

Abstract
Eotaxin-2 is a potent chemoattractant. High concentration of eotaxin-2 triggers the inflammation and tumor metastasis. Inhibition of eotaxin-2 may protect experimental atherogenesis although the mechanism is still unclear. Toll-like receptor 4 (TLR4) plays a major role mediating vascular inflammation, which is related to atherogenesis. In the results, we demonstrated that eotaxin-2 potentially impairs the tube formation capacity of human coronary artery endothelial cells (HCAECs). Eotaxin-2 augments the monocytic adhesion in lipopolysaccharides (LPS)-induced HCAECs, and which were reversed by TLR4 siRNA. Thus this study was conducted to investigate whether eotaxin-2 increases TLR4 expression, and then enhances the sensitivity of cells to antigen stimulation in HCAECs, which mediates the increasing of the development of serious atherosclerosis. In fact, we showed that JNK/SAPK, p38 MAPK, and ERK1/2 activation contribute to the transcriptional signaling pathway, JNK/SAPK and p38 MAPK regulate post-transcriptional modification, as well as protein-trafficking pathway in eotaxin-2-treated HCAECs TLR4 expression. RNA binding proteins, such as human antigen R (HuR) and tristetraprolin (TTP) mediate stability of TLR4 mRNA and chaperone, such as PRAT4A (a protein associated with TLR4) regulate trafficking of TLR4 protein might confer eotaxin-2 responsiveness. Eotaxin-2 administration led to a significant elevation of high cholesterol diet-induced atherosclerosis, and of TLR4 expression in B6.129S7-Ldlrtm1Her/J but not Ldlr-/--/-/ Tlr4-/- mice. Our results revealed that eotaxin-2 induced overexpression TLR4 via mitogen-activated protein kinases (MAPK) signaling pathways, RNA binding proteins-mediated mRNA stabilization, and PRAT4A-regulated trafficking in HCAECs. These effects may lead to amplification of inflammatory responses contribute to the pathogenesis of cardiovascular disorders.
Keywords: Eotaxin-2, toll-like receptor 4, atherosclerosis, endothelial cells
Introduction
Eotaxin-2 is a potent chemoattractant and binds to C-C chemokine receptor-3 (CCR3) for intracellular messaging, and quickly accumulate the eosinophils. Additionally, respiratory epithelial cells, bronchial smooth muscle cells, vascular endothelial cells, fibroblasts, monocytes, helper T cells, basophils et al. also express CCR3 [1,2] and response to eotaxin-2 stimulation [3,4], suggesting eotaxin-2 involves several cellular responses. Eotaxin-2 production is differentially regulated in monocytes and macrophages, resulting from monocytes-derived eotaxin-2 and macrophages-derived eotaxin-2 are implicated in innate and adaptive immunity, respectively [5]. Epithelial eotaxin-2 expression may contribute to eosinophils migration in asthma onset, control, and severity [6]. However, susceptibility of patients with severe asthma may be due to high plasma level of eotaxin-2, which may be associated with the +1272 A to G polymorphism and ht2 as well as ht6 haplotypes in the eotaxin-2 gene [7]. monocytes-derived CD16+ macrophages produce eotaxin-2 and then activate T cells for HIV infection [8], and eotaxin-2 involves in the mechanisms of CD4+ lymphocytes activation induced by lentiviral protein [9]. High concentration of eotaxin-2 strongly triggers T cells migration and associates with metastatic tumor of colorectal origin [10]. Interestingly, inhibition of eotaxin-2 by antibodies has an efficient protection in experimental atherosclerosis and arthritis [11,12] although the pathogenic mechanism is still unclear.
Toll-like receptors 4 (TLR4) are type I transmembrane receptors that expressed on the cell membrane and response to lipopolysaccharide (LPS) stimulation [13]. Previous evidence has demonstrated that the expression of TLR4 is abundantly in endothelial cells in macrophages infiltrating lipid-rich atherosclerotic lesions [14] and that a repertoire of TLR4 is associated with augmentation of intimal hyperplasia [15,16]. Endogenous and pathogenic heatshock protein also activate endothelial cells through TLR4 sequentially induce vascular disturbance [17,18]. Additionally, TLR4 signaling augmented TLR2 expression, resulting in the intracellular adhesion molecule-1 expression in endothelial cells [19]. Even though upregulation of TLR4 enhances by endothelial cell expression, which accelerates atherogenesis in the presence of hypercholesterolemia [18,20], we hypothesized that eotaxin-2 may increase TLR4 expression in the endothelium, which mediates the increasing of inflammatory response and accelerating the development of serious atherosclerosis. Thus, the aim of this study was to explore the cellular events and the underlying mechanisms involved in eotaxin-2-induced TLR4 expression in human coronary endothelial cells (HCAECs) in vitro. Furthermore, we also examined whether eotaxin-2 increased neointimal hyperplasia and TLR4 expression in high cholesterol diet-feed mouse aorta. Our findings indicated that MAPK signaling pathways, RNA binding proteins-mediated mRNA stabilization, and intracellular chaperone PRAT4A may play critical roles in eotaxin-2-enhanced TLR4 expression in HCAECs, which contributes to enhancing of sensitivity for LPS stimulation and inflammatory responses. Additionally, we also demonstrated that eotaxin-2 exacerbates high-cholesterol diet-induced atherosclerotic lesion formation mediating by the presenting of TLR4 in experimental B6.129S7-Ldlrtm1Her/J mice.
Materials and methods
In vitro study
Construction of eotaxin-2 expression vectors
The open reading frame of eotaxin-2 (CCL24) was originally PCR-amplified using THP-1 cells cDNA as a template, 0.1 mM dNTPs, 0.2 mM each of gene specific primers and 1 U Pfu DNA polymerase (Promega, WI, USA) with the following program: one cycle of 95°C for 5 min; 35 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec; and 1 cycle of 60°C for 30 sec and 72°C for 10 min with a final incubation at 72°C for 10 min with 1 U Taq DNA polymerase. The eotaxin-2-specific forward and reverse primers used in the PCR reaction are Pr-CCL24-BamHI-F2: 5’aag gat cca agt ggt cat ccc ctc tcc ctg ct 3’ and Pr-CCL24-R1: 5’ ccc tcg agt tag cag gtg gtt tgg ttg cca g 3’. The amplified eotaxin-2 (CCL24) fragment was then cloned into the pCR2.1-TOPO vector (Invitrogen, CA, USA) and subsequently cloned in-frame into the BamHI and XhoI sites of the pGEX-5X-1 expression vector (GE Healthcare Amersham Biosciences, CA, USA) for expression in E. coli (DH5a).
Purification of recombinant eotaxin-2 protein
BL21 cells were transformed with the pGEX-5X-1-eotaxin-2 expression vector, and the recombinant eotaxin-2 protein was purified. Briefly, BL21 (DE3) pLysS cells (RBC Bioscience, New Taipei City, Taiwan) containing the plasmid pGEX-5X-1-eotaxin-2 were grown overnight at 37°C in 50 mL of LB medium supplemented with 100 μg/mL ampicillin. Then, 50 mL of overnight culture was transferred into 1000 mL of LB medium and grown at 16°C to an A600 of 0.6-0.8 (approximately 2 h). Expression of the fusion protein was then induced by adding IPTG to a final concentration of 1 mM at 16°C for 6 h. The bacteria were pelleted by centrifugation for 10 min at 8000 rpm, and recombinant GroEL was extracted under native conditions using the GST Gene Fusion System according to the manufacturer’s instructions (GE Healthcare Amersham Biosciences, CA, USA). Finally, the recombinant eotaxin-2 protein was purified with elution buffer containing 50 mM Tris-HCl and 10 mM reduced glutathione (pH 8.0). The quantity of recombinant eotaxin-2 protein was measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA). The fusion protein was detected by SDS gel electrophoresis and identified by immunoblotting with a GST antibody (GE Healthcare Amersham Biosciences, CA, USA). The endotoxin levels in the recombinant eotaxin-2 protein preparation were measured using a Limulus Amebocyte Lysate kit from Cambrex Inc. in the USA. The LPS levels were below 1 pg/mL.
Cell culture
Human coronary arterial endothelial cells (HCAECs) were purchased from Cascade Biologics, Inc. (Portland, OR, USA). Human monocytic THP-1 cells were purchased from American Type Culture Collection (Manassas, VA, USA). Cell culturing and passages were performed according to the manufacturer’s instructions.
Western blotting analysis
Total cell lysate and membrane proteins were processed according to previous reports [21]. The protein concentration in the supernatant was measured using a Bio-Rad protein determination kit (Bio-Rad, CA, USA). The supernatants were subjected to 8% SDS-PAGE and transferred for 1 hour at room temperature to polyvinylidene difluoride membranes. The membranes were treated for 1 hour at room temperature with PBS containing 0.05% Tween-20 and 2% skimmed milk and incubated separately for 1 hour at room temperature with primary antibodies. The membranes were then incubated with horseradish peroxidase-conjugated IgG. Immunodetection was performed using a chemiluminescence reagent and with exposure to VersaDoc Imaging System 5000 MP (Bio-Rad, CA, USA).
Measurement of cytotoxicity by MTT assay
The cytotoxicity of eotaxin-2 was analyzed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HCAECs (2×104 cells) were grown in 96-well plates and incubated with eotaxin-2 at 1-10 ng/mL for 8-24 hours. Subsequently, MTT (0.5 µg/mL) was applied to the cells for 4 hours to allow the conversion of MTT into formazan crystals. After washing with PBS, the cells were lysed with dimethyl sulfoxide, and the absorbance was read at 530 nm with a DIAS Microplate Reader (Dynex Technologies, VA, USA).
Tube formation assay
Tube formation assays were performed on HCAECs to assess angiogenic capacity, which is believed to be important for endothelium function. The in vitro tube formation assays were performed using the Angiogenesis Assay Kit (Chemicon, CA, USA) [22] according to the manufacturer’s protocol. Briefly, ECMatrix gel solution was thawed at 4°C overnight, mixed with ECMatrix diluent buffer, and placed in a 96-well plate at 37°C for 1 hour to allow the matrix solution to solidify. HCAECs were treated with eotaxin-2 for 24 hours and then harvested. A total of 104 cells were placed on the matrix solution, and the samples were incubated at 37°C for 8 hours. Tubule formation was inspected under an inverted light microscope, and five representative fields were taken. The average of the total intersection of three tubes formed by cells was calculated.
HCAECs/THP-1 cells adhesion assay
HCAECs (5×105) were distributed into 24-well plates before the assay. Then, the growth medium was supplemented with 1-10 ng/mL eotaxin-2 for 18 hours followed by 10 ng/mL LPS treatment for 8 hours. THP-1 cells were labeled for 1 h at 37°C with 10 μM of 2,7,-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF/AM, Boehringer-Mannheim) in serum-free RPMI 1640 medium; they were then washed with PBS to remove free dye and resuspended in RPMI 1640 containing 2% FBS. One million labeled THP-1 cells were added to each HCAEC-containing well, and incubation continued for 1 h. Non-adherent cells were removed by three gentle washes with HBSS. The degree of THP-1 cells adhered to the HCAECs was observed using inverted fluorescent microscopy and counted using a Multilabel Counter Victor2 (Wallace, CA, USA) at an emission of 530 nm and an absorption of 435 nm after the cells were lysed with DMSO.
Quantitative real time polymerase chain reaction and actinomycin D chase experiment
Total RNA was isolated using a TRIZOL reagent kit (Invitrogen, CA, USA). cDNA was synthesized from total RNA using Superscript® II reverse transcriptase. Quantitative real time polymerase chain reaction (PCR) was performed using a FastStart DNA Master SYBR Green I kit and LightCycler (Roche, CA, USA). FastStart Taq DNA polymerase was activated by incubation at 95°C for 2 min before 40 cycles of 95°C for 1 s, 60°C for 5 s, and 72°C for 7 s. Fluorescence was measured at 86°C after the 72°C extension step. The level of TLR4 mRNA expression was determined in arbitrary units by comparison with an external DNA standard that was amplified by the TLR4 primers. Actinomycin D (20 μg/mL) was added to cells for 1 h following their treatment under 5 and 10 ng/mL of eotaxin-2 for 4 hours. Total RNA was extracted at 0, 30, 60, 120, 240, and 300 min after the addition of actinomycin D, and quantitative real time PCR was performed. The half-life (t1/2) of the TLR4 mRNA was calculated according to the following formula t1/2 = 0.693/k, where k = ln (N0/Nt)/t in which N0 is the cross-point of real-time PCR at t = 0 and Nt is the cross-point at time t. The PCR primers used for amplification of TLR4 and GAPDH were: TLR4 forward primer: 5’-AAG CCG AAA GGT GAT TGT TG-3’, reverse primer: 5’-CTG TCC TCC CAC TCC AGG TA-3’; GAPDH forward primer: 5’-TGC CCC CTC TGC TGA TGC C-3’, reverse primer: 5’-CCT CCG ACG CCT GCT TCA CCA C-3’. All specific primers were synthesized by Sigma-Aldrich (MO, USA).
Immunofluorescent staining
For immunofluorescent staining, cells were plated on cover slips, grown to confluence, and then treated with 10 ng/mL of eotaxin-2 for 4 hours. After the treatment, the cells were fixed with 4% formaldehyde. Cell membranes were fenestrated using 0.4% Triton-100, and nonspecific binding sites were blocked with 2% BSA-Tween 20 (0.1% v/v). The cells were incubated with mouse anti-human antigen R (HuR), AU-rich element RNA-binding protein 1 (AUF1) or tristetraprolin (TTP) antibodies (Chemicon, CA, USA) and then incubated with the secondary antibody conjugated to fluorescein isothiocyanate (FITC). The 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) was used to identify the nuclei. The slides were observed with fluorescent microscopy.
Luciferase reporter assay
Functional analysis of the 3’UTR of TLR4 mRNA was performed using plasmids containing the 3’UTR and a luciferase reporter gene (from pGL-Basic vector (Promega, USA). we had generated a construction of luciferase reporter plasmids containing the 3’ UTR of TLR4 mRNA for expression in mammalian cells [23]. The following specific primers were used in the PCR reaction: TLR4-3’UTR Pr-forward: TGACC CACAA GTBAA AAAGG and TLR4-3’UTR Pr-reverse: TCCCA GCCAT CTGTG TCTC. A total of 106 cells were trypsinized and resuspended in 100 μl of Nucleofector solution; 1 μg of the reporter plasmid (CMV-Luciferase-TLR4 3’UTR sense, and CMV-Luciferase-TLR4 3’UTR antisense) was transfected using the Nucleofector electroporation device according to the manufacturer’s instructions. Equal amounts of the luciferase reporter gene containing pcDNA3.1 plasmid (CMV-Luciferase) were used as a control group. Transfection efficiency was normalized to uniformity using a β-galactosidase reporter plasmid. Cell extracts were prepared with reporter lysis buffer (300 µl/well), protein concentrations were determined, and the luciferase activity was quantified by luminometry (Wallac Victor2, Finland) using the luciferase assay system (Promega, CA, USA). β-galactosidase activity was measured using a β-galactosidase enzyme assay kit (Stratagene, CA, USA).
Cross-linking Immunoprecipitation for RNA-protein Interaction
To determine whether TTP interacts directly with the 3’UTR of TLR4 mRNA, we carried out immunoprecipitation and RT-PCR with modifications based on a previous report [24]. To induce cross-linking, we irradiated cells in ice-cold PBS directly with 4000 mJ of ultraviolet B light three times. The cells were lysed with cold cell lysis buffer, and RNA-protein complexes were extracted by centrifugation. For immunoprecipitation, 500 μg of cytoplasmic fraction aliquots was incubated with protein G-sepharose and 10 μg of an antibody that recognizes TTP. Western blot analysis was conducted to determine the levels of TTP in HCAECs. Protein extracts were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene difluoride membrane. The membrane was probed with a rabbit anti-TTP antibody and then incubated with horseradish peroxidase-conjugated secondary antibody. The blot was developed using enzyme-linked chemiluminescence detection reagents. The RNA in the immunoprecipitated material was used in quantitative real-time PCR reactions to detect the presence of the 3’ UTR of TLR4 mRNA. The mRNA was reverse transcribed using a Reverse-iTi 1st Strand Synthesis Kit (ABgene, Epsom, UK), followed by quantitative real-time PCR to measure the 3’ UTR transcript levels. The PCR primers designed for the 3’UTR of TLR4 mRNA were 5’-GAA CTG GGT GTT CAC TTT TTC C-3’ and 5’-ATC CCA GCC ATC TGT GTC TC-3’.
Animal study
Ethics statement
All animals were treated according to protocols approved by the Institutional Animal Care Committee of the National Defense Medical Center (certificate No.: IACUC-07-172). Experimental procedures and animal care conformed to the “Guide for the Care and Use of Laboratory Animals” published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Animal grouping and experiment
All animals were kept in microisolator cages on a 12-h day/night cycle and fed normal mouse chow diet (Scientific Diet Services, Essex, UK) or DIO rodent purified high cholesterol (HC) diet (TestDiet, MO, USA) with water ad libitum. Twenty male B6.129S7-Ldlrtm1Her/J mice (a homozygous Ldlrtm1Her mutation mouse have an elevated cholesterol level when fed a high fat diet; JAX®, 002207, Jackson Laboratory, ME, USA) were used. B6.B10ScN-Tlr4lps-del/JthJ females (a TLR4-knockout mouse homozygous for the defective LPS-response deletion allele Tlr4lps-del, Jackson Laboratory, ME, USA) were bred with B6.129S7-Ldlrtm1Her/J males to obtain mice lacking both Ldlr and Tlr4 (here after collectively referred to as Ldlr-/-/Tlr4-/- double knockout mice). Five male Ldlr-/-/Tlr4-/- mice were included in this study. The animals were divided into five groups (5 animals/per group). B6.129S7-Ldlrtm1Her/J mice were included in groups 1-4 and Ldlr-/-/Tlr4-/- mice were included in group 5. Group 1 (naïve control): B6.129S7-Ldlrtm1Her/J mice fed with normal chow diet; group 2: B6.129S7-Ldlrtm1Her/J mice fed with high cholesterol (HC) diet; group 3: B6.129S7-Ldlrtm1Her/J mice fed with HC diet and receiving a tail vein injection of 5 ng/kg body weight (BW) of eotaxin-2 twice a week throughout the experiment (42 days); group 4: B6.129S7-Ldlrtm1Her/J mice fed with HC diet and receiving a tail vein injection of 10 ng/kg BW of eotaxin-2; group 5: Ldlr-/-/ Tlr4-/- mice fed with HC diet and receiving a tail vein injection of 10 ng/kg BW of eotaxin-2; At the end of the experiment (day 42), the mice were sacrificed and the thoracic aorta were removed.
Immunohistochemistry
The animals were sacrificed at the end of week 6, and the thoracic aortas were harvested, gently dissected free of adherent tissues, rinsed with ice-cold phosphate buffered saline, immersion-fixed with 4% buffered paraformaldehyde, paraffin-embedded, and 5-µm-thick paraffin-embedded cross-sections of mouse aortas were stained. Immunohistochemical staining used host anti-TLR4 (Biossusa, MA, USA), host anti-HuR (abcam, CA, USA), host anti-TTP (Novusbio, CA, USA), and host anti-PRAT4A (Santa Cruz, CA, USA) antibodies. The slides were observed via microscopy.
Statistical analysis
Results were expressed as the mean ± SEM. Data were analyzed using ANOVA followed by the Dunnett’s test. A p value less of than 0.05 was considered statistically significant.
Results
HCAECs expressed CCR3 and eotaxin-2 decreased tube formation in non-impaired situation of cell viability in HCAECs: CCR3 is the specific receptor of eotaxin-2, therefore to confirm its expression before eotaxin-2 stimulation was necessary in HCAECs. Indeed, the western blotting evidenced that CCR3 was expressed in HCAECs (Figure 1A). An MTT assay was performed to analyze cell viability and the cytotoxicity of eotaxin-2. HCAECs were treated with 1-10 ng/mL of eotaxin-2 for 8, 12, or 24 hours. The results showed that treatment with eotaxin-2 did not affect cell viability (Figure 1B). The deficiency of capillary network formation of endothelial cells is believed to be important issues during atherogenesis. Therefore, in vitro tube formation assays were performed. After 24 hours of culture in 5 or 10 ng/mL eotaxin-2, the functional capacity for tube formation of HCAECs on ECMatrix gel was significantly decreased compared to the control group (5 ng/mL eotaxin-2: 45.7±9.8% of the control, and 10 ng/mL eotaxin-2: 26.4±5.9% of the control) (Figure 1C and 1D). These results indicate that eotaxin-2 (approximately 5-10 ng/mL) potentially decreases the tube formation capacity of HCAECs, which are involved in the lining of endothelium, whereas 1-10 ng/mL eotaxin-2 did not induce endothelial cell cytotoxicity.
Figure 1.
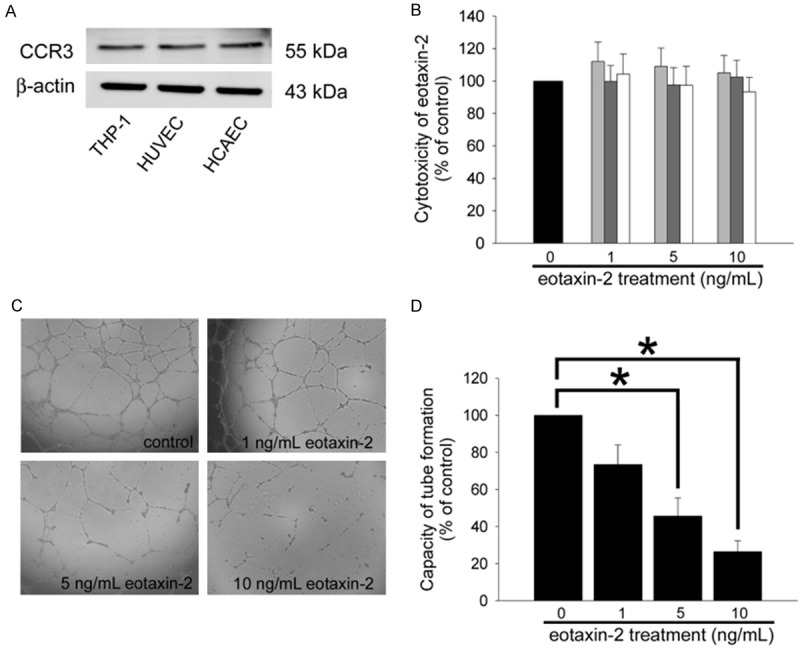
Eotaxin-2 impaired the function of HCAECs without impairing their viability. A: The total protein was extracted from THP-1 cells, HCAECs, and human umbilical vein endothelial cells (HUVECs), CCR3 were analyzed by western blot. β-actin protein levels were used as a loading control. B: Treatment of HCAECs with eotaxin-2 protein for 8-24 hours; cell cytotoxicity of eotaxin-2 was analyzed by MTT assay, and the absorbance was recorded using a microplate reader. C: HCAECs were pretreated with eotaxin-2 for 24 hours. An in vitro tube formation assay was performed using ECMatrix gel to investigate the effect of eotaxin-2 on the HCAECs’ lining function. D: The photographs were taken 24 hours after seeding of HCAECs. All data are expressed as the mean ± SEM of five experiments performed in triplicate and as a percentage of the control. Statistical evaluation was performed using one-way ANOVA followed by a Dunnett’s test. *P<0.05 was considered significant.
Eotaxin-2 affects HCAEC functions mediated by enhancing of TLR4 expression: Lipopolysaccharides (LPS) induces endothelial cells inflammation which resulting from atherosclerosis. We analyzed whether eotaxin-2 aggravates the sensitivity to LPS in HCAECs. In Figure 2A, the confluent control HCAECs showed minimal adhesiveness to THP-1 cells, but adhesion was substantially increased when the HCAECs were treated with 10 ng/mL of LPS for 8 hours (146.8±18.4% of the control). However, HCAECs were pretreated with 5 and 10 ng/mL of eotaxin-2 for 18 hours followed by LPS treatment, the adhesiveness of THP-1 cells were sever increased than that in LPS-treatment group (199.4±14.0% of the control, and 221.4±19.7% of the control, respectively). In order confirm the fact that eotaxin-2 affects HCAECs function mediating by regulation of TLR4 expression, TLR4 expression on HCAECs were blocked by TLR4 siRNA before eotaxin-2 and LPS treatment. The result showed that TLR4 siRNA may reverse the increasing of THP-1 cells/HCAECs adhesiveness in 10 ng/mL eotaxin-2 plus LPS treatment HCAECs (TLR4 siRNA+eotaxin-2+LPS: 95.0±15.9 of control, eotaxin-2+LPS: 230.5±24.6% of the control). This effect was not observed with the validate siRNA (NC siRNA) (Figure 2B). Additionally, adhesion molecules (such as ICAM-1 and VCAM-1) mediate the adhesion of monocytes to the endothelium to control the initial process of atherosclerosis. Therefore, we hypothesized that expression of VCAM-1 and ICAM-1 may be regulated by eotaxin-2 in LPS-treatment HCAECs. The western blot analysis evidenced that ICAM-1 and VCAM-1 were spontaneously expressed at a basal level in the control (untreated) and LPS treatment HCAECs. Eotaxin-2 treatment for 24 hours caused significant up-regulation of the expression of VCAM-1 (10 ng/mL of eotaxin-2) and ICAM-1 (5 and 10 ng/mL of eotaxin-2) in LPS-stimulated HCAECs (Figure 2C). Previous evidences had demonstrated that TLR4 plays critical roles in atherogenesis. As the result of TLR4 specific to LPS stimulation, to explore the expression of TLR4 in this study is necessary. The western blotting showed 10 ng/mL LPS and 1 ng/mL eotaxin-2 slightly, and 5-10 ng/mL eotaxin-2 significantly induce TLR4 expression in endothelial cells (Figure 2D). Similarly, TLR4 mRNA expression presented a significant dose- and time-dependent increase in eotaxin-2-induced HCAECs compared to naïve HCAECs (Figure 2E). Controlling the stability of the TLR4 mRNA modulates gene expression and efficiently adjusts inflammatory responses [25]. To determine whether eotaxin-2 affects the steady-state dynamic balance between the rate of transcription and the message stability of the TLR4 mRNA, an actinomycin D chase experiment was conducted. The mRNA half-life, deduced under various conditions according to the curve, indicated that 5-10 ng/mL of eotaxin-2 stimulation for 4 hours rapidly increased the stability of the TLR4 mRNA in HCAECs (half life of TLR4 mRNA: control group, 145.3±24.7 minutes; 5 ng/mL eotaxin-2 group, 229.7±20.3 minutes; 10 ng/mL eotaxin-2 group, 269.8±40.9 minutes; Figure 2F). These results indicate that eotaxin-2 aggregates atherogenic phenomena in endothelial cells mediated by upregulating TLR4 expression in transcriptional regulation and post transcriptional modification pathway.
Figure 2.
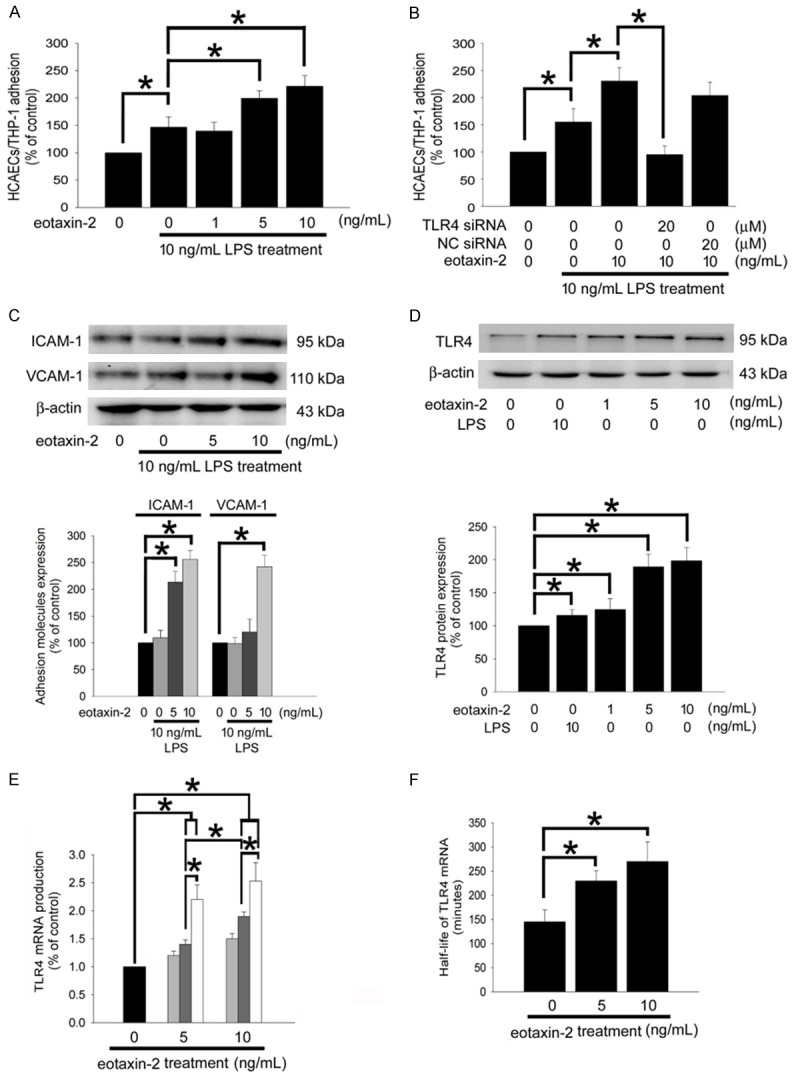
Eotaxin-2 affects HCAEC functions mediated by enhancing of TLR4 expression. A: HCAECs were pretreated with eotaxin-2 for 18 hours, followed by LPS treatment for 8 hours and then co-cultured with THP-1 cells for 1 hour. B: HCAECs were transfected with TLR4 siRNA or validate (as a negative control knockdown; NC) siRNA for 24 hours then eotaxin-2 plus LPS stimulation for 8 hours. The degree of THP-1 adhesion to the HCAECs was counted using a Multilabel Counter Victor2. C: HCAECs were stimulated for 18 hours without or with eotaxin-2 followed by LPS treatment for 8 hours. The total ICAM-1 and VCAM-1 expression were analysis using western blotting. D: HCAECs were stimulated for 18 hours with eotaxin-2 or LPS. Western blot analyses of TLR4 protein were performed. Total β-actin protein was used as a loading control. The amount of proteins expression was quantified using densitometry and presented as bar graph. E: HCAECs were stimulated for 0 (black), 4 (gray), 8 (dark gray), or 12 (white) hours with eotaxin-2. The production levels of TLR4 mRNA were analyzed by quantitative real time PCR. F: HCAECs were stimulated for 4 hours with eotaxin-2. The half-life of TLR4 mRNA was analyzed by actinomycin D chase experiment. The half-life of TLR4 mRNA was calculated according to the mRNA decay rate. All bars represent the results of three independent experiments. The data are presented as the mean ± SEM and statistical evaluation was performed using one-way ANOVA followed by a Dunnett’s test. *P<0.05 was considered significant.
Eotaxin-2 prolongs TLR4 mRNA stability and promotes TLR4 expression: Transcriptional signaling pathway and post-transcriptional modification were reported to involve in TLR4 expression. Therefore, western blotting analysis and Quantitative Real Time PCR were done to assess the activation of related signal pathways in HCAECs treated with eotaxin-2. In Figure 3A, the activation of p38 MAPK, JNK/SAPK, and ERK1/2 were significantly and quickly increased by treatment of 10 ng/mL of eotaxin-2, suggesting that the MAPKs signal pathways play potential roles in eotaxin-2-induced HCAECs. Pretreatment with SB203580 (a p38 MAPK inhibitor), SP600125 (a JNK/SAPK inhibitor), and PD98059 (an ERK1/2 inhibitor) for 1 hour may have ameliorated the positive effects of 10 ng/mL eotaxin-2 on HCAECs TLR4 mRNA production. Interestingly, exposure to PD98059 did not change the half-life of TLR4 mRNA in eotaxin-2-induced HCAECs (control group, 145.3±24.7 minutes; 10 ng/mL eotaxin-2 group, 246.9±30.1 minutes; SB203580 plus eotaxin-2 group, 163.2±29.7 minutes; SP600125 plus eotaxin-2 group, 79.4±21.5 minutes; PD98059 plus eotaxin-2 group, 199.6±23.5 minutes; Figure 3C). Finally, the western blotting demonstrated that TLR4 protein production induced by 10 ng/mL of eotaxin-2 was also reduced by SP600125 and PD98059 (Figure 3D), indicating JNK/SAPK and ERK1/2 regulates the TLR4 protein production in eotaxin-2 stimulation HCAECs. These results suggest that JNK/SAPK, p38 MAPK, and ERK1/2 activation contribute to the transcriptional signaling pathway, JNK/SAPK and p38 MAPK regulate post-transcriptional modification, as well as JNK/SAPK and ERK1/2 regulate translational signaling pathway in HCAECs treated with 10 ng/mL eotaxin-2.
Figure 3.
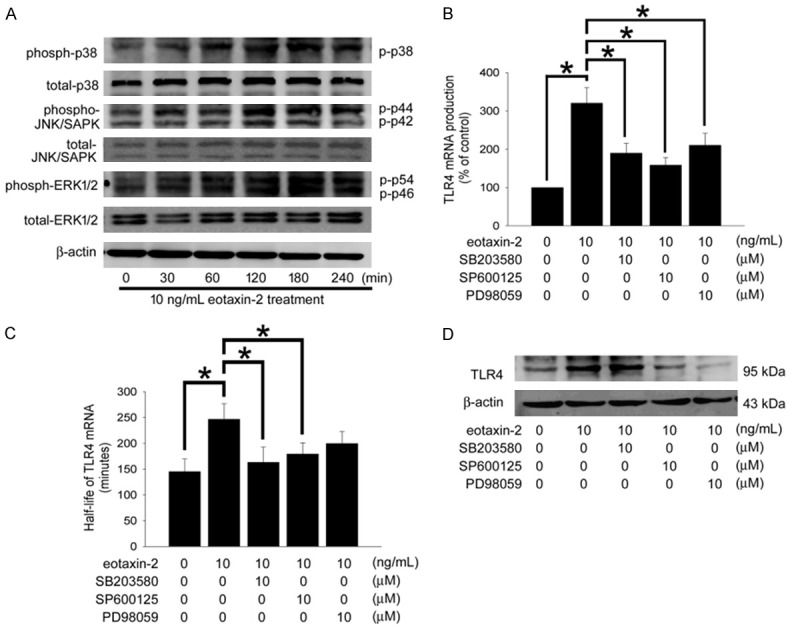
Eotaxin-2 Prolongs TLR4 mRNA stability and promotes TLR4 expression. A: Following treatment of HCAECs with eotaxin-2 for 30-240 minutes, the phosphorylation of p38 MAPK, JNK/SAPK and ERK1/2 were analyzed by Western blot. The total p38 MAPK, JNK/SAPK, ERK1/2, and β-actin protein levels were used as loading controls. B: HCAECs were pretreated with SB203580, SP600125, or PD98059 for 1 hour prior to treatment with eotaxin-2 for 8 hours. The expression levels of TLR4 were analyzed by quantitative real time PCR. C: HCAECs were pretreated with SB203580, SP600125, or PD98059 for 1 hour prior to treatment with eotaxin-2 for 4 hours. The half-life of TLR4 mRNA were analyzed by actinomycin D chase experiment. D: HCAECs were pretreated with SB203580, SP600125, or PD98059 for 1 hour prior to treatment with eotaxin-2 for 18 hours. The total TLR4 expression was analysis using western blotting. All bars represent the results of three independent experiments. The data are presented as the mean ± SEM and statistical evaluation was performed using one-way ANOVA followed by a Dunnett’s test. *P<0.05 was considered significant.
HuR activation and TTP expression mediate TLR4 mRNA stability confer eotaxin-2 responsiveness: The RNA binding proteins, such as HuR, AUF1, and TTP regulate TLR4 mRNA stability. In Western blot analysis, eotaxin-2 markedly increased the cytoplasmic level of HuR but not AUF1 (Figure 4A); in contrast, TTP protein expression in HCAECs after 10 ng/mL eotaxin-2 treatment was decreased. HuR and AUF1 were found predominantly in the nucleus in nontreated endothelial cells. Treatment with 10 ng/mL eotaxin-2 for 4 hours caused a marked accumulation of cytoplasmic HuR; in contrast, AUF1 and TTP expression was found predominantly in the nucleus, and its distribution remained unchanged following eotaxin-2 treatment (Figure 4B). To investigate the fact that the 3’UTR of TLR4 mRNA confers eotaxin-2 responsiveness and that HuR modulates the 3’UTR-mediated gene expression in HCAECs, a reporter plasmid containing the 3’UTR and luciferase reporter gene were transfected into cells. The CMV-luciferase-TLR4 3’UTR sense plasmid-transfected group had a higher basal luciferase activity than the CMV-luciferase plasmid-transfected group. Treatment with 10 ng/mL eotaxin-2 for 4 hours caused a significant increase in luciferase activity in the CMV-luciferase-TLR4 3’UTR sense plasmid-transfected group (Figure 4C). HCAECs were cotransfected with the 25 μM HuR siRNA and CMV-luciferase-TLR4 3’UTR sense plasmid followed by eotaxin-2 treatment for 4 hours. HuR-specific siRNA effectively blocked the luciferase activity in CMV-luciferase-TLR4 3’UTR sense plasmid-transfected cells stimulated with eotaxin-2. In contrast, The CMV-luciferase-TLR4 3’UTR antisense plasmid-transfected group did not have a higher basal luciferase activity than the CMV-luciferase plasmid-transfected group. Additionally, on the basis of the specific region of the ARE of TLR4 recognized by TTP [24], we postulated that eotaxin-2 might affect the interaction of TTP and 3’UTR of TLR4 mRNA, and we assessed this possibility using immunoprecipitation and quantitative real-time PCR. Protein fractions were subjected to immunoprecipitation with a rabbit anti-TTP antibody or control preimmune rabbit serum and were subjected to polyacrylamide gel electrophoresis. The mouse anti-TTP antibody was efficient in the immunoprecipitation process. Treatment with eotaxin-2 markedly decreased TTP interaction with the 3’UTR of TLR4 mRNA (Figure 4D). To assess the importance of the MAPKs in eotaxin-2 mediated activation of HuR and TTP signaling, we used the MAPK inhibitors to identify whether the expression of HuR and TTP as well as TTP interaction with the 3’UTR of TLR4 mRNA are regulated by the MAPK-signaling pathways. We found that SB23580 and SP600125 significantly reduced cytoplasmic HuR expression, and SP600125 decreased TTP production in HCAECs treated with eotaxin-2 (Figure 4E); the interaction with the 3’UTR of TLR4 mRNA is blocked partially by SP600125 (Figure 4F). These findings suggest that the 3’UTR of TLR4 mRNA confers eotaxin-2 responsiveness and eotaxin-2 affected HuR and TTP expression is mediated by the p38 MAPK and JNK/SAPK signaling pathway in HCAECs.
Figure 4.
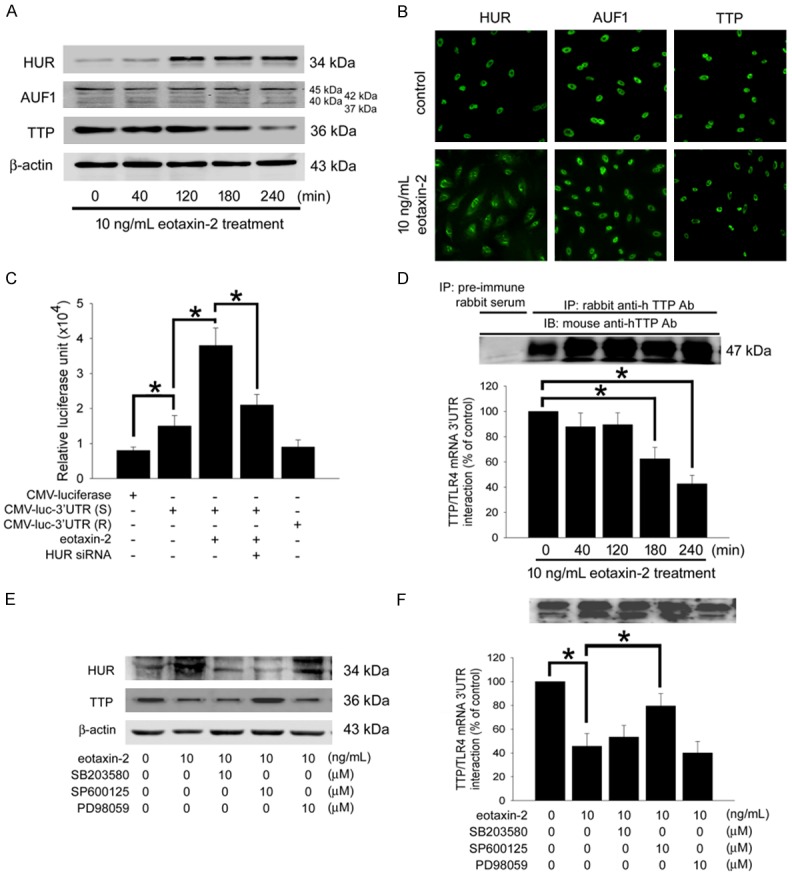
The 3’UTR flanking sequence of TLR4 mRNA, HuR and TTP conferred eotaxin-2-responsiveness in HCAECs, which mediates by the activation of p38 MAPK and JNK/SAPK. A: HCAECs were treated with eotaxin-2 for 40-240 minutes. HuR, AUF1, and TTP protein expression in the cells cytoplasm were detected by western blot analysis. β-actin was used as internal controls. B: Subcellular distribution of HuR, AUF1, and TTP after eotaxin-2 treatment for 4 hours in HCAECs detected by immunofluorescence and observed by confocal microscope. C: HCAECs were transfected with CMV-luciferase-TLR4 3’UTR sense (CMV-luc-3’UTR (S)) or CMV-luciferase-TLR4 3’UTR antisense CMV-luc-3’UTR (R) plasmids. Equal amounts of luciferase reporter gene containing plasmid (CMV-Luciferase) were used as controls. Uniform transfection efficiencies were confirmed using a β-galactosidase reporter plasmid. The luciferase activity was quantified by luminometry. Data are expressed as relative luciferase units. D: The cytoplasmic fractions in 4 hours of eotaxin-2 treatment HCAECs were analyzed by immunoprecipitation. Immunoprecipitated material (5 μL) was used in quantitative real-time PCR reactions to detect the presence of the 3’ UTR of TLR4 mRNA. TTP interaction with the 3’UTR of TLR4 mRNA was identified by quantitative real-time PCR. E: HCAECs were pretreated with SB203580, SP600125, or PD98059 for 1 hour prior to treatment with eotaxin-2 for 4 hours. The expression of cytoplasmic HuR and TTP were assessed by western blot analysis. β-actin was used as internal controls. F: HCAECs were pretreated with PD98059, SB203580, and SP600125 after eotaxin-2 treatment. TTP interaction with the 3’UTR of TLR4 mRNA was identified by immunoprecipitation and quantitative real-time PCR. The data are presented as mean ± SEM and represent the results of three separate experiments and statistical evaluation was performed using one-way ANOVA followed by a Dunnett’s test. *P<0.05 was considered significant.
Eotaxin-2 markedly induce PRAT4A expression but blurry for gp96: Previous evidence showed that chaperones, such as gp96 and PRAT4A regulate TLR4 localization and surface expression [26], therefore we examined if their expressions changed with eotaxin-2 exposure. In western blotting that PRAT4A expression is obviously increased both in time- or dose-dependent manner under 10 ng/mL of eotaxin-2 stimulation. Unlike PRAT4A, the expressions of gp96 were erratic in eotaxin-2 treatment HCAECs (Figure 5A and 5B). According to the results, we predict that PRAT4A might regulate the trafficking of TLR4 in eotaxin-2-stimulated HCAECs.
Figure 5.
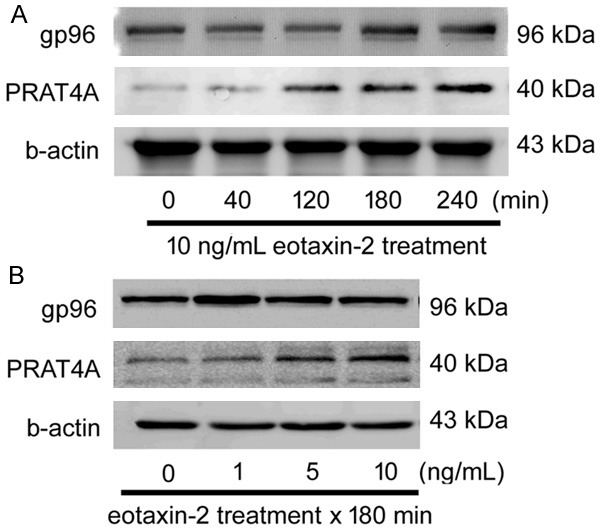
Eotaxin-2 Markedly Induce PRAT4A Expression. A: HCAECs were treated with eotaxin-2 for 40-240 minutes. The gp96 and PRAT4A protein expression in the cells were detected by western blot analysis. B: HCAECs were treated with eotaxin-2 for 180 minutes. The gp96 and PRAT4A protein expression were detected by western blot analysis. b-actin was used as internal controls.
Eotaxin-2 enhanced and aggravated neointima formation in HC diet-fed B6.129S7-Ldlrtm1Her/J but not Ldlr-/-/ Tlr4-/- mice: mice which fed with normal chaw diet. In contrast, fed HC diet may induce the lesion formation in B6.129S7-Ldlrtm1Her/J mice. Interestingly, the administration of 10 ng/kg BW eotaxin-2 may more serious the neointima formation in HC diet-fed B6.129S7-Ldlrtm1Her/J mice but not Ldlr-/-/Tlr4-/- mice. According to present in vitro results in this study, immunohistochemical staining was performed using antibodies against TLR4, HuR, TTP, and PRAT4A on sections of the aortas. Compared with sections from the control (normal diet) group, the sections of the HC diet-fed B6.129S7-Ldlrtm1Her/J mouse aortas did not showed changed expression levels of TLR4, HuR, TTP, and PRAT4A on the vessel wall. Indeed, B6.129S7-Ldlrtm1Her/J mouse treatment with 5 ng/kg BW of eotaxin-2 slightly or 10 ng/kg BW of eotaxin-2 markedly strengthened TLR4, HuR, and PRAT4A expression in the HC diet-fed group. Obviously, 10 ng/kg BW of eotaxin-2 administration did decrease TTP expression in the aortas of the HC diet-fed B6.129S7-Ldlrtm1Her/J mouse. Additionally, compared with sections from the eotaxin-2 plus HC diet treatment groups, the administration of 10 ng/kg BW eotaxin-2 in HC diet-fed Ldlr-/-/Tlr4-/- mice may emerge the restricted neointima formation, HuR, PRAT4A, and TTP expression (Figure 6). These results demonstrate that eotaxin-2 administration significantly exacerbated atherogenesis and aggregative TLR4, HuR, PRAT4A expression in the aorta in HC diet-fed B6.129S7-Ldlrtm1Her/J mice, as mediated by TLR4.
Figure 6.
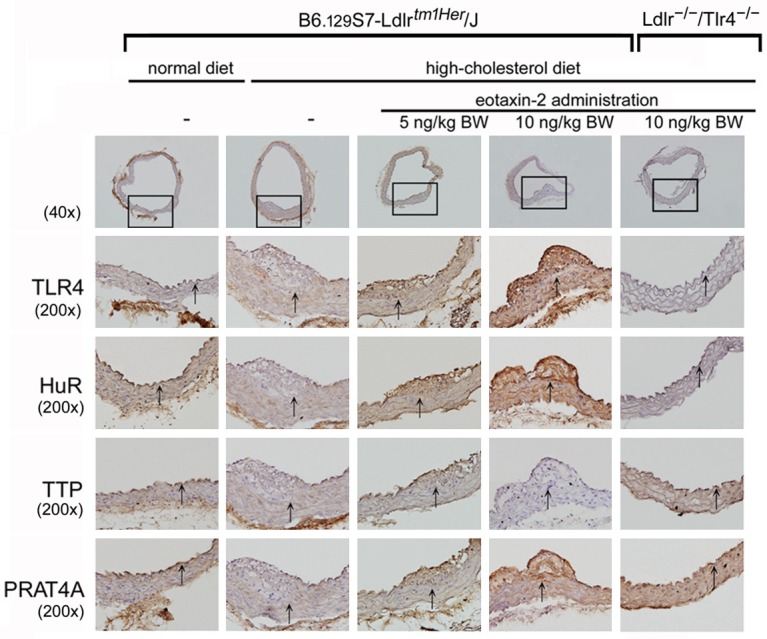
Eotaxin-2 protein significantly exacerbated neointimal formation in HC diet-fed B6.129S7-Ldlrtm1Her/J mice, which is mediated by TLR4 expression. Histopathological features of cross-sections of aortas were stained using hematoxylin. There were no atherosclerotic lesions in the aortas of normal diet-fed B6.129S7-Ldlrtm1Her/J mice group and HC diet-fed Ldlr-/-/Tlr4-/- mice. The graphs show 40× or 200× magnification of the slide. The lumen is uppermost in all sections, and corresponding hematoxylin staining was used to identify the nucleus. The arrows indicated the internal elastic laminae on the vessel walls.
Discussion
TLR4 are type I transmembrane receptors that are expressed on the endothelial cells, macrophages, neutrophils, platelet, keratinocytes, dendritic cells, and epithelial cells [27-29], and are critical for the induction of downstream signals during innate immune responses to bacterial components [13]. Recent findings have demonstrated that a repertoire of TLR4 is associated with atherosclerotic lesions and that the expression of TLR4 is upregulated in endothelial cells in lesions [16]. Of clinical and animal experimental relevance, our group had highlight the level of TLR4 expression on platelets [30] and monocytes [24] are associated with platelet function and early outcomes in experimental C57BL/6 mouse and coronary artery bypass graft (CABG) surgery patients. In 2002, Dybdahl B. et al. demonstrated that open heart surgery induces an inflammatory response and the release of heat-shock protein 70 (HSP70) via TLR4 signaling [31], and the level of TLR4 expression on cells is associated with the early outcome of patients [24]. TLR4 is well known as a receptor for LPS may contribute to cellular inflammation. LPS treatment increases TLR4 expression in HCAECs, and directly contributes to the progression of heart failure and coronary artery disease [32]. However, recently studies have explored that TLR4 also responses to several molecules stimulation. Endogenous HSP60 as a danger signal for neuron injury via TLR4 [33]. GroEL protein of Porphyromonas gingivalis binds to TLR4, then accelerates tumor growth by enhancing neovascularization [34,35]. HSP60 of Chlamydia pneumoniae induces lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) expression and increases the uptake of oxdized low density lipoprotein (oxLDL), which results from TLR4-axis signaling pathway and plays a critical role in atherogenesis [18]. Base on these evidences, we predict that increased presence of TLR4 on vessel walls is likely to cause the hypersensitivity of vascular cells to both endogenous factors and exogenous antigen, and so exacerbate and worsen the inflammation. It also provides a basis for further controlling TLR4 expression as a therapeutic strategy to avoid vascular disorders.
Even though the TLR4-mediated signaling pathway may contribute to inflammation including cardiovascular diseases, the underlying mechanisms involved in regulation of TLR4 expression on the surface of cells are still confusion and remain to be explored. However, according to the previous evidences, we predict the signaling pathways associated with TLR4 expression are diversity in different cell types and situation. LPS enhanced TLR4 expression in human aortic smooth muscle cell via SAPK/JNK and ERK1/2-mediated transcriptional pathway [36], and p38 MAPK-HuR axis-mediated post-transcriptional modification [23]. HuR with a high affinity for the 3’UTR of TLR4 mRNA, contributes to the up-regulation of TLR4 induced by GroEL in HCAECs [37]. Bone component exposure downregulated TLR4 expression in a gp96-related manner in monocytic U937 cells [38], in contrast eotaxin-2 increased PRAT4A-mediated TLR4 expression in HCAECs in this study. Of clinical relevance, we also explored novel roles for monocyte and platelet TLR4 that are associated with early outcomes in cardiac surgery. The oxidative stress from cardiopulmonary bypass technique mediated decrease of SAPK/JNK, ERK1/2, and calpain activity may cripple the TLR4 expression in monocytes and platelets [24,30]. Knockout of TTP may cause a severe inflammatory syndrome in vivo [39], and up-regulation of TTP on monocytes resulting from cardiac surgery may induce immune suppression via regulation of TLR4 expression [40]. Additionally, the activation of AUF1 four isoforms (p37AUF1, p40AUF1, p42AUF1, and p45AUF1) causes diverse impacts on inflammation during atherogenesis. Rab7b is a negatively modulator of TLR4 signaling by leading the translocation of TLR4 into lysosomes for degradation in mouse macrophage [41,42]. Recently, we demonstrated that calpain may cleavage myosin-9 which coordinate with Rab7b, and let to a positively TLR4 trafficking as well as hence TLR4 expression in thrombin treated platelets [43]. Although we have explored that transcriptional signaling pathways, post-transcriptional modification, and protein trafficking may regulate the expression of TLR4, the detailed underlying mechanisms of TLR4 expression in else cells remain to be studied continuously in the future. These preliminary results also provide a basis for further controlling TLR4 expression as controlling and therapeutic strategies to avoid inflammation and cardiovascular disorders.
In this in vitro study, the cells were treated with LPS to as the positive control group and which compared with eotaxin-2 treatment group. Indeed, this situation did not match with the animal study that B6.129S7-Ldlrtm1Her/J mice were fed with HC diet. However, our in vitro approach was only to cause cellular inflammatory responses and mimic the functional changing situation of endothelial cells. In short, eotaxin-2 is similar to inflammatory substances, can cause increasing of TLR4 expression thereby increasing the sensitivity of cells to inflammatory substances, subsequently also amplify the inflammatory responses. In the other hand, cells with relatively low level of protein in terms of performance which related to inflammatory responses, immunoregulation, oncogenesis, and cell growth [44]. The cells always use post-transcriptional modification, well known as possibly because of repression of transcription through the potentially unstable expression of mRNAs [25] to regulate the proteins expression and result in coping with the rapidly changing environment and external stimulation. Even though the Figure 2E showed 4 hours stimulation of eotaxin-2 did not significantly induce TLR4 mRNA expression, but the prolong stability of TLR4 mRNA was observed after 4 hours treatment in endothelial cells, indicating that regulation of transcriptional modification plays critical roles and allows endothelial cells to react to eotaxin-2 stimulation in a short period of time. Previous studies have demonstrated that TLR4 is expressed abundantly in macrophage-infiltrated lipid-rich atherosclerotic lesions [14], which may augment the intimal hyperplasia [15]. Although TLR4 deficiency decreases atherosclerosis but does not protect against inflammation in obese LDL receptor-deficient mice had been reported [45], HC diet-fed Ldlr-/-/Tlr4-/- mice did not present markedly atherosclerotic lesion in our study. We also predict that diversity may result from insufficient experimental duration and indicating that eotaxin-2 may accelerate the program of atheroma formation. This provides a basis for further investigation of eotaxin-2 modulation as a preventing strategy for atherogenesis.
Conclusion in this study, we found that eotaxin-2 impaired the tube formation function in HCAECs. Additionally, eotaxin-2 augmented TLR4 and VCAM-1 expression in LPS-stimulated HCAECs, which may result in increasing sensitivity of response to antigen and mediates sever monocytes adhesion, respectively. The regulation of TLR4 expression in eotaxin-2-stimulated HCAECs might via both transcriptional signaling pathway (JNK/SAPK, p38 MAPK, and ERK1/2 activation were involved) post-transcriptional modification (JNK/SAPK and p38 MAPK mediated HuR and TTP expression were involved), and chaperone (PRAT4A)-regulated trafficking Mechanisms. Finally, the administration of the eotaxin-2 also led to a significant elevation of lesion formation in high cholesterol diet-induced B6.129S7-Ldlrtm1Her/J than that in eotaxin-2 plus high cholesterol diet treatment Ldlr-/-/Tlr4-/- and high cholesterol diet treatment B6.129S7-Ldlrtm1Her/J mice. The TLR4, HuR, TTP, and PRAT4A expression also enhanced in the vessel wall of eotaxin-2 induced hypercholesterolemic B6.129S7-Ldlrtm1Her/J mice. The data provide evidence for a direct involvement of eotaxin-2, which may impair the endothelial function and accelerate atherosclerosis progression.
Acknowledgements
We thank Tze-Liang Yang, Min-Yu Lo and Ya-Yu Jan for excellent technical assistance. This work was supported by National Ministry of Science and Technology (NSC 101-2314-B-038 -041-MY3, MOST103-2314-B-016 -022-MY3 and MOST 104-2314-B-038-074) and Taipei Medical University (103TMU-TMUH-10) in Taiwan.
Disclosure of conflict of interest
None.
References
- 1.de Paulis A, Annunziato F, Di Gioia L, Romagnani S, Carfora M, Beltrame C, Marone G, Romagnani P. Expression of the chemokine receptor ccr3 on human mast cells. Int Arch Allergy Immunol. 2001;124:146–150. doi: 10.1159/000053694. [DOI] [PubMed] [Google Scholar]
- 2.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor ccr3 by human t helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 3.Forssmann U, Uguccioni M, Loetscher P, Dahinden CA, Langen H, Thelen M, Baggiolini M. Eotaxin-2, a novel cc chemokine that is selective for the chemokine receptor ccr3, and acts like eotaxin on human eosinophil and basophil leukocytes. J Exp Med. 1997;185:2171–2176. doi: 10.1084/jem.185.12.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, Williams TJ, Kay AB. Eotaxin (ccl11) and eotaxin-2 (ccl24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol. 2002;169:2712–2718. doi: 10.4049/jimmunol.169.5.2712. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe K, Jose PJ, Rankin SM. Eotaxin-2 generation is differentially regulated by lipopolysaccharide and il-4 in monocytes and macrophages. J Immunol. 2002;168:1911–1918. doi: 10.4049/jimmunol.168.4.1911. [DOI] [PubMed] [Google Scholar]
- 6.Coleman JM, Naik C, Holguin F, Ray A, Ray P, Trudeau JB, Wenzel SE. Epithelial eotaxin-2 and eotaxin-3 expression: Relation to asthma severity, luminal eosinophilia and age at onset. Thorax. 2012;67:1061–1066. doi: 10.1136/thoraxjnl-2012-201634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Min JW, Lee JH, Park CS, Chang HS, Rhim TY, Park SW, Jang AS, Shin HD. Association of eotaxin-2 gene polymorphisms with plasma eotaxin-2 concentration. J Hum Genet. 2005;50:118–123. doi: 10.1007/s10038-005-0230-3. [DOI] [PubMed] [Google Scholar]
- 8.Ancuta P, Autissier P, Wurcel A, Zaman T, Stone D, Gabuzda D. Cd16+ monocyte-derived macrophages activate resting t cells for hiv infection by producing ccr3 and ccr4 ligands. J Immunol. 2006;176:5760–5771. doi: 10.4049/jimmunol.176.10.5760. [DOI] [PubMed] [Google Scholar]
- 9.Fiorucci G, Olivetta E, Chiantore MV, Federico M. Microarray analysis reveals ccl24/eotaxin-2 as an effector of the pathogenetic effects induced by hiv-1 nef. Curr Drug Discov Technol. 2007;4:12–23. doi: 10.2174/157016307781115502. [DOI] [PubMed] [Google Scholar]
- 10.Cheadle EJ, Riyad K, Subar D, Rothwell DG, Ashton G, Batha H, Sherlock DJ, Hawkins RE, Gilham DE. Eotaxin-2 and colorectal cancer: A potential target for immune therapy. Clin Cancer Res. 2007;13:5719–5728. doi: 10.1158/1078-0432.CCR-07-1145. [DOI] [PubMed] [Google Scholar]
- 11.Ablin JN, Entin-Meer M, Aloush V, Oren S, Elkayam O, George J, Barshack I. Protective effect of eotaxin-2 inhibition in adjuvant-induced arthritis. Clin Exp Immunol. 2010;161:276–283. doi: 10.1111/j.1365-2249.2010.04172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adi M, Arnon A, M ME, Gad K, Jacob G. Anti eotaxin-2 antibodies attenuate the initiation and progression of experimental atherosclerosis. World J Cardiovas Dis. 2013;3:339–346. [Google Scholar]
- 13.Heumann D, Roger T. Initial responses to endotoxins and gram-negative bacteria. Clin Chim Acta. 2002;323:59–72. doi: 10.1016/s0009-8981(02)00180-8. [DOI] [PubMed] [Google Scholar]
- 14.Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, Kaul S, Arditi M. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized ldl. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 15.Vink A, Schoneveld AH, van der Meer JJ, van Middelaar BJ, Sluijter JP, Smeets MB, Quax PH, Lim SK, Borst C, Pasterkamp G, de Kleijn DP. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation. 2002;106:1985–1990. doi: 10.1161/01.cir.0000032146.75113.ee. [DOI] [PubMed] [Google Scholar]
- 16.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 17.Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through toll-like receptor 4 and md2 in a myd88-dependent pathway. J Immunol. 2002;168:1435–1440. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 18.Lin FY, Lin YW, Huang CY, Chang YJ, Tsao NW, Chang NC, Ou KL, Chen TL, Shih CM, Chen YH. Groel1, a heat shock protein 60 of chlamydia pneumoniae, induces lectin-like oxidized low-density lipoprotein receptor 1 expression in endothelial cells and enhances atherogenesis in hypercholesterolemic rabbits. J Immunol. 2011;186:4405–4414. doi: 10.4049/jimmunol.1003116. [DOI] [PubMed] [Google Scholar]
- 19.Fan J, Frey RS, Malik AB. Tlr4 signaling induces tlr2 expression in endothelial cells via neutrophil nadph oxidase. J Clin Invest. 2003;112:1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karper JC, de Vries MR, van den Brand BT, Hoefer IE, Fischer JW, Jukema JW, Niessen HW, Quax PH. Toll-like receptor 4 is involved in human and mouse vein graft remodeling, and local gene silencing reduces vein graft disease in hypercholesterolemic apoe*3leiden mice. Arterioscler Thromb Vasc Biol. 2011;31:1033–1040. doi: 10.1161/ATVBAHA.111.223271. [DOI] [PubMed] [Google Scholar]
- 21.Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of bax and bcl-x(l) during apoptosis. Proc Natl Acad Sci U S A. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen JZ, Zhu JH, Wang XX, Xie XD, Sun J, Shang YP, Guo XG, Dai HM, Hu SJ. Effects of homocysteine on number and activity of endothelial progenitor cells from peripheral blood. J Mol Cell Cardiol. 2004;36:233–239. doi: 10.1016/j.yjmcc.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Lin FY, Chen YH, Lin YW, Tsai JS, Chen JW, Wang HJ, Chen YL, Li CY, Lin SJ. The role of human antigen r, an rna-binding protein, in mediating the stabilization of toll-like receptor 4 mrna induced by endotoxin: A novel mechanism involved in vascular inflammation. Arterioscler Thromb Vasc Biol. 2006;26:2622–2629. doi: 10.1161/01.ATV.0000246779.78003.cf. [DOI] [PubMed] [Google Scholar]
- 24.Tsai CS, Chen DL, Lin SJ, Tsai JC, Lin TC, Lin CY, Chen YH, Huang GS, Tsai HY, Lin FY, Li CY. Tnf-alpha inhibits toll-like receptor 4 expression on monocytic cells via tristetraprolin during cardiopulmonary bypass. Shock. 2009;32:40–48. doi: 10.1097/SHK.0b013e318199608d. [DOI] [PubMed] [Google Scholar]
- 25.Wen X, Wu GD. Evidence for epigenetic mechanisms that silence both basal and immune-stimulated transcription of the il-8 gene. J Immunol. 2001;166:7290–7299. doi: 10.4049/jimmunol.166.12.7290. [DOI] [PubMed] [Google Scholar]
- 26.Saitoh S. Chaperones and transport proteins regulate tlr4 trafficking and activation. Immunobiology. 2009;214:594–600. doi: 10.1016/j.imbio.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi T, Sakurai Y, Fukuda K, Yada K, Ogiwara K, Matsumoto T, Yoshizawa H, Takahashi Y, Yoshikawa Y, Hayata Y, Taniguchi S, Shima M. Correlations between global clotting function tests, duration of operation, and postoperative chest tube drainage in pediatric cardiac surgery. Paediatr Anaesth. 2011;21:865–871. doi: 10.1111/j.1460-9592.2011.03524.x. [DOI] [PubMed] [Google Scholar]
- 28.Beaulieu LM, Freedman JE. Inflammation & the platelet histone trap. Blood. 2011;118:1714–1715. doi: 10.1182/blood-2011-06-362764. [DOI] [PubMed] [Google Scholar]
- 29.Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. 2010;67:525–544. doi: 10.1007/s00018-009-0210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai JC, Lin YW, Huang CY, Lin FY, Tsai CS. Calpain activity and toll-like receptor 4 expression in platelet regulate haemostatic situation in patients undergoing cardiac surgery and coagulation in mice. Mediators Inflamm. 2014;2014:484510. doi: 10.1155/2014/484510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dybdahl B, Wahba A, Lien E, Flo TH, Waage A, Qureshi N, Sellevold OF, Espevik T, Sundan A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation. 2002;105:685–690. doi: 10.1161/hc0602.103617. [DOI] [PubMed] [Google Scholar]
- 32.Zeuke S, Ulmer AJ, Kusumoto S, Katus HA, Heine H. Tlr4-mediated inflammatory activation of human coronary artery endothelial cells by lps. Cardiovasc Res. 2002;56:126–134. doi: 10.1016/s0008-6363(02)00512-6. [DOI] [PubMed] [Google Scholar]
- 33.Lehnardt S, Schott E, Trimbuch T, Laubisch D, Krueger C, Wulczyn G, Nitsch R, Weber JR. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the cns. J Neurosci. 2008;28:2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Argueta JG, Shiota S, Yamaguchi N, Masuhiro Y, Hanazawa S. Induction of porphyromonas gingivalis groel signaling via binding to toll-like receptors 2 and 4. Oral Microbiol Immunol. 2006;21:245–251. doi: 10.1111/j.1399-302X.2006.00286.x. [DOI] [PubMed] [Google Scholar]
- 35.Lin FY, Huang CY, Lu HY, Shih CM, Tsao NW, Shyue SK, Lin CY, Chang YJ, Tsai CS, Lin YW, Lin SJ. The groel protein of porphyromonas gingivalis accelerates tumor growth by enhancing endothelial progenitor cell function and neovascularization. Mol Oral Microbiol. 2015;30:198–216. doi: 10.1111/omi.12083. [DOI] [PubMed] [Google Scholar]
- 36.Lin FY, Chen YH, Tasi JS, Chen JW, Yang TL, Wang HJ, Li CY, Chen YL, Lin SJ. Endotoxin induces toll-like receptor 4 expression in vascular smooth muscle cells via nadph oxidase activation and mitogen-activated protein kinase signaling pathways. Arterioscler Thromb Vasc Biol. 2006;26:2630–2637. doi: 10.1161/01.ATV.0000247259.01257.b3. [DOI] [PubMed] [Google Scholar]
- 37.Huang CY, Shih CM, Tsao NW, Lin YW, Shih CC, Chiang KH, Shyue SK, Chang YJ, Hsieh CK, Lin FY. The groel protein of porphyromonas gingivalis regulates atherogenic phenomena in endothelial cells mediated by upregulating toll-like receptor 4 expression. Am J Transl Res. 2016;8:384–404. [PMC free article] [PubMed] [Google Scholar]
- 38.Lin JA, Lin FY, Chen TL. Bone components downregulate expression of toll-like receptor 4 on the surface of human monocytic u937 cells: A cell model for postfracture immune dysfunction. Mediators Inflamm. 2015;2015:896576. doi: 10.1155/2015/896576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 40.Lin FY, Tsai YT, Lee CY, Lin CY, Lin YW, Li CY, Shih CM, Huang CY, Chang NC, Tsai JC, Chen TL, Tsai CS. Tnf-alpha-decreased thrombomodulin expression in monocytes is inhibited by propofol through regulation of tristetraprolin and human antigen r activities. Shock. 2011;36:279–288. doi: 10.1097/SHK.0b013e3182236e7e. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Chen T, Han C, He D, Liu H, An H, Cai Z, Cao X. Lysosome-associated small rab gtpase rab7b negatively regulates tlr4 signaling in macrophages by promoting lysosomal degradation of tlr4. Blood. 2007;110:962–971. doi: 10.1182/blood-2007-01-066027. [DOI] [PubMed] [Google Scholar]
- 42.Yao M, Liu X, Li D, Chen T, Cai Z, Cao X. Late endosome/lysosome-localized rab7b suppresses tlr9-initiated proinflammatory cytokine and type i ifn production in macrophages. J Immunol. 2009;183:1751–1758. doi: 10.4049/jimmunol.0900249. [DOI] [PubMed] [Google Scholar]
- 43.Tsai JC, Lin YW, Huang CY, Lin CY, Tsai YT, Shih CM, Lee CY, Chen YH, Li CY, Chang NC, Lin FY, Tsai CS. The role of calpain-myosin 9-rab7b pathway in mediating the expression of toll-like receptor 4 in platelets: A novel mechanism involved in alpha-granules trafficking. PLoS One. 2014;9:e85833. doi: 10.1371/journal.pone.0085833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. Ared: Human au-rich element-containing mrna database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding Y, Subramanian S, Montes VN, Goodspeed L, Wang S, Han C, Teresa AS 3rd, Kim J, O’Brien KD, Chait A. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:1596–1604. doi: 10.1161/ATVBAHA.112.249847. [DOI] [PMC free article] [PubMed] [Google Scholar]