

Abstract
Hyperinsulinemia, which is associated with aging and metabolic disease, may lead to defective protein homeostasis (proteostasis) due to hyperactivation of insulin-sensitive pathways such as protein synthesis. We investigated the effect of chronic hyperinsulinemia on proteostasis by generating a time-resolved map of insulin-regulated protein turnover in adipocytes using metabolic pulse-chase labeling and high resolution mass spectrometry. Hyperinsulinemia increased the synthesis of nearly half of all detected proteins and did not affect protein degradation despite suppressing autophagy. Unexpectedly, this marked elevation in protein synthesis was accompanied by enhanced protein stability and folding and not by markers of proteostasis stress such as protein carbonylation and aggregation. The improvement in proteostasis was attributed to a coordinate up-regulation of proteins in the global proteostasis network, including ribosomal, proteasomal, chaperone, and endoplasmic reticulum/mitochondrial unfolded protein response proteins. We conclude that defects associated with hyperactivation of the insulin signaling pathway are unlikely attributed to defective proteostasis because up-regulation of protein synthesis by insulin is accompanied by up-regulation of proteostatic machinery.
Keywords: adipocyte, insulin, insulin resistance, proteomics, proteostasis, pulse-chase SILAC
Introduction
The insulin signaling pathway (ISP)4 is a master regulator of protein metabolism. Many of its effects, such as increased protein synthesis and reduced autophagy, are orchestrated via mTORC1 (1, 2). The ISP is hyperactivated by chronic hyperinsulinemia, a common consequence of insulin resistance and aging (3). This in turn exacerbates insulin resistance, obesity (4–6), and aging (7–10). Chronic ISP activity may partially cause these detrimental effects by dysregulating protein homeostasis (proteostasis) (11, 12). Supporting this view, genetic manipulations that lead to either endoplasmic reticulum (ER) stress, reduced proteasomal activity, or reduced autophagy are associated with insulin resistance in mice (13–15). Moreover, the mTORC1 inhibitor rapamycin extends lifespan in a range of animals, and in Drosophila these effects are at least in part due to reduced protein synthesis and enhanced autophagy (16).
Proteins are made via a carefully orchestrated process that minimizes misfolding. This involves chaperones that ensure efficient folding of newly synthesized proteins, the ubiquitin-proteasome system that degrades misfolded proteins, and autophagy for removal of protein aggregates (17, 18). It has been proposed that chronic insulin signaling impairs protein folding and promotes aggregation because it simultaneously activates protein synthesis, suppresses autophagy, and inhibits the activity of FOXO, a transcription factor that regulates chaperone gene expression (11, 12, 19–21).
The effect of loss of function of daf-2/ISP activity on global proteostasis was recently studied in Caenorhabditis elegans (22), but few studies have examined the effects of hyperactivation of the ISP on proteostasis, and none have examined the effects at a systems-wide level. Furthermore, the full complement of proteins that is synthesized and/or degraded in response to insulin has not been characterized. Here we investigated the global effects of hyperactive insulin signaling on protein synthesis and degradation using pulse-chase labeling with stable isotopes (stable isotope-labeled amino acids in cell culture (SILAC)) (23–28) and high resolution mass spectrometry (MS)-based proteomics.
Our studies focus on postmitotic 3T3-L1 adipocytes because exposure of these cells to chronic hyperinsulinemia results in insulin resistance (29), and defects in insulin action in adipocytes contribute to whole body insulin resistance (30). Moreover, although primary mouse/rat adipocytes lose their insulin sensitivity upon long term culturing (31), 3T3-L1 adipocytes are highly insulin-responsive for long periods (32).
Unexpectedly, although insulin potently stimulated protein synthesis and suppressed autophagy, there was no evidence of protein aggregation or carbonylation. Rather, insulin induced the synthesis of cytosolic chaperones and ER unfolded protein response (UPR) and mitochondrial UPR proteins, and this was associated with improved rather than impaired protein folding. We therefore suggest that hyperactivation of the ISP does not contribute to impaired proteostasis as insulin coordinated protein biosynthesis with the up-regulation of the proteostasis network.
Results
Mapping Large Scale Protein Synthesis and Degradation
To assess the effect of chronic insulin signaling on global proteostasis, we exposed 3T3-L1 adipocytes to insulin over 4 days and analyzed protein turnover by pulse-chase metabolic labeling and mass spectrometry (Fig. 1A). Insulin caused sustained stimulation of protein biosynthetic pathways as indicated by a marked increase in total cellular protein content and S6 phosphorylation (Fig. 1, B and C). To measure global protein turnover, we metabolically labeled 3T3-L1 fibroblasts with “heavy” isotopes (H) of arginine and lysine (Arg +10/Lys +8) until complete incorporation had occurred and then differentiated the fibroblasts into adipocytes. Heavy labeled cells were then switched into culture medium containing “medium” isotopes (M) of Arg and Lys (Arg +6/Lys +4) and harvested over a time series. Consequently, a decay in heavy labeled proteins is indicative of protein degradation, and an increase in medium proteins reflects protein synthesis (Fig. 1A) (24).
FIGURE 1.
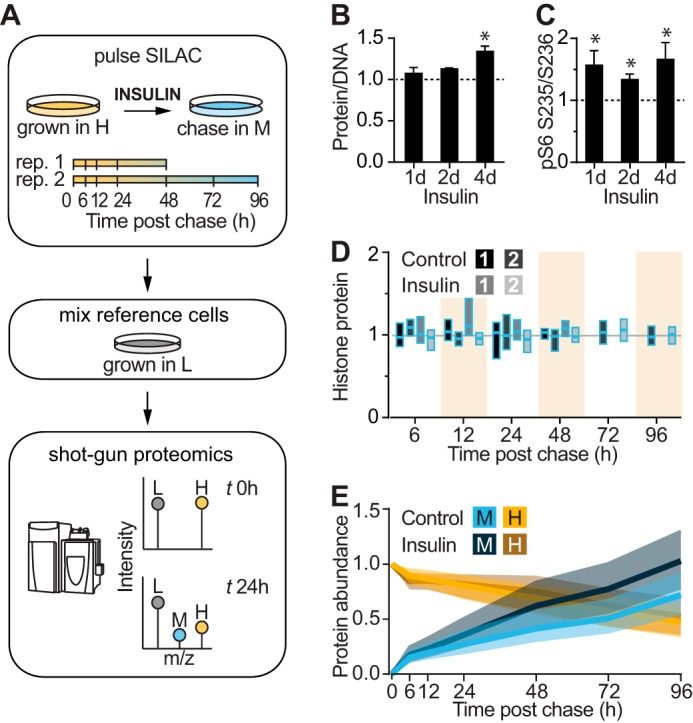
Analysis of insulin-stimulated protein synthesis and degradation by pulse-chase SILAC and mass spectrometry. A, schematic of pulse-chase SILAC experiment. 3T3-L1 adipocytes were labeled with heavy and pulsed with medium isotopes of arginine and lysine. After switching to medium isotopes, cells were harvested after 6, 12, 24, and 48 h in the first experiment and after 6, 12, 24, 48, 72, and 96 h in the second experiment. Adipocytes were then mixed with L-labeled adipocytes, and the amount of H-, M-, and L-labeled proteins was analyzed by shotgun proteomics. B, protein/DNA ratio was quantified in 3T3-L1 adipocytes treated with insulin for the indicated days (d) and normalized to untreated cells (indicated by the dotted line) (n = 4; error bars represent S.E.; one-sample t test; *, p < 0.05). C, 3T3-L1 adipocytes were treated with insulin for the indicated days, and lysates were immunoblotted with phospho-S6 (pS6) (Ser-235/Ser-236) and 14-3-3 (loading control) antibodies. Immunoblots were quantified and normalized to the loading control and untreated cells (indicated by the dotted line) (n = 6; error bars represent S.E.; one-sample t test; *, p < 0.05). D, box plots of histone protein abundance quantified in pulse-chase SILAC experiment relative to time 0 for control and insulin-treated cells (n = 9 proteins) of two independent experiments. E, global change in M and H proteins in control and insulin-treated samples during pulse-chase SILAC experiment. Shading indicates interquartile range, and the line indicates median.
The cell lysates were mixed with a reference sample of “light” isotope (L)-labeled adipocytes for proteomic analysis. The experimental and reference samples were mixed such that DNA rather than protein content was constant because altered protein synthesis might affect normalization by protein mass. The samples contained an equal number of cells as histone abundance, which is proportional to DNA mass, was highly consistent between samples (Fig. 1D).
We identified a total of 6,550 proteins of which 2,560 newly synthesized medium proteins and 2,321 degraded heavy proteins were quantified across at least 50% of the time series (supplemental Table S1). Medium and heavy labeled proteins increased and decreased, respectively, over the time course and intersected at ∼72 h, suggesting that half the pool of proteins turns over after 3 days (Fig. 1E).
Insulin treatment commenced 1 day prior to switching to medium isotopes (the start of the chase). Insulin-treated adipocytes demonstrated a similar decrease in heavy labeled proteins as compared with untreated cells but a greater increase in medium labeled proteins (Fig. 1E). These changes correspond to increased protein synthesis with no change in protein degradation rates in response to insulin treatment.
Insulin Enhances the Synthesis and Stability of Newly Made Proteins
To identify the primary insulin-responsive proteins, we derived synthesis rates by fitting the increase in medium proteins to a variation of a kinetic model described previously (27, 33). In this model, proteins are synthesized linearly and, once made, are subject to degradation (Fig. 2, A and B). An example of the model fit for two proteins is shown in Fig. 2B. The initial increase in medium labeled squalene epoxidase was due to proteins synthesis (msyn), and this eventually reached a plateau (steady state) due to degradation of the newly synthesized protein (mdeg). In contrast, GLUT4 was synthesized more slowly and did not exhibit a plateau, so the degradation component was not apparent (Fig. 2B). We modeled the degradation rate of medium and heavy labeled proteins separately because the degradation of newly synthesized proteins (mdeg) may differ from pre-existing proteins (hdeg).
FIGURE 2.

Insulin up-regulates synthesis and stability of newly made proteins. A, number of proteins that were quantified, curve fitted, or insulin-regulated in protein synthesis data. B, graph of abundance of representative medium labeled proteins over the time course in control (Con) and insulin (Ins)-treated cells. The line indicates the best fit synthesis curve. The bar graph indicates mean and S.D. of the curve parameters msyn for synthesis and mdeg for degradation of newly synthesized protein (*, p < 0.05). C, relative abundance of significantly insulin-regulated proteins relative to control samples 48 h postchase. D, bar chart of insulin-regulated proteins with significantly increased or decreased (p < 0.05) msyn or mdeg. B and D, statistics were performed on linearized model as described under “Experimental Procedures.”
Of the initial 2,560 quantified medium proteins, 1,445 fit the model with adj. R2 >0.8, and 752 fit with a more stringent adj. R2 >0.9 (Fig. 2A and supplemental Table S1). Manual inspection revealed that a cutoff of adj. R2 >0.8 was sufficient to eliminate proteins with irregular profiles over the time course. Proteins may have exhibited irregular time profiles because of inaccurate quantification due to low abundance, slow protein synthesis, or cycling of protein abundance such as occurs under circadian control (34). Although proteins with irregular profiles may be of interest, here we focused on proteins that fit our basic model of turnover. We classified proteins as insulin-regulated if the control and insulin-treated samples fit separate models better than one model with both data combined (adj. p < 0.0005). Using this criterion, several proteins with synthesis rates known to be regulated by insulin were identified, including GLUT4 and GAPDH (35–37). In total, insulin regulated the levels of around 46% of the proteins modeled (663 of 1,445 proteins).
Some studies of mTORC1-mediated protein synthesis have suggested that chronic activation of mTORC1 reduces protein synthesis as either inhibitory proteostatic pathways are activated or there is increased degradation of newly made proteins due to increased misfolding (38–40). In contrast to this view, in insulin-stimulated 3T3-L1 adipocytes we observed that all but one of the 663 insulin-regulated proteins increased in abundance (median increase of 49% after 48 h) (Fig. 2C). To determine whether this was due to increased synthesis (msyn) and/or decreased degradation of newly made proteins (mdeg), we compared the probable parameter values for msyn and mdeg and found that protein synthesis rates were specifically increased for 468 and decreased for 22 proteins, whereas degradation rates were decreased for 197 and increased only for 20 proteins (Fig. 2D). These data suggest that chronic insulin treatment enhances both the synthesis and stability of newly made proteins.
Insulin Minimally Affects Protein Degradation Despite Inhibiting Autophagy
We next examined the degradation of the premade heavy proteins for evidence of insulin-regulated protein degradation or proteome destabilization (Fig. 3A). To achieve this, heavy proteins were fitted to an exponential decay model. Interestingly, 27% of proteins approached a plateau greater than one-quarter of their starting abundance after 96 h and thus did not completely degrade. To allow the model to approach a non-zero plateau, we included the hsyn parameter so that the plateau is given by hsyn/hdeg. The incomplete protein degradation may reflect discrete pools of proteins with different turnovers. This is not surprising because the majority of proteins exist both in protein complexes and in isolation (41). Alternatively, heavy amino acids liberated by proteolysis may be reincorporated into protein. However, this seems unlikely because hsyn varied among proteins. By allowing incomplete degradation in our model, we estimated the half-life of the insulin receptor to be 11.7 h (S.D. 8.76–15.7), which is similar to previously observed values (42).
FIGURE 3.
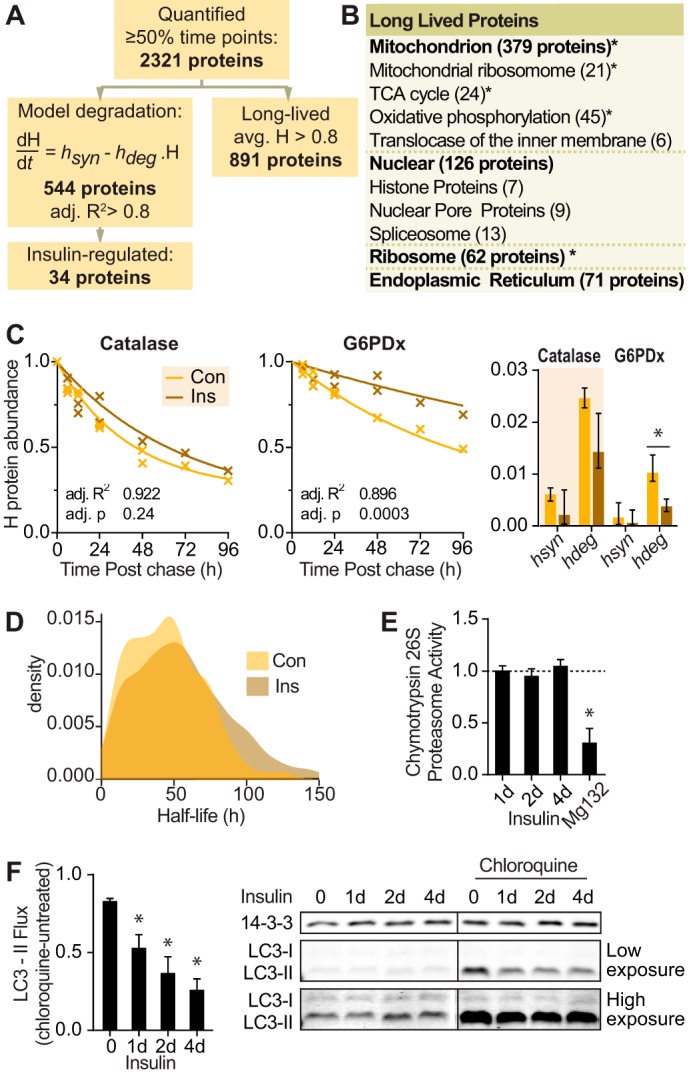
Insulin minimally affects protein degradation despite suppression of autophagy. A, number of proteins that were quantified, curve fitted, insulin-regulated, or long lived in protein degradation data. B, gene ontology terms of long lived proteins from proteomic data with number of proteins in each category in parentheses. Significantly enriched gene ontology terms are indicated (*, p < 0.05). TCA, tricarboxylic acid. C, graph of abundance of representative heavy labeled proteins in control (Con) and insulin (Ins)-treated cells over the time course. The line indicates the best fit degradation curve. The bar graph indicates mean and S.D. of the curve parameters hdeg for degradation and hsyn for reincorporation of heavy amino acids into proteins. Statistics were performed on a linearized model as described under “Experimental Procedures” (*, p < 0.05). D, density graph of proteome half-lives calculated from protein degradation data. E, 3T3-L1 adipocytes were treated with insulin for the indicated times or MG132 for 2 h. Chymotrypsin activity of 26S proteasome was quantified and normalized to untreated cells (indicated by the dotted line) (n = 4; error bars represent S.E.; one-sample t test; *, p < 0.05). F, 3T3-L1 adipocytes were treated with insulin for the indicated days (d) and/or 400 μm chloroquine for 15 min. Cell lysates were immunoblotted with the indicated antibodies. Vertical lines on immunoblots indicate where the blot was spliced. Immunoblots were quantified, and LC3-II was normalized to the 14-3-3 loading control. LC3-II flux was calculated as the difference of LC3-II with chloroquine minus without chloroquine for each time point (n = 5; error bars represent S.E.; one-sample t test; *, p < 0.05).
In contrast to earlier pulse-chase SILAC studies, we found that many proteins were long lived with 38% of proteins (891 proteins) exhibiting no detectable degradation over a 96-h period (Fig. 3B and supplemental Table S2). Insulin had no detectable effect on the degradation of long lived proteins. These included nuclear pore proteins and histones, consistent with a recent report in vivo (43), as well as mitochondrial electron transport chain proteins.
Among the heavy labeled proteins, 544 proteins fit our model of protein degradation with adj. R2 >0.8 (Fig. 3, A and C, and supplemental Table S1). For these proteins, the median half-life was 47 h (Fig. 3D). There was more variability in protein degradation compared with synthesis. Because both processes were measured using the same samples, it is likely that protein degradation occurs with greater biological variability in 3T3-L1 adipocytes. In comparison, earlier pulse-chase SILAC studies in dividing cells reported less variable protein degradation. This may be because cell division is associated with activation of ubiquitin ligases (44) and higher rates of protein turnover (23–28).
In contrast to the effects of insulin on protein synthesis, insulin modulated the degradation of only 34 proteins using an adj. p <0.01, and nine proteins using a more stringent cutoff of adj. p <0.005. Consistent with this, insulin did not significantly modulate proteasomal activity in adipocytes (Fig. 3E). However, insulin suppressed autophagy as indicated by the significant reduction in autophagic flux (Fig. 3F). Autophagic flux was measured by the difference in LC3-II, a marker of autophagosomes, with and without an inhibitor of autophagic degradation, chloroquine. Hence, these data indicate that insulin does not regulate global protein degradation, and by extension insulin-mediated inhibition of autophagy does not have a major impact on the adipocyte proteome.
Insulin Coordinates Protein Synthesis with the Proteostasis Network
Insulin increased the synthesis of proteins involved in glucose metabolism, including glycolysis, tricarboxylic acid cycle, and pentose phosphate pathway (Fig. 4A and supplemental Table S3). Insulin also increased the synthesis of proteins that support protein synthesis and the clearance of misfolded proteins. These included ribosomal, tRNA biosynthetic, proteasomal, ER, and Golgi proteins that regulate homeostasis of the secretory/biosynthetic tract as well as proteins involved in heat shock and mitochondrial and ER UPR. The expression of these proteins was proportional to the overall increase in cellular protein synthesis. As protein synthesis was not impaired but rather significantly enriched in the cytosol, ER, and Golgi compartments, it seems likely that elevated expression of these proteins is part of a coordinated response to support protein synthesis.
FIGURE 4.

Insulin coordinates synthesis of proteins across multiple compartments with proteostasis network. A, gene ontology of newly made proteins significantly increased in response to insulin are highlighted in blue and depicted in the illustration of a cell. TCA, tricarboxylic acid; PPP, pentose phosphate pathway. Bar graphs depict the percent increase in abundance of ER UPR, mitochondrial (Mito) UPR, or cytosolic chaperone proteins synthesized after 48-h incubation with insulin compared with control. Black bars indicate significantly insulin-regulated proteins. The blue horizontal bar indicates the interquartile range of the percent increase in insulin-regulated proteins. B, 3T3-L1 adipocytes were treated with insulin (Ins) for 24 h, and cell lysates were immunoblotted for the indicated proteins. Con, control. Immunoblots were quantified and normalized to loading control and untreated samples (n = 5 for phosphorylated IRE1 (pIRE1) and EIF2α (pEIF2α); n = 3 for phosphorylated PERK (pPERK); error bars represent S.E.; one-sample t test; *, p < 0.05). C and D, rats were cannulated and infused with 40 mg/kg/min glucose for the indicated periods or saline (Sal) for 4 days (d). C, plasma glucose and insulin levels are reproduced from Laybutt et al. (46). D, adipose tissue of rats was immunoblotted for the indicated proteins. Immunoblots were quantified and normalized to loading control and saline samples (n = 4; error bars represent S.E.; two-sample t test; *, p < 0.05). E, 3T3-L1 adipocytes were serum-starved for 1.5 h and treated with insulin for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. Immunoblots were quantified and normalized to 14-3-3. Data were scaled so that basal is set to 0, and the maximal response set to 1 (n = 4; error bars represent S.E.; one-sample t test; *, p < 0.05). F and G, 3T3-L1 adipocytes were serum-starved for 1.5 h and treated with the indicated inhibitors for 30 min followed by insulin for 30 min. Cell lysates were immunoblotted for the indicated proteins. Vertical lines on the immunoblots indicate where the blot was spliced. Immunoblots were quantified and normalized (norm) to 14-3-3. Insulin-stimulated phosphorylated IRE1 is expressed as -fold change relative to DMSO-treated samples (indicated by the dotted line) (F, n = 4; G, n = 5; error bars represent S.E.; one-sample t test; *, p < 0.05). Rap, rapamycin; CHX, cycloheximide.
The up-regulation of ER UPR proteins was not associated with PERK activation as phosphorylation of neither PERK nor its substrate EIF2α was increased, but rather IRE1 phosphorylation was increased (Fig. 4B). These data are interesting as PERK reportedly promotes apoptosis, whereas IRE1 is cytoprotective (45). We examined whether hyperinsulinemic rats also exhibited ER UPR activation in adipose tissue. Previously, rats were cannulated and infused with glucose (40 mg/kg/min) for 1 or 4 days, resulting in a state of chronic in vivo hyperinsulinemia (Fig. 4C) (46) and chronic mTORC1 signaling in adipose as indicated by elevated S6 phosphorylation after 1- and 4-day glucose infusion, although changes after 4 days were insignificant due to high variation (Fig. 4D). Akt phosphorylation was unchanged probably because of negative feedback in response to chronic insulin signaling. In adipose tissue from 1- or 4-day hyperglycemic hyperinsulinemic rats, phosphorylation of IRE1, but not EIF2α, was increased compared with saline-infused rats (Fig. 4D). IRE1 phosphorylation was not induced by a proteostasis stress as IRE1 phosphorylation was increased after 30-s insulin stimulation in 3T3-L1 adipocytes (Fig. 4E), well before mTORC1 activation (47). In addition, IRE1 phosphorylation was not inhibited by cycloheximide, an inhibitor of protein synthesis, or by the mTORC1 inhibitor rapamycin (Fig. 4F). Rather, IRE1 phosphorylation was induced by PI3K/Akt activity because it was blocked by inhibitors of Akt (MK2206) and PI3K/mTOR (LY294002) but not by inhibitors of MAPK (U0126) (Fig. 4G). Therefore, we speculate that the activation of UPR proteins by insulin is not indicative of protein misfolding or cellular stress but likely represents part of a coordinate proteostatic response, which may be regulated by IRE1 in preparation for a burst of insulin-regulated protein synthesis.
Protein Synthesis via Insulin and mTORC1 Does Not Increase Protein Misfolding
To test the hypothesis that a burst of protein synthesis in response to insulin is accompanied by a coordinate proteostatic response, we next examined protein folding. Misfolded proteins often assemble into large insoluble aggregates that can be readily detected using a detergent solubility assay. As expected, we observed a 4-fold increase in detergent-insoluble proteins in cells treated with the proteasomal inhibitor MG132. However, we found no detectable increase in detergent-insoluble proteins in cells treated with insulin for 4 days (Fig. 5A). Similarly, there was no change in detergent-insoluble protein in adipose tissue from glucose-infused hyperinsulinemic rats (Fig. 5B). Next, we examined protein carbonylation, a measure of cellular oxidative damage (Fig. 5C). Hydrogen peroxide caused a 2-fold increase in protein carbonylation in 3T3-L1 adipocytes, whereas no detectable increase was observed with insulin. Rather, insulin induced a small but significant reduction in protein oxidation, indicating an adaptive response against not only protein folding stress but also oxidative stress. Evidence for an adaptive antioxidant response was also found in our proteomic data: insulin up-regulated the antioxidants peroxiredoxin 4 and carbonyl reductases 1 and 3, and a transcription factor enrichment analysis predicted activation of NRF2, which is involved in oxidative stress tolerance (supplemental Table S4). There was also no effect of in vivo hyperinsulinemia on protein carbonylation levels in rat adipose tissue (Fig. 5D). Collectively, these data indicate that chronic activation of insulin signaling is not accompanied by compromised protein synthesis or degradation or a demonstrable change in markers of protein misfolding.
FIGURE 5.

Insulin preserves protein folding. A, 3T3-L1 adipocytes were treated with insulin for the indicated periods or MG132 for 24 h. Triton X-100-insoluble protein was measured and normalized to untreated cells (indicated by the dotted line) (n = 3; error bars represent S.E.; one-sample t test; *, p < 0.05). B, Triton X-100-insoluble protein relative to total protein was measured in adipose tissue of rats (Fig. 4C) (saline (Sal), n = 6; 1-day (d) glucose, n = 8; 4-day glucose, n = 7; error bars represent S.E.; two-sample t test; *, p < 0.05). C, 3T3-L1 adipocytes were treated with insulin for the indicated periods or H2O2 for 30 min. Carbonylation was measured and normalized to untreated cells (n = 3; error bars represent S.E.; one-sample t test; *, p < 0.05). D, carbonylation was measured in rats (Fig. 4C) (saline, n = 6; 1-day glucose, n = 8; 4-day glucose, n = 7; error bars represent S.E.; two-sample t test; *, p < 0.05). E, 3T3-L1 adipocytes stably expressing WT Fluc or DM Fluc were assayed for luminescence. Luminescence (Lum.) was normalized to cells expressing WT Fluc (n = 4; error bars represent S.E.; one-sample t test; *, p < 0.05). F and G, 3T3-L1 adipocytes stably expressing WT Fluc (F) or DM Fluc (G) were treated with insulin for the indicated periods and assayed for luminescence. Luminescence was normalized to untreated cells (indicated by the dotted line) (F, n = 4; G, n = 6; error bars represent S.E.; one-sample t test; *, p < 0.05). H, 3T3-L1 adipocytes stably expressing DM Fluc were treated with insulin and/or rapamycin for 24 h and assayed for luminescence. Luminescence was normalized to untreated cells (indicated by the dotted line) (n = 3; error bars represent S.E.; one-way analysis of variance; *, p < 0.05). I–K, HEK293 cells were transiently co-transfected with WT Fluc and either enhanced GFP (EGFP) or Rheb-Myc and treated with insulin and/or rapamycin for 24 h. Lysates were immunoblotted with the indicated antibodies (I), quantified, and normalized to 14-3-3 and untreated sample expressing enhanced GFP (indicated by the dotted line) (J) (n = 3; error bars represent S.E.; one-sample t test; *, p < 0.05). Luminescence was measured in separate cells and normalized to untreated sample expressing enhanced GFP (K) (n = 4; error bars represent S.E.; one-sample t test; *, p < 0.05). pS6, phospho-S6.
Because previous studies suggested that accelerated protein synthesis via mTORC1 causes protein misfolding, we reasoned that although the endogenous proteome was not detectably misfolded insulin treatment might reduce folding capacity under stress. To test this hypothesis, we assayed folding in insulin-treated adipocytes using firefly luciferase (Fluc). Luciferase is commonly used for measuring protein folding capacity as it folds rapidly upon translation and does not require post-translational modifications for its activity (48). The folding efficiency of wild type (WT) Fluc was not reduced by insulin treatment but instead demonstrated a trend toward increased synthesis (Fig. 5F). To further challenge the folding environment, we expressed a destabilized luciferase construct (DM Fluc), which contains two point mutations that impair the ability of the protein to fold (49). DM Fluc had 5% of the activity of WT Fluc in control cells (Fig. 5E). However, insulin treatment improved folding of DM Fluc by 50% (Fig. 5G). This increase was mTORC1-dependent because rapamycin abrogated the increase in DM Fluc folding (Fig. 5H). These data contrast with a report by Conn and Qian (38) where either chronic mTORC1 activation in TSC2−/− mouse embryonic fibroblasts or Rheb overexpression in HEK293 cells inhibited WT Fluc folding. To test whether our results were cell type-dependent or whether insulin differs in its response compared with selective and chronic mTORC1 activation, we expressed WT Fluc in HEK293 cells and measured folding in response to chronic insulin or Rheb overexpression. Both insulin and Rheb overexpression caused activation of mTORC1 but in neither case was folding significantly inhibited (Fig. 5, I–K). Rather, Rheb overexpression led to an increase in WT Fluc activity, which was rapamycin-insensitive. Based on our data, we suggest that increased translation associated with either chronic insulin or mTORC1 activation does not cause a reduction in proteins synthesized due to elevated misfolding as has been proposed previously. Rather, both insulin and mTORC1 activity concurrently increase protein synthesis with the ability to fold proteins.
Discussion
The ISP is a major regulator of protein metabolism, but how hyperactivation of ISP, which occurs with aging and metabolic diseases, affects global proteostasis is unclear. To assess the effects of insulin on the composition, stability, and folding of the proteome, we undertook a global analysis of protein synthesis and degradation followed by assays to assess protein quality in 3T3-L1 adipocytes exposed chronically to insulin. Surprisingly, we found that insulin does not affect protein degradation despite suppressing autophagy. Additionally, insulin increased synthesis of 46% of the proteome without evidence of protein damage. Among the up-regulated proteins were many belonging to the proteostasis machinery, including metabolic enzymes, protein chaperones, protein synthesis machinery, and ER/Golgi proteins. Hence, this demonstrates that in response to chronic elevation of protein synthesis cells possess adaptive mechanisms to cope with the additional protein folding burden by coordinately up-regulating the proteostatic machinery.
The concept that the insulin signaling pathway is detrimental to health due to impaired protein misfolding stems largely from loss of function studies (22, 50–53). Most notably, rapamycin is thought to extend lifespan in part by reducing protein synthesis and thus improving proteostasis (16). Based on these studies, it has been inferred that increased insulin signaling would compromise cellular proteostasis. In the present study, we tested the consequences of chronic hyperactivation of the insulin signaling pathway under well controlled experimental conditions using both adipocytes grown in culture and adipose tissue from hyperglycemic hyperinsulinemic rats, and in neither case could we find evidence that hyperactivation of this pathway compromises cellular proteostasis. We assessed proteostasis by measuring insoluble proteins (Fig. 5, A and B), protein carbonylation (Fig. 5, C and D), synthesis of a destabilized luciferase construct (Fig. 5G), and stability of existing or newly made proteins in the MS-based map of proteome turnover (Figs. 2D and 3A). It has been shown that hyperactivation of the ISP reduces toxicity of misfolding-prone neurodegenerative proteins that have been expressed in various cell types, and so these studies are in general agreement with our findings that the ISP in fact promotes proteostasis (54–57). In contrast, it has been shown that constitutive hyperactivation of mTORC1 via either TSC2 deletion or overexpression of Rheb in HEK293 cells resulted in impaired protein folding, although we have been unable to replicate the latter observation in the current study. The basis for why these findings contrast with the present findings remains unclear. However, aspects of proteostasis do appear to be inconsistent across studies using the chronic mTORC1 activation cell models (38–40, 58). One possibility is that long term overexpression of these hyperactivating mutants in cells over many generations results in ill-defined adaptive changes, and it could be these changes that drive the proteostasis defect. Regardless, in our study, increased protein synthesis via either insulin or mTORC1 activation does not impair cellular proteostasis.
One of the reasons insulin was thought to cause protein misfolding is because it suppresses autophagy. Genetically inhibiting autophagy reduces protein degradation, thereby increasing protein aggregation (59, 60). Here, however, insulin had a relatively modest effect on overall protein degradation in adipocytes, indicating that insulin-dependent reduction in autophagy does not affect protein degradation.
We propose that cells accommodate the increased synthetic demand imposed by insulin by increasing synthesis of proteins involved in proteostasis, including chaperones and vesicle transport proteins. Initially, we expected that cytosolic chaperones would be down-regulated by chronic insulin as many of these are regulated by FOXO, a transcription factor that is suppressed by insulin. The insulin-dependent synthesis of chaperone proteins may instead be regulated via a pathway such as HSF1 or HIF1α. HSF1 was reported to be activated by mTORC1 following heat stress (61). Furthermore, HSF1 is induced during protein translation in various cancer cell lines (62). Although it is possible that HSF1 may mediate the induction of the proteostasis network here, HSF1 was not significantly enriched in our transcription factor enrichment analysis (supplemental Table S4). Future studies examining this will be of considerable interest.
Insulin also up-regulated components of the mitochondrial and ER unfolded protein response. Previously, activation of the ER UPR in adipose tissue in obese hyperinsulinemic subjects as well as in healthy subjects rendered acutely hyperinsulinemic was assumed to indicate compromised ER protein synthesis and stress and as a result proposed to cause insulin resistance (13, 63–65). This is largely because the ER UPR was initially characterized as a response to agents such as tunicamycin and thapsigargin, which have a profound inhibitory effect on ER protein synthesis and cause insulin resistance (66). Our proteomic data reveal that insulin does not compromise ER protein synthesis. Rather, we propose that the induction of the ER UPR by insulin is part of a protective response and may be preemptively induced by Akt-mediated IRE1 phosphorylation in anticipation of a burst of protein synthesis. These observations are supported by other studies, which demonstrated that IGF-1 stimulates IRE1 phosphorylation to protect against ER stress and that the PI3K subunit p85α stimulates XBP1 splicing downstream of IRE1 phosphorylation, leading to resistance to ER stress (67, 68). As such, activation of the ER UPR should not be used alone to indicate ER stress. Our data indicate that, because hyperinsulinemia does not induce ER stress, ER stress is not likely to cause hyperinsulinemia-induced insulin resistance. Additionally, ER stress during obesity is likely caused by factors aside from hyperinsulinemia.
In addition to examining the effects of insulin on proteostasis, we also explored the stability of the endogenous proteome of adipocytes. Proteins that were degraded in adipocytes had an average half-life of 47 h, which is similar to HeLa and NIH3T3 cells, which are reported to have half-lives of 20 and 46 h, respectively (33, 69). However, compared with these mitotic cells, we found that many proteins in adipocytes are highly stable: 24% of quantified proteins did not turn over during the 96-h period of investigation, and other proteins comprised a pool of protein that was resistant to degradation. This may suggest that proteins in postmitotic cells, such as adipocytes, are particularly stable. This slow turnover of proteins may explain why knockdown of proteins is difficult using siRNA in adipocytes; cells are typically exposed to siRNA for 2–3 days. We suggest that our global protein turnover data will be an invaluable guide for such experiments. The long lived proteins are also of interest in the study of proteostasis as specialized repair mechanisms for the maintenance of long lived proteins have not yet been identified.
The current study suggests that cellular proteostasis is a highly regulated process that can accommodate dynamic changes in protein turnover. Hyperinsulinemia increases protein biosynthesis, which has the potential to burden the proteostasis machinery. Here we show that insulin coordinately up-regulates the proteostasis network concomitant with protein synthesis. Therefore, the metabolic and health consequences of hyperinsulinemia are unlikely to be mediated by compromised proteostasis. Rather, it seems likely that proteostasis defects are triggered by alternative mechanisms. In view of these data, it seems unlikely that reduced protein synthesis accounts for the lifespan-extending effects of loss of function mutations in the insulin signaling pathway or drugs such as rapamycin that dampen signaling downstream of the insulin receptor.
Experimental Procedures
Cell Culture
3T3-L1 adipocytes were cultured as described previously (47). For chronic insulin treatments, adipocytes were incubated with 10 nm insulin for 1–5 days with medium replenished every 12 h. For acute insulin treatments, 3T3-L1 adipocytes were serum-starved in DMEM supplemented with 0.2% BSA for 2 h before incubation with 10 nm insulin. Inhibitors were added 30 min prior to insulin stimulation at the following concentrations: 10 μm cycloheximide (Sigma-Aldrich), 500 nm rapamycin (LC Laboratories), 10 μm MK2206 (Sellekchem), 50 μm LY294002 (Tocris Bioscience), and 10 μm U0126 (Cell Signaling Technology). H2O2 (BDH) was used at 10 mm.
Pulse-Chase Using SILAC
SILAC DMEM (deficient in lysine, arginine, and leucine; Thermo Fisher) was supplemented with 10% dialyzed FCS (Hyclone Laboratories), GlutaMAX (Life Technologies), and leucine. Different isotopes of arginine and lysine were added to the DMEM to result in either light DMEM (Arg and Lys; Sigma), medium DMEM (Arg +6, Lys +4; Silantes), or heavy DMEM (Arg +10, Lys +8; Silantes). 3T3-L1 fibroblasts were passaged for six doublings in light or heavy SILAC DMEM and differentiated into adipocytes. On day 8 of differentiation to commence the pulse-chase, heavy labeled adipocytes were washed three times in warm PBS and switched to medium SILAC DMEM. Subsequently, medium DMEM was replenished every 12 h. Cells were harvested at the following times after switching to medium SILAC DMEM: 0, 6, 12, 24, and 48 h in the first experiment and 0, 6, 12, 24, 48, 72, and 96 h in the second experiment. Insulin-treated cells were exposed to 10 nm insulin for 24 h prior to start of the chase and thereafter.
Harvested cells were washed three times with ice-cold PBS and solubilized by sonication in HES buffer (20 mm HEPES, pH 7.4, 1 mm EDTA, 250 mm sucrose) and protease inhibitors (Roche Applied Science). DNA content was measured by SYBR Green I nucleic acid stain (S-7585, Thermo Fisher) assay using salmon sperm DNA (D1626, Sigma) as a standard. Samples were mixed with light labeled adipocytes (reference cells) at a ratio of 1:1 based on DNA content, and SDS was added (final concentration, 2% (w/v)). Following mixing, proteins were processed by filter-aided sample preparation and trypsin-digested (Promega), and peptides were fractionated by strong anion exchange as described previously (70).
Mass Spectrometry
LC-MS/MS analysis was carried out on an LTQ-Orbitrap Velos Pro (Thermo Fisher Scientific) as described previously (47). Raw mass spectrometry data were processed using MaxQuant (71) version 1.3.0.5 using default settings except for oxidized methionine and acetylation (protein N terminus), which were selected as variable modifications, and carbamidomethyl (Cys) and triple SILAC labels, which were set as fixed modifications. A maximum of two missed cleavages was permitted, 10 peaks per 100 Da, MS/MS tolerance was 20 ppm, and minimum peptide length was 7 for analysis. Database searching was performed using the Andromeda search engine integrated into the MaxQuant environment (72) against the mouse UniProt database (July 2013). Protein, peptide, and site false discovery rate thresholds in MaxQuant were each set to a maximum of 1%.
Modeling of Synthesis and Degradation Rates
The MS data set contained two time courses of H/L and M/L proteins both under control and insulin conditions. Each time course was normalized to starting amounts of H/L in control, log2 transformed, and normalized using LOESS (locally weighted scatter plot smooth) curve fitting. Proteins with ≥50% missing values in a time course were removed. The resulting data were fitted to a kinetic model of protein synthesis and degradation described by the following ordinary differential equations.
| (Eq. 1) |
| (Eq. 2) |
where [Ṁ] is the rate of change of the concentration of M-labeled proteins of interest, and msyn and mdeg are their synthesis and degradation rate constants, respectively. Similarly, [Ḣ] is the rate of change of the concentration of H-labeled proteins, and hsyn and hdeg are their corresponding synthesis and degradation rate constants. The differential equations were solved with the initial conditions [M](0) = 0 and [H](0) = 1 to obtain the following models.
| (Eq. 3) |
| (Eq. 4) |
The data were fitted to the models, and optimal parameters were obtained by minimizing the weighted least square residual (ϵ) using the Nelder-Mead simplex method in MATLAB version R2015a (The MathWorks Inc.). The half-life of proteins was determined as ln 2/mdeg.
Statistical Identification of Insulin-regulated Proteins
The analysis of M-labeled proteins is described here in detail and the same analysis was applied to H-labeled proteins. The kinetic parameters were modeled as a linear model in logarithmic space using the following formulas for control (Equations 5 and 6) and insulin (Equations 7 and 8).
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
| (Eq. 8) |
Next, each protein was tested for whether it agreed with the hypothesis that there was a change in [Ṁ] when insulin was present (H1) and therefore a change in msyn or mdeg or whether it agreed with the null hypothesis (H0).
| (Eq. 9) |
| (Eq. 10) |
Proteins passed H0 if the adjusted coefficient of determination (adj. R2) of the null hypothesis (adj. R02) is larger than that of the alternative hypothesis (adj. R12) because if adj. R02 > adj. R12 then it implies that H0 is a better fit to the data than H1. This step also removes bias for H1 generated by the increased degree of freedom in the model. For the remaining proteins, an F-statistic was obtained for the two models (H1 and H0) using degrees of freedom df1 = 2 and df2 = 15. The F-statistic was converted to a p value, which was adjusted for multiple comparison testing using the Benjamini-Hochberg algorithm in R (73, 74). H0 was rejected for proteins with a cutoff of p < 0.0005 for analysis of medium and p < 0.01 for heavy based on manual inspection for irregular time profiles of the resulting ranked list.
Statistical Comparison of Insulin and Control msyn and mdeg Rates
Of the insulin-regulated proteins, which are those that rejected the null hypothesis above (9), we determined whether msyn, mdeg, or both parameters contributed to the changes with insulin. Expressed mathematically, we tested whether α1, β1, or both were non-zero (5–8). We evaluated this hypothesis by examining the joint probability distributions of α1 and β1; this distribution illustrates the likely values of α1 and β1 (Fig. 6C). The joint probability distributions of α1 and β1 were generated by the following steps. Initially, the joint likelihood distributions of α0 and β0 in the control conditions were constructed (Fig. 6A) from the following likelihood estimator equation.
| (Eq. 11) |
where ϵcon is the sum of the squared residuals weighted to the standard deviation. Similarly, the joint likelihood distribution of α0 + α1 and β0 + β1 under insulin conditions is given by the following equation.
| (Eq. 12) |
FIGURE 6.
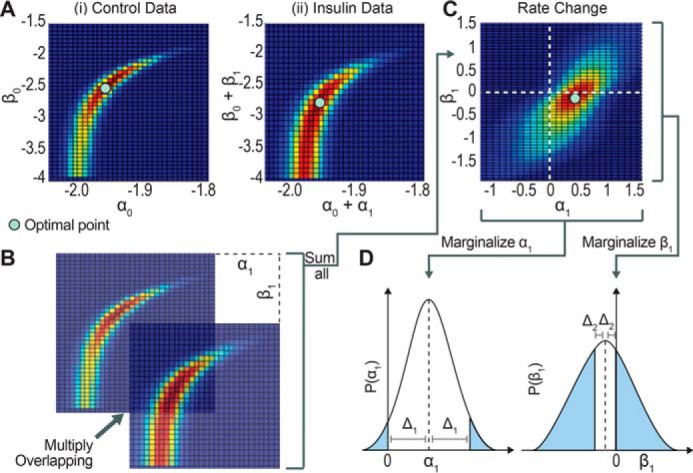
Sensitivity analysis of synthesis and degradation parameters to determine significance of their change using maximum likelihood. A, the joint likelihood distribution of control with respect to α0 and β0, corresponding to msyn_con and mdeg_con, respectively (panel i), and insulin with respect to α0 + α1 and β0 +β1, corresponding to msyn_ins and mdeg_ins, respectively (panel ii). B, the joint likelihoods for control and insulin are superimposed and their likelihoods are multiplied and then summed across the entire landscape to form one grid point in the joint likelihood distribution for α1 and β1 (Equation 13). Each subsequent grid point in the joint likelihood distribution for α1 and β1 is determined by repeating the process but shifting the joint likelihood for insulin in the x and y directions. C, after performing the operation described in B for all possible offsets between the control and insulin joint likelihoods, the joint likelihood for the parameter change is formed. D, the marginalized probability distribution for α1 and β1, the change in the synthesis and degradation rate, respectively, are obtained by first marginalizing the joint likelihood distribution in C and then normalizing the integral to 1. The p value is represented by the blue area under the curve and is a measure of the distance of the distribution away from zero.
Finally, the joint likelihood distributions of α0 and β0 and of α0 + α1 and β0 + β1 were then used to construct the joint probability distributions for α1 and β1 (Fig. 6, B and C) using the following equation.
| (Eq. 13) |
where E, a normalization factor, is defined as follows.
| (Eq. 14) |
Next, the marginal probability distribution for just the α1 or β1 variable was obtained by numerically integrating with respect to β1 and α1, respectively (Fig. 6D). The p values for the marginal variables α1 and β1 were found by first calculating the distance between the mean and zero (Δ) and then calculating the area under the curve outside the interval mean − Δ and mean + Δ (Fig. 6D, shaded area). The p values were used to test the significance of the deviation of the marginal probabilities of α1 and β1 from zero.
Gene Ontology and Transcription Factor Analysis
Insulin-regulated proteins or long lived proteins were analyzed for enriched gene ontology categories (p < 0.01) using DAVID (75). Enriched transcription factors were identified through a hypergeometric test using the Molecular Signatures Database version 4.0 (76) and the “C3 motif gene set” (which is based on conserved cis-regulatory motifs between humans and other model organisms) curated from the TRANSFAC (77) database. Proteins that fit the models of synthesis with adj. R2 >0.8 were used as the background for this, and p value significance was defined by controlling for 5% false discovery rate in this test.
Data Quantification and General Statistical Analysis
Immunoblots were quantified by densitometry analysis using ImageJ software. t tests and analyses of variance were done in GraphPad Prism version 6.01 for Windows (GraphPad Software).
Luciferase Plasmids, Transfection, and Luciferase Assay
pCIneo-WT-Fluc and pCIneo-DM-Fluc were kindly provided by Dr. Ulrich Hartl and Dr. Swasti Raychaudhuri (Max Planck Institute) (49). pEGFP-N1 was from Clontech. pRK7-myc-Rheb was kindly provided by Dr. John Blenis (Weill Cornell Medical College) (78). WT Fluc and DM Fluc were PCR-amplified and subcloned into pBabepuro. For stable overexpression, 3T3-L1 fibroblasts were infected with pBabepuro-WT-Fluc or pBabepuro-DM-Fluc retrovirus. HEK293 cells were co-transfected with pEGFP-N1 (Clontech) or pRK7-myc-Rheb in combination with pCIneo-WT-Fluc at a 1:1 molar ratio using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer's instructions for 6 h. After transfection, the culture medium was replenished with DMEM/FCS, and cells were treated with the indicated drugs for 16 h and then harvested. To assay luciferase activity, cells were lysed in 100 mm potassium phosphate, pH 7.8, and 0.2% Triton X-100 by passing through a 27-gauge needle. Equivalent amounts of protein were assayed for luciferase activity with the Luciferase Assay System (Promega).
Detergent-insoluble Proteins
Cells were lysed by passing through a 27-gauge needle in Triton buffer (1% Triton X-100, 100 mm NaCl, 10 mm Tris, 10 μm MG132, and 1 mm N-ethylmaleimide). Cell lysates were spun at 20,000 × g for 30 min at 4 °C. The resulting pellet was transferred to a new Eppendorf tube, resuspended in Triton buffer containing 30% sucrose by passing through a 27-gauge needle, and then spun for 100,000 × g for 30 min at 4 °C. This step was repeated twice more to the cellular pellet. The final pellet was solubilized in 8 m urea, 2% SDS, and 50 mm Tris by sonication, and protein content was determined by BCA assay.
Carbonylation
Protein carbonylation in freshly prepared extracts was measured by derivatization of carbonyl groups with 2,4-dinitrophenyl (DNP) hydrazine followed by detection of DNP-derivatized protein by immunoblotting using an OxyBlot Protein Oxidation Detection kit (S7150, Millipore). Membranes were then probed for tubulin, and DNP-derivatized protein levels were normalized to tubulin levels.
Proteasome Activity
Cells were solubilized by 5-min gentle agitation in proteasome lysis buffer (50 mm Tris, pH 7.5, 250 mm sucrose, 5 mm MgCl2, 2 mm ATP, 1 mm DTT, 0.5 mm EDTA, and 250 μg/ml digitonin). Samples with equivalent amounts of protein were assayed for proteasome activity with Proteasome-GloTM Chymotrypsin-Like Cell-Based Reagent (G8660, Promega).
Animals
Rat tissue was generated previously (46). Briefly, adult male Wistar rats were anesthetized, and cannulas were implanted into the right jugular vein and left carotid artery and exteriorized at the back of the neck. After surgery, rats were housed in separate cages, and 3 days after surgery rats were infused with 0.9% saline or 50% glucose in water at 40 mg/kg/min via the carotid cannula using a peristaltic roller pump. After 1 and 4 days of glucose infusion, rats were sacrificed, and tissues were rapidly dissected, frozen in liquid nitrogen, and stored at −80 °C. Animal experiments were approved by the Garvan Institute/St. Vincent's Hospital Animal Ethics Committee.
Data Accessibility
All raw and processed data associated with the manuscript have been deposited in PRIDE ProteomeXchange (project accession PXD003696).
Author Contributions
A. Y. M. and D. E. J. designed experiments and wrote the manuscript. A. Y. M. performed the majority of the experiments. M. K. L. W. designed and performed the majority of the statistical analysis of proteomic data. R. C., J. Y. Y., and A. C. F. C. contributed to the statistical analysis of protein turnover data. S.-X. T. and J. S. assisted with study design. J. S. cloned DNA constructs. S. J. H. established MS methods. B. L. P. and S. J. H. ran MS samples. D. R. L. and G. J. C. performed experiments on rats. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank members of the James laboratory, in particular Daniel Fazakerley, for valuable discussions and Swasti Raychaudhuri for luciferase constructs.
This work was supported in part by National Health and Medical Research Council (NHMRC) Project Grants GNT1061122 and GNT1086850 (to D. E. J.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S4.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession number PXD003696.
- ISP
- insulin signaling pathway
- ER
- endoplasmic reticulum
- SILAC
- stable isotope-labeled amino acids in cell culture
- M
- medium isotopes
- L
- light isotopes
- H
- heavy isotopes
- msyn
- synthesis rate of M
- mdeg
- degradation rate of M
- hsyn
- synthesis rate of H
- hdeg
- degradation rate of H
- UPR
- unfolded protein response
- mTOR
- mechanistic target of rapamycin
- adj.
- adjusted
- Fluc
- firefly luciferase
- df
- degrees of freedom
- DNP
- 2,4-dinitrophenyl
- DM
- destabilized mutant
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- FOXO
- Forkhead box class O family member protein.
References
- 1. Proud C. G. (2006) Regulation of protein synthesis by insulin. Biochem. Soc. Trans. 34, 213–216 [DOI] [PubMed] [Google Scholar]
- 2. Ma X. M., and Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 3. Shanik M. H., Xu Y., Skrha J., Dankner R., Zick Y., and Roth J. (2008) Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31, Suppl. 2, S262–S268 [DOI] [PubMed] [Google Scholar]
- 4. Mehran A. E., Templeman N. M., Brigidi G. S., Lim G. E., Chu K. Y., Hu X., Botezelli J. D., Asadi A., Hoffman B. G., Kieffer T. J., Bamji S. X., Clee S. M., and Johnson J. D. (2012) Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 16, 723–737 [DOI] [PubMed] [Google Scholar]
- 5. Ortega-Molina A., Lopez-Guadamillas E., Mattison J. A., Mitchell S. J., Muñoz-Martin M., Iglesias G., Gutierrez V. M., Vaughan K. L., Szarowicz M. D., González-García I., López M., Cebrián D., Martinez S., Pastor J., de Cabo R., et al. (2015) Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell Metab. 21, 558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang X., Mei S., Gu H., Guo H., Zha L., Cai J., Li X., Liu Z., and Cao W. (2014) Exposure to excess insulin (glargine) induces type 2 diabetes mellitus in mice fed on a chow diet. J. Endocrinol. 221, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Selman C., Lingard S., Choudhury A. I., Batterham R. L., Claret M., Clements M., Ramadani F., Okkenhaug K., Schuster E., Blanc E., Piper M. D., Al-Qassab H., Speakman J. R., Carmignac D., Robinson I. C., et al. (2008) Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 22, 807–818 [DOI] [PubMed] [Google Scholar]
- 8. Holzenberger M., Dupont J., Ducos B., Leneuve P., Géloën A., Even P. C., Cervera P., and Le Bouc Y. (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187 [DOI] [PubMed] [Google Scholar]
- 9. Tatar M., Kopelman A., Epstein D., Tu M. P., Yin C. M., and Garofalo R. S. (2001) A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292, 107–110 [DOI] [PubMed] [Google Scholar]
- 10. Finkel T. (2015) The metabolic regulation of aging. Nat. Med. 21, 1416–1423 [DOI] [PubMed] [Google Scholar]
- 11. Hands S. L., Proud C. G., and Wyttenbach A. (2009) mTOR's role in ageing: protein synthesis or autophagy? Aging 1, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen E., and Dillin A. (2008) The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 9, 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 14. Otoda T., Takamura T., Misu H., Ota T., Murata S., Hayashi H., Takayama H., Kikuchi A., Kanamori T., Shima K. R., Lan F., Takeda T., Kurita S., Ishikura K., Kita Y., et al. (2013) Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes 62, 811–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang L., Li P., Fu S., Calay E. S., and Hotamisligil G. S. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bjedov I., Toivonen J. M., Kerr F., Slack C., Jacobson J., Foley A., and Partridge L. (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 11, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hipp M. S., Park S. H., and Hartl F. U. (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514 [DOI] [PubMed] [Google Scholar]
- 18. Kaushik S., and Cuervo A. M. (2015) Proteostasis and aging. Nat. Med. 21, 1406–1415 [DOI] [PubMed] [Google Scholar]
- 19. Demontis F., and Perrimon N. (2010) FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143, 813–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taylor R. C., Berendzen K. M., and Dillin A. (2014) Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat. Rev. Mol. Cell Biol. 15, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan S. X., Fisher-Wellman K. H., Fazakerley D. J., Ng Y., Pant H., Li J., Meoli C. C., Coster A. C., Stöckli J., and James D. E. (2015) Selective insulin resistance in adipocytes. J. Biol. Chem. 290, 11337–11348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walther D. M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R. I., Dobson C. M., Vendruscolo M., Mann M., and Hartl F. U. (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 24. Schwanhäusser B., Gossen M., Dittmar G., and Selbach M. (2009) Global analysis of cellular protein translation by pulsed SILAC. Proteomics 9, 205–209 [DOI] [PubMed] [Google Scholar]
- 25. Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., and Rajewsky N. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 [DOI] [PubMed] [Google Scholar]
- 26. Iadevaia V., Huo Y., Zhang Z., Foster L. J., and Proud C. G. (2012) Roles of the mammalian target of rapamycin, mTOR, in controlling ribosome biogenesis and protein synthesis. Biochem. Soc. Trans. 40, 168–172 [DOI] [PubMed] [Google Scholar]
- 27. Kristensen A. R., Gsponer J., and Foster L. J. (2013) Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 9, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gawron D., Ndah E., Gevaert K., and Van Damme P. (2016) Positional proteomics reveals differences in N-terminal proteoform stability. Mol. Syst. Biol. 12, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., and James D. E. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abel E. D., Peroni O., Kim J. K., Kim Y. B., Boss O., Hadro E., Minnemann T., Shulman G. I., and Kahn B. B. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409, 729–733 [DOI] [PubMed] [Google Scholar]
- 31. Ruan H., Zarnowski M. J., Cushman S. W., and Lodish H. F. (2003) Standard isolation of primary adipose cells from mouse epididymal fat pads induces inflammatory mediators and down-regulates adipocyte genes. J. Biol. Chem. 278, 47585–47593 [DOI] [PubMed] [Google Scholar]
- 32. Govers R., Coster A. C., and James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boisvert F. M., Ahmad Y., Gierliński M., Charriere F., Lamont D., Scott M., Barton G., and Lamond A. I. (2012) A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell. Proteomics 11, M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robles M. S., Cox J., and Mann M. (2014) In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet. 10, e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander M., Curtis G., Avruch J., and Goodman H. M. (1985) Insulin regulation of protein biosynthesis in differentiated 3T3 adipocytes. Regulation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 260, 11978–11985 [PubMed] [Google Scholar]
- 36. Ong J. M., Kirchgessner T. G., Schotz M. C., and Kern P. A. (1988) Insulin increases the synthetic rate and messenger RNA level of lipoprotein lipase in isolated rat adipocytes. J. Biol. Chem. 263, 12933–12938 [PubMed] [Google Scholar]
- 37. Sargeant R. J., and Pâquet M. R. (1993) Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem. J. 290, 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conn C. S., and Qian S. B. (2013) Nutrient signaling in protein homeostasis: an increase in quantity at the expense of quality. Sci. Signal. 6, ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ozcan U., Ozcan L., Yilmaz E., Düvel K., Sahin M., Manning B. D., and Hotamisligil G. S. (2008) Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 29, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tyagi R., Shahani N., Gorgen L., Ferretti M., Pryor W., Chen P. Y., Swarnkar S., Worley P. F., Karbstein K., Snyder S. H., and Subramaniam S. (2015) Rheb inhibits protein synthesis by activating the PERK-eIF2α signaling cascade. Cell Rep. 10, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hein M. Y., Hubner N. C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I. A., Weisswange I., Mansfeld J., Buchholz F., Hyman A. A., and Mann M. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 [DOI] [PubMed] [Google Scholar]
- 42. Okabayashi Y., Maddux B. A., McDonald A. R., Logsdon C. D., Williams J. A., and Goldfine I. D. (1989) Mechanisms of insulin-induced insulin-receptor downregulation. Decrease of receptor biosynthesis and mRNA levels. Diabetes 38, 182–187 [DOI] [PubMed] [Google Scholar]
- 43. Toyama B. H., Savas J. N., Park S. K., Harris M. S., Ingolia N. T., Yates J. R. 3rd, and Hetzer M. W. (2013) Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teixeira L. K., and Reed S. I. (2013) Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 82, 387–414 [DOI] [PubMed] [Google Scholar]
- 45. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., and Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laybutt D. R., Chisholm D. J., and Kraegen E. W. (1997) Specific adaptations in muscle and adipose tissue in response to chronic systemic glucose oversupply in rats. Am. J. Physiol. Endocrinol. Metab. 273, E1–E9 [DOI] [PubMed] [Google Scholar]
- 47. Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frydman J., Erdjument-Bromage H., Tempst P., and Hartl F. U. (1999) Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat. Struct. Biol. 6, 697–705 [DOI] [PubMed] [Google Scholar]
- 49. Gupta R., Kasturi P., Bracher A., Loew C., Zheng M., Villella A., Garza D., Hartl F. U., and Raychaudhuri S. (2011) Firefly luciferase mutants as sensors of proteome stress. Nat. Methods 8, 879–884 [DOI] [PubMed] [Google Scholar]
- 50. Cohen E., Paulsson J. F., Blinder P., Burstyn-Cohen T., Du D., Estepa G., Adame A., Pham H. M., Holzenberger M., Kelly J. W., Masliah E., and Dillin A. (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. King M. A., Hands S., Hafiz F., Mizushima N., Tolkovsky A. M., and Wyttenbach A. (2008) Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol. 73, 1052–1063 [DOI] [PubMed] [Google Scholar]
- 52. Cohen E., Bieschke J., Perciavalle R. M., Kelly J. W., and Dillin A. (2006) Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610 [DOI] [PubMed] [Google Scholar]
- 53. David D. C., Ollikainen N., Trinidad J. C., Cary M. P., Burlingame A. L., and Kenyon C. (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 8, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Novosyadlyy R., Kurshan N., Lann D., Vijayakumar A., Yakar S., and LeRoith D. (2008) Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 15, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 55. Humbert S., Bryson E. A., Cordelières F. P., Connors N. C., Datta S. R., Finkbeiner S., Greenberg M. E., and Saudou F. (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 [DOI] [PubMed] [Google Scholar]
- 56. Di Carlo M., Picone P., Carrotta R., Giacomazza D., and San Biagio P. L. (2010) Insulin promotes survival of amyloid-beta oligomers neuroblastoma damaged cells via caspase 9 inhibition and Hsp70 upregulation. J. Biomed. Biotechnol. 2010, 147835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carro E., Trejo J. L., Gomez-Isla T., LeRoith D., and Torres-Aleman I. (2002) Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 8, 1390–1397 [DOI] [PubMed] [Google Scholar]
- 58. Zhang Y., Nicholatos J., Dreier J. R., Ricoult S. J., Widenmaier S. B., Hotamisligil G. S., Kwiatkowski D. J., and Manning B. D. (2014) Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Riley B. E., Kaiser S. E., Shaler T. A., Ng A. C., Hara T., Hipp M. S., Lage K., Xavier R. J., Ryu K. Y., Taguchi K., Yamamoto M., Tanaka K., Mizushima N., Komatsu M., and Kopito R. R. (2010) Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 191, 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Simonsen A., Cumming R. C., Brech A., Isakson P., Schubert D. R., and Finley K. D. (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 [DOI] [PubMed] [Google Scholar]
- 61. Chou S. D., Prince T., Gong J., and Calderwood S. K. (2012) mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One 7, e39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Santagata S., Mendillo M. L., Tang Y. C., Subramanian A., Perley C. C., Roche S. P., Wong B., Narayan R., Kwon H., Koeva M., Amon A., Golub T. R., Porco J. A. Jr., Whitesell L., and Lindquist S. (2013) Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341, 1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boden G., and Merali S. (2011) Measurement of the increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Methods Enzymol. 489, 67–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boden G., Cheung P., Salehi S., Homko C., Loveland-Jones C., Jayarajan S., Stein T. P., Williams K. J., Liu M. L., Barrero C. A., and Merali S. (2014) Insulin regulates the unfolded protein response in human adipose tissue. Diabetes 63, 912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gregor M. F., and Hotamisligil G. S. (2007) Thematic review series: adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J. Lipid Res. 48, 1905–1914 [DOI] [PubMed] [Google Scholar]
- 66. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., and Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 67. Winnay J. N., Boucher J., Mori M. A., Ueki K., and Kahn C. R. (2010) A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 16, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park S. W., Zhou Y., Lee J., Lu A., Sun C., Chung J., Ueki K., and Ozcan U. (2010) The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 70. Wiśniewski J. R., Zougman A., and Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 [DOI] [PubMed] [Google Scholar]
- 71. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 72. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 73. Benjamini Y., Drai D., Elmer G., Kafkafi N., and Golani I. (2001) Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284 [DOI] [PubMed] [Google Scholar]
- 74. R Development Core Team (2008) R: a Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 75. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 76. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Matys V., Kel-Margoulis O. V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K., Voss N., Stegmaier P., Lewicki-Potapov B., Saxel H., Kel A. E., et al. (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tee A. R., Manning B. D., Roux P. P., Cantley L. C., and Blenis J. (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed data associated with the manuscript have been deposited in PRIDE ProteomeXchange (project accession PXD003696).