H
- endoglycosidase H
- DMPG
- 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- HFIP
- hexafluoroisopropanol
- RM
- rough microsomes
- HSQC
- heteronuclear single quantum coherence
- OST
- oligosaccharyl transferase.
References
- 1. Lefkowitz R. J. (2013) A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 52, 6366–6378 [DOI] [PubMed] [Google Scholar]
- 2. Kobilka B. (2013) The structural basis of G-protein-coupled receptor signaling (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 52, 6380–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., and Miyano M. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 4. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., and Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 5. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., and Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 6. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., and Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H., Zhang C., et al. (2015) Crystal structure of rhodopsin bound to arrestin by fs X-ray laser. Nature 523, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelman D. M., Chen Y., Chin C. N., Curran A. R., Dixon A. M., Dupuy A. D., Lee A. S., Lehnert U., Matthews E. E., Reshetnyak Y. K., Senes A., and Popot J. L. (2003) Membrane protein folding: beyond the two stage model. FEBS Lett. 555, 122–125 [DOI] [PubMed] [Google Scholar]
- 10. Popot J. L., and Engelman D. M. (1990) Membrane protein folding and oligomerization: the two-stage model. Biochemistry 29, 4031–4037 [DOI] [PubMed] [Google Scholar]
- 11. Popot J. L., and Engelman D. M. (2000) Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69, 881–922 [DOI] [PubMed] [Google Scholar]
- 12. White S. H., and von Heijne G. (2008) How translocons select transmembrane helices. Annu. Rev. Biophys. 37, 23–42 [DOI] [PubMed] [Google Scholar]
- 13. Martínez-Gil L., Saurí A., Marti-Renom M. A., and Mingarro I. (2011) Membrane protein integration into the endoplasmic reticulum. FEBS J. 278, 3846–3858 [DOI] [PubMed] [Google Scholar]
- 14. Bhushan S., Gartmann M., Halic M., Armache J. P., Jarasch A., Mielke T., Berninghausen O., Wilson D. N., and Beckmann R. (2010) α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 17, 313–317 [DOI] [PubMed] [Google Scholar]
- 15. Mingarro I., Nilsson I., Whitley P., and von Heijne G. (2000) Different conformations of nascent polypeptides during translocation across the ER membrane. BMC Cell Biol. 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Woolhead C. A., McCormick P. J., and Johnson A. E. (2004) Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell 116, 725–736 [DOI] [PubMed] [Google Scholar]
- 17. Wimley W. C., and White S. H. (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nature Struct. Biol. 3, 842–848 [DOI] [PubMed] [Google Scholar]
- 18. Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., and von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 [DOI] [PubMed] [Google Scholar]
- 19. Baeza-Delgado C., von Heijne G., Marti-Renom M. A., and Mingarro I. (2016) Biological insertion of computationally designed short transmembrane segments. Sci. Rep. 6, 23397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White S. H. (2003) Translocons, thermodynamics, and the folding of membrane proteins. FEBS Lett. 555, 116–121 [DOI] [PubMed] [Google Scholar]
- 21. Whitley P., and Mingarro I. (2014) Stitching proteins into membranes, not sew simple. Biol. Chem. 395, 1417–1424 [DOI] [PubMed] [Google Scholar]
- 22. Bowie J. U. (2013) Structural biology: membrane protein twists and turns. Science 339, 398–399 [DOI] [PubMed] [Google Scholar]
- 23. Lu Y., Turnbull I. R., Bragin A., Carveth K., Verkman A. S., and Skach W. R. (2000) Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol. Biol. Cell 11, 2973–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Virkki M. T., Agrawal N., Edsbäcker E., Cristobal S., Elofsson A., and Kauko A. (2014) Folding of Aquaporin 1: multiple evidence that helix 3 can shift out of the membrane core. Protein Sci. 23, 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bañó-Polo M., Martínez-Gil L., Wallner B., Nieva J. L., Elofsson A., and Mingarro I. (2013) Charge pair interactions in transmembrane helices and turn propensity of the connecting sequence promote helical hairpin insertion. J. Mol. Biol. 425, 830–840 [DOI] [PubMed] [Google Scholar]
- 26. Chamberlain A. K., Faham S., Yohannan S., and Bowie J. U. (2003) Construction of helix-bundle membrane proteins. Adv. Protein Chem. 63, 19–46 [DOI] [PubMed] [Google Scholar]
- 27. Gratkowski H., Lear J. D., and DeGrado W. F. (2001) Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. U.S.A. 98, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Senes A., Engel D. E., and DeGrado W. F. (2004) Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 14, 465–479 [DOI] [PubMed] [Google Scholar]
- 29. Marino J., Geertsma E. R., and Zerbe O. (2012) Topogenesis of heterologously expressed fragments of the human Y4 GPCR. Biochim. Biophys. Acta 1818, 3055–3063 [DOI] [PubMed] [Google Scholar]
- 30. Shao X., Zou C., Naider F., and Zerbe O. (2012) Comparison of fragments comprising the first two helices of the human Y4 and the yeast Ste2p G-protein-coupled receptors. Biophys. J. 103, 817–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kahn T. W., and Engelman D. M. (1992) Bacteriorhodopsin can be refolded from two independently stable transmembrane helices and the complementary five-helix fragment. Biochemistry 31, 6144–6151 [DOI] [PubMed] [Google Scholar]
- 32. Drew D., Sjöstrand D., Nilsson J., Urbig T., Chin C. N., de Gier J. W., and von Heijne G. (2002) Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc. Natl. Acad. Sci. U.S.A. 99, 2690–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miozzari G. F., and Yanofsky C. (1978) Translation of the leader region of the Escherichia coli tryptophan operon. J. Bacteriol. 133, 1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohen L. S., Becker J. M., and Naider F. (2010) Biosynthesis of peptide fragments of eukaryotic GPCRs in Escherichia coli by directing expression into inclusion bodies. J. Pept. Sci. 16, 213–218 [DOI] [PubMed] [Google Scholar]
- 35. Martin N. P., Celić A., and Dumont M. E. (2002) Mutagenic mapping of helical structures in the transmembrane segments of the yeast α-factor receptor. J. Mol. Biol. 317, 765–788 [DOI] [PubMed] [Google Scholar]
- 36. Caroccia K. E., Estephan R., Cohen L. S., Arshava B., Hauser M., Zerbe O., Becker J. M., and Naider F. (2011) Expression and biophysical analysis of a triple-transmembrane domain-containing fragment from a yeast G protein-coupled receptor. Biopolymers 96, 757–771 [DOI] [PubMed] [Google Scholar]
- 37. Neumoin A., Cohen L. S., Arshava B., Tantry S., Becker J. M., Zerbe O., and Naider F. (2009) Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophys. J. 96, 3187–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zou C., Kumaran S., Markovic S., Walser R., and Zerbe O. (2008) Studies of the structure of the N-terminal domain from the Y4 receptor, a G-protein coupled receptor, and its interaction with hormones from the NPY family. ChemBioChem 9, 2276–2284 [DOI] [PubMed] [Google Scholar]
- 40. Eilers M., Hornak V., Smith S. O., and Konopka J. B. (2005) Comparison of class A and D G protein-coupled receptors: common features in structure and activation. Biochemistry 44, 8959–8975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fitzkee N. C., and Bax A. (2010) Facile measurement of 1H-15N residual dipolar couplings in larger perdeuterated proteins. J. Biomol. NMR 48, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hagn F., Etzkorn M., Raschle T., and Wagner G. (2013) Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 135, 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Chapter 11: reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van der Rest M. E., Kamminga A. H., Nakano A., Anraku Y., Poolman B., and Konings W. N. (1995) The plasma membrane of Saccharomyces cerevisiae: structure, function, and biogenesis. Microbiol. Rev. 59, 304–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 [DOI] [PubMed] [Google Scholar]
- 46. Tamborero S., Vilar M., Martínez-Gil L., Johnson A. E., and Mingarro I. (2011) Membrane insertion and topology of the translocating chain-associating membrane protein (TRAM). J. Mol. Biol. 406, 571–582 [DOI] [PubMed] [Google Scholar]
- 47. Nilsson I., and von Heijne G. (1990) Fine-tuning the topology of a polytopic membrane protein: role of positively and negatively charged amino acids. Cell 62, 1135–1141 [DOI] [PubMed] [Google Scholar]
- 48. Nilsson I., Kelleher D. J., Miao Y., Shao Y., Kreibich G., Gilmore R., von Heijne G., and Johnson A. E. (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell Biol. 161, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruiz-Canada C., Kelleher D. J., and Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., and Förster F. (2014) Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072. [DOI] [PubMed] [Google Scholar]
- 51. Bañó-Polo M., Baldin F., Tamborero S., Marti-Renom M. A., and Mingarro I. (2011) N-Glycosylation efficiency is determined by the distance to the C-terminus and the amino acid preceding an Asn-Ser-Thr sequon. Protein Sci. 20, 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martínez-Gil L., Saurí A., Vilar M., Pallás V., and Mingarro I. (2007) Membrane insertion and topology of the p7B movement protein of Melon Necrotic Spot Virus (MNSV). Virology 367, 348–357 [DOI] [PubMed] [Google Scholar]
- 53. Nilsson I. M., and von Heijne G. (1993) Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 268, 5798–5801 [PubMed] [Google Scholar]
- 54. Martínez-Gil L., Pérez-Gil J., and Mingarro I. (2008) The surfactant peptide KL4 sequence is inserted with a transmembrane orientation into the endoplasmic reticulum membrane. Biophys. J. 95, L36–L38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albert A. D., and Yeagle P. L. (2000) Domain approach to three-dimensional structure of rhodopsin using high-resolution nuclear magnetic resonance. Methods Enzymol. 315, 107–115 [DOI] [PubMed] [Google Scholar]
- 56. Katragadda M., Chopra A., Bennett M., Alderfer J. L., Yeagle P. L., and Albert A. D. (2001) Structures of the transmembrane helices of the G-protein coupled receptor, rhodopsin. J. Pept. Res. 58, 79–89 [DOI] [PubMed] [Google Scholar]
- 57. Pervushin K. V., Arseniev A. S., Kozhich A. T., and Ivanov V. T. (1991) Two-dimensional NMR study of the conformation of (34–65)bacterioopsin polypeptide in SDS micelles. J. Biomol. NMR 1, 313–322 [DOI] [PubMed] [Google Scholar]
- 58. Pervushin K. V., and Arseniev A. S. (1992) Three-dimensional structure of (1–36)bacterioopsin in methanol-chloroform mixture and SDS micelles determined by 2D 1H-NMR spectroscopy. FEBS Lett. 308, 190–196 [DOI] [PubMed] [Google Scholar]
- 59. Pervushin K. V., Orekhov V. Yu., Popov A. I., Musina L. Yu., and Arseniev A. S. (1994) Three-dimensional structure of (1–71)bacterioopsin solubilized in methanol/chloroform and SDS micelles determined by 15N-1H heteronuclear NMR spectroscopy. Eur. J. Biochem. 219, 571–583 [DOI] [PubMed] [Google Scholar]
- 60. Liao M. J., London E., and Khorana H. G. (1983) Regeneration of the native bacteriorhodopsin structure from two chymotryptic fragments. J. Biol. Chem. 258, 9949–9955 [PubMed] [Google Scholar]
- 61. Martin N. P., Leavitt L. M., Sommers C. M., and Dumont M. E. (1999) Assembly of G protein-coupled receptors from fragments: identification of functional receptors with discontinuities in each of the loops connecting transmembrane segments. Biochemistry 38, 682–695 [DOI] [PubMed] [Google Scholar]
- 62. Tastan O., Dutta A., Booth P., and Klein-Seetharaman J. (2014) Retinal proteins as model systems for membrane protein folding. Biochim. Biophys. Acta. 1837, 656–663 [DOI] [PubMed] [Google Scholar]
- 63. Finger C., Volkmer T., Prodöhl A., Otzen D. E., Engelman D. M., and Schneider D. (2006) The stability of transmembrane helix interactions measured in a biological membrane. J. Mol. Biol. 358, 1221–1228 [DOI] [PubMed] [Google Scholar]
- 64. MacKenzie K. R., and Engelman D. M. (1998) Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization. Proc. Natl. Acad. Sci. U.S.A. 95, 3583–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xie H., Ding F. X., Schreiber D., Eng G., Liu S. F., Arshava B., Arevalo E., Becker J. M., and Naider F. (2000) Synthesis and biophysical analysis of transmembrane domains of a Saccharomyces cerevisiae G protein-coupled receptor. Biochemistry 39, 15462–15474 [DOI] [PubMed] [Google Scholar]
- 66. Popot J. L., and Engelman D. M. (2016) Membranes do not tell proteins how to fold. Biochemistry 55, 5–18 [DOI] [PubMed] [Google Scholar]
- 67. Choma C., Gratkowski H., Lear J. D., and DeGrado W. F. (2000) Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7, 161–166 [DOI] [PubMed] [Google Scholar]
- 68. Zhou F. X., Merianos H. J., Brunger A. T., and Engelman D. M. (2001) Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. U.S.A. 98, 2250–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krishnamani V., and Lanyi J. K. (2012) Molecular dynamics simulation of the unfolding of individual bacteriorhodopsin helices in sodium dodecyl sulfate micelles. Biochemistry 51, 1061–1069 [DOI] [PubMed] [Google Scholar]
- 70. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 71. Skach W. R. (2009) Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 16, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McCormick P. J., Miao Y., Shao Y., Lin J., and Johnson A. E. (2003) Cotranslational protein integration into the ER membrane is mediated by the binding of nascent chains to translocon proteins. Mol. Cell. 12, 329–341 [DOI] [PubMed] [Google Scholar]
- 73. Pitonzo D., Yang Z., Matsumura Y., Johnson A. E., and Skach W. R. (2009) Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol. Biol. Cell 20, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sadlish H., Pitonzo D., Johnson A. E., and Skach W. R. (2005) Sequential triage of transmembrane segments by Sec61α during biogenesis of a native multispanning membrane protein. Nat. Struct. Mol. Biol. 12, 870–878 [DOI] [PubMed] [Google Scholar]
- 75. Saurí A., Saksena S., Salgado J., Johnson A. E., and Mingarro I. (2005) Double-spanning plant viral movement protein integration into the endoplasmic reticulum membrane is signal recognition particle-dependent, translocon-mediated, and concerted. J. Biol. Chem. 280, 25907–25912 [DOI] [PubMed] [Google Scholar]
- 76. Saurí A., McCormick P. J., Johnson A. E., and Mingarro I. (2007) Sec61α and TRAM are sequentially adjacent to a nascent viral membrane protein during its ER integration. J. Mol. Biol. 366, 366–374 [DOI] [PubMed] [Google Scholar]
- 77. Ismail N., Crawshaw S. G., Cross B. C., Haagsma A. C., and High S. (2008) Specific transmembrane segments are selectively delayed at the ER translocon during opsin biogenesis. Biochem. J. 411, 495–506 [DOI] [PubMed] [Google Scholar]
- 78. Staley J. P., and Kim P. S. (1994) Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 3, 1822–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Glück J. M., Wittlich M., Feuerstein S., Hoffmann S., Willbold D., and Koenig B. W. (2009) Integral membrane proteins in nanodiscs can be studied by solution NMR spectroscopy. J. Am. Chem. Soc. 131, 12060–12061 [DOI] [PubMed] [Google Scholar]
- 80. Shenkarev Z. O., Lyukmanova E. N., Butenko I. O., Petrovskaya L. E., Paramonov A. S., Shulepko M. A., Nekrasova O. V., Kirpichnikov M. P., and Arseniev A. S. (2013) Lipid-protein nanodiscs promote in vitro folding of transmembrane domains of multi-helical and multimeric membrane proteins. Biochim. Biophys. Acta 1828, 776–784 [DOI] [PubMed] [Google Scholar]
- 81. Killian J. A., Trouard T. P., Greathouse D. V., Chupin V., and Lindblom G. (1994) A general method for the preparation of mixed micelles of hydrophobic peptides and sodium dodecyl sulphate. FEBS Lett. 348, 161–165 [DOI] [PubMed] [Google Scholar]
- 82. Fesik S. W., Luly J. R., Erickson J. W., and Abad-Zapatero C. (1988) Isotope-edited proton NMR study on the structure of a pepsin/inhibitor complex. Biochemistry 27, 8297–8301 [DOI] [PubMed] [Google Scholar]
- 83. Muto Y., Yamasaki K., Ito Y., Yajima S., Masaki H., Uozumi T., Wälchli M., Nishimura S., Miyazawa T., and Yokoyama S. (1993) Sequence specific H-1 and N-15 resonance assignments and secondary structure of GDP-bound human c-Ha-Ras protein in solution. J. Biomol. NMR 3, 165–184 [DOI] [PubMed] [Google Scholar]
- 84. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Buchner L., and Güntert P. (2015) Automated NMR structure calculation with CYANA. J. Biomol. NMR 62, 81–95 [DOI] [PubMed] [Google Scholar]
- 86. Doñate-Macian P., Bañó-Polo M., Vazquez-Ibar J. L., Mingarro I., and Perálvarez-Marín A. (2015) Molecular and topological membrane folding determinants of transient receptor potential vanilloid 2 channel. Biochem. Biophys. Res. Commun. 462, 221–226 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.