

Abstract
This study aimed to investigate the potential role of lncRNA CCAT1 in the progression of pancreatic cancer (PC) and to reveal its possible molecular mechanism. The expression of CCAT1 was analyzed in PC tissues and their adjacent normal tissues from patients diagnosed with PC and in two pancreas cancer cell lines, namely PANC-1 and Aspc-1 using real-time polymerase chain reaction (qRT-PCR) and western blot, respectively. The effects of CCAT1 expression on cell proliferation, cell cycle, and migration were analyzed using MTT assay, flow cytometry, and transwell assay, respectively. The effects of c-Myc expression on the expression of CCAT1 and E-box were also analyzed using RNA immunoprecipitation (RIP) and chromatin immunoprecipitation (ChIP) assays, respectively. The results showed that CCAT1 was highly expressed in PC tissues compared to the adjacent tissues (P<0.01) and was also overexpressed in PANC-1 and Aspc-1 cells (P<0.05). The silencing of CCAT1 significantly inhibited cell proliferation and migration (P<0.05), arrested cell cycle at G0/G1 stage, and decreased cyclin D1 expression (P<0.05). An increased expression of c-Myc was observed in the PC tissues compared to the adjacent tissues. We found that suppression of c-Myc altered CCAT1 expression by targeting its promoter at E-box. This study demonstrated that c-Myc-activated CCAT1 may contribute to tumorigenesis and metastasis of PC, which may serve as a potential target for the therapy of PC.
Keywords: Pancreatic cancer, lncRNA CCAT1, cell proliferation, metastasis, c-Myc
Introduction
Pancreatic cancer (PC) remains to be one of the most common digestive system malignancies. It is usually diagnosed at an advanced state, which results in unavailability of effective therapies [1]. Moreover, it is a highly lethal disease with the worst prognosis among all the major malignancies such that the patients with metastatic PC exhibit a five-year survival rate of only 25% [2,3]. Since 1998, the incidence and mortality rate of PC have been on the rise [4], leading to an estimated 227,000 deaths per year worldwide [5]. Therefore, a better understanding of the molecular mechanisms underlying PC tumorigenesis is the need of the hour to discover novel therapeutic targets for patients with PC.
Recently, some reports have highlighted that non-coding RNAs (ncRNAs) are closely implicated in tumorigenesis [6,7]. For instance, microRNAs such as miR-1290 and miR-150 have been found to play important roles in PC development and progression [8,9]. Long non-coding RNAs (lncRNAs) are newly known members of the ncRNA family, which have also been proved to be associated with carcinogenesis and tumor growth in various kinds of tumors, such as cervical, colon, and thyroid cancer [10-13]. Importantly, MALAT1 and HULC, two lncRNAs, have been found to play pivotal roles in the biology of PC [14,15].
Colon Cancer-Associated Transcript 1 (CCAT1), a recently discovered lncRNA, is located in the vicinity of a well-known transcription factor, c-Myc. Previous studies have revealed that CCAT1 is up-regulated in colon gastric cancer tissues as compared to the normal tissues [16,17]. However, the role of CCAT1 in PC tumorigenesis is still not well documented and needs to be investigated.
Therefore, in the current study, we investigated the regulation of CCAT1 in PC tissues and cell lines. The effects of CCAT1 on PC cell proliferation, cell migration, cell cycle, and cell migration-associated protein expressions were determined using two types of PC cells lines. In addition, the interaction between CCAT1 and c-Myc was also studied. Thus, the present study aimed to investigate the potential roles of CCAT1 in the PC development and to elucidate the underlying mechanism.
Materials and methods
Patients
A total of 26 PC patients were included in this study, who provided prior informed consents. The PC diagnosis was pathologically confirmed, and cancer tissues and their adjacent normal tissues were obtained from clinically ongoing surgical specimens. Tissues were snap-frozen in liquid nitrogen and stored at -80°C till used for RNA extraction. All procedures in this study were approved by the Human Ethics Committee of Ning Bo NO.2 Hospital hospital.
Cell culture
The human PC cell lines, PANC-1 and Aspc-1 (obtained from the American Type Culture Collection), were cultured in the Dulbecco’s Modified Eagle Medium (DMEM) medium and maintained in a humidified incubator at 37°C with 5% CO2. The normal pancreatic ductal epithelial cell line HPDE6-C7 (obtained from the American Type Culture Collection) was grown in keratinocyte growth medium (KGM) (Invitrogen, Carlsbad, CA, USA) supplemented with human epidermal growth factor and bovine pituitary extract.
Cell transfection
The siRNAs specifically targeting c-Myc (si-c-Myc) and CCAT1 (si-CCAT1) were constructed based on the full-length, wild-type c-Myc, and CCAT1 coding sequences, respectively, by Sangon Biotech (Shanghai, China). The siRNA sequences used were CCAT1: 5’-CCATTCCATTCATTTCTCTTTCCTA-3’ and c-Myc: 5’-GGUGAUCCAGACUCUGACCUU-3’. The cell transfections were conducted using Lipofectamine 2000 reagent following the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). The siRNA vector with no silenced CCAT1 or c-Myc sequence was transfected into cancer cells as a control.
RNA isolation and qRT-PCR
Total RNA from cancer tissues and cells was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). After treatment with RNase-free DNase I (Promega Biotech, USA), 0.5 μg/μL purified RNA was reverse-transcribed into cDNA with the PrimeScript 1st strand cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA). The expression of targets in tissues or cells was measured in an Eppendorf Mastercycler (Brinkman Instruments, Westbury, NY) using the SYBR ExScript qRT-PCR Kit (Takara, China) at a final volume of 20 μL with a standard protocol. Each reaction was performed in triplicate, and the 2-ΔΔCT method was used to determine the relative gene expression level. Melting curves of amplified products were analyzed at the end of each PCR to confirm that only one product was amplified and detected. Phosphoglyceraldehyde dehydrogenase (GAPDH) was chosen as the internal control. Primers used for amplification of targets are shown in Table 1.
Table 1.
The primers used for targets amplification
| Name | Forward primer | Reverse primer |
|---|---|---|
| CCAT1 | CATTGGGAAAGGTGCCGAGA | ACGCTTAGCCATACAGAGCC |
| C-myc | CCACAGCAAACCTCCTCACA | TCCAACTTGACCCTCTTGGC |
| Cyclin D1 | CAAATGGAGCTGCTCCTGGTG | CTTCGATCTGCTCCTGGCAGG |
| Cyclin E1 | CCTCCAAAGTTGCACCAGTT | GGACGCACAGGTCTAGAAGC |
| Cyclin A1 | CAAGGTCCTGATGCTTGTCA | CCCATGGTCAGAGAGCACTT |
| E-cadherin | AACGCATTGCCACATACAC | AACGCATTGCCACATACAC |
| N-cadherin | AACTCCAGGGGACCTTTTC | CAAATGAAACCGGGCTATC |
| Vimentin | TCCAAGTTGCTGACCTCTC | TCAACGGCAAAGTTCTCTTC |
| GAPDH | GGGAGCCAAAAGGGTCAT | GAGTCCTTCCACGATACCAA |
Western blot analysis
Cells were lysed in RIPA lysis and extraction buffer (Sangon Biotech, Shanghai, China). The supernatant was collected for the measurement of protein concentration using BCA protein assay kit (Pierce, Rochford, IL, USA). Then 20 μg of protein per cell lysate was subjected to a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to the polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The membrane was blocked with Tris Buffered Saline-Tween (TBST) containing 5% non-fat milk at room temperature for 1 h. After incubation with antibodies specific for c-Myc, cyclin D1, cyclin E1, cyclin A1, E-cadherin, N-cadherin, and vimentin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), the membrane was incubated with horseradish peroxidase-labeled goat anti-rat secondary antibody (1:1000). Then the PVDF membrane was washed three times with 1 × TBST buffer and visualized with an enhanced chemiluminescence method. GAPDH served as the internal control.
Cell proliferation assay
Cell proliferation was assessed using MTT (3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyltetrazolium bromide) assay. Cells were seeded in 96-well culture plates at a density of 5 × 103 cells per well. Each group had five repeats. Then 20 μL of 10 mg/mL MTT was added to each well, followed by incubation for 3 h at 37°C. Then 150 μL of dimethyl sulfoxide (DMSO) was added to each well, and the optical density was measured at 590 nm using a Multiskan EX (Thermo, Vantaa, Finland).
Clonogenic assay
The clonogenic assay was performed according to a previously published method [18] with a modification. Briefly, after transfection, cells were plated into 6-cm tissue culture dishes in triplicate at a density of 100 cells/dish. The cells were grown in RPMI-1640 medium containing 10% fetal bovine serum (FBS) for 14 days. The cells were then fixed and stained with Diff-Quick. After air-drying, colonies were counted under the microscope, and at least 30 cells were included in each colony.
Cell cycle assay
After transfection, the cells were cultured in RPMI-1640 containing 10% FBS for another 48 h. The cells were then seeded into 6-cm dishes until the concentration reached 80%. The cells were washed with ice-cold PBS followed by fixation with methanol at 4°C for 30 min. The cells were then stained with propidium iodide solution for 30 min. Finally, cell cycle and DNA content were analyzed using flow cytometry.
Cell migration assay
The cell migration was determined using wound healing assay. The cells were cultured in 6-well plates at a density of 5 × 105 cells per well. After the formation of a confluent monolayer, a wound was made across the well with a 200-μL pipette tip. The wound was photographed immediately and the migration of cells across the gap wound was observed using an inverted microscope (× 10 magnification). The width of the gap was detected in five different visual fields, and the average width was calculated.
RNA immunoprecipitation assay
RNA immunoprecipitation (RIP) assay was conducted using Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Massachusetts, USA) following the manufacturer’s instructions. Anti-c-Myc antibody (BD Biosciences, San Jose, CA, USA) and IgG (control) were used for RIP. Co-precipitated RNAs were detected by qRT-PCR analysis. Total RNAs (input controls) and isotype controls were detected to demonstrate that the detected signals were the results of RNAs specifically binding to the E-box.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed using an EZ-ChIP Chromatin Immunoprecipitation Kit (Millipore, Massachusetts, USA) according to the manufacturer’s protocol. Cross-linked chromatin was sonicated to obtain 200 bp to 1000 bp fragments. The chromatin was immunoprecipitated using anti-c-Myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Normal mouse immunoglobulin G was used as the negative control. Quantitative PCR was conducted using SYBR Green Mix (Roche, Mannheim, Germany) to quantify ChIP-derived DNA.
Statistical analysis
The statistical analyses were carried out using Graph Prism 5.0 software (GraphPad Prism, San Diego, CA). The data were expressed as mean ± standard deviation (SD). Independent samples t-test was used for the calculation of significance of paired data. Post-hoc Tukey’s test was employed to calculate the difference among groups. P<0.05 was considered as statistically significant.
Results
CCAT1 was up-regulated in PC tissues and cells
To determine whether CCAT1 was dysregulated in PC, we examined the expression levels of CCAT1 in PC tissues and pair-matched adjacent normal pancreatic tissues, as well as PC cells and normal pancreatic cells using qRT-PCR. As presented in Figure 1A, CCAT1 expression was significantly increased in PC tissues in comparison with the adjacent normal pancreatic tissues (P<0.01). Additionally, the expression levels of CCAT1 in PC cells (PANC-1 and Aspec-1) were significantly higher than that in normal pancreatic cells (HPDE6-C7) (P<0.05).
Figure 1.

The mRNA expression of lncRNA CCAT1 in pancreatic cancer tissues (A) and cell lines (B) as assayed by qRT-PCR. *: P<0.05, **: P<0.01.
siRNA-CCAT1 inhibited cell proliferation
The expression of CCAT1 decreased significantly after transfection with siRNA-CCAT1 in two types of PC cells (Figure 2A). To investigate the effects of CCAT1 on cell proliferation, we performed MTT assays which revealed that suppression of CCAT1 expression by si-CCAT1 could inhibit cell viability of PANC-1 and Aspec-1 cells significantly (P<0.05) (Figure 2B). In accordance with the results of MTT assay, the colony-formation assays showed that clonogenic survival was significantly decreased in PANC-1 and Aspec-1 cells after CCAT1 suppression (P<0.01) (Figure 2C and 2D).
Figure 2.

The mRNA expression of lncRNA CCAT1 in pancreatic cancer cells after transfection with si-CCAT1 (A); lncRNA CCAT1 suppression significantly inhibited the cells proliferation of two types of pancreatic cancer cells assayed by MTT assay (B); lncRNA CCAT1 suppression significantly decreased the number of pancreatic cancer cell colonies assayed by clonogenic assay (C and D). *: P<0.05, **: P<0.01.
CCAT1 expression modulated cell cycle
In order to explore the mechanism of CCAT1 suppression-mediated regulation of PC cell proliferation, flow cytometry was employed to study the cellular DNA content and cell cycle profile. As shown in Figure 3A, the percentage of cells at G0/G1 stage was significantly increased when cells (PANC-1 and Aspc-1) were transfected with siRNA-CCAT1 (P<0.05) suggesting a possible arrest at G0/G1 stage after the suppression of CCAT1.
Figure 3.
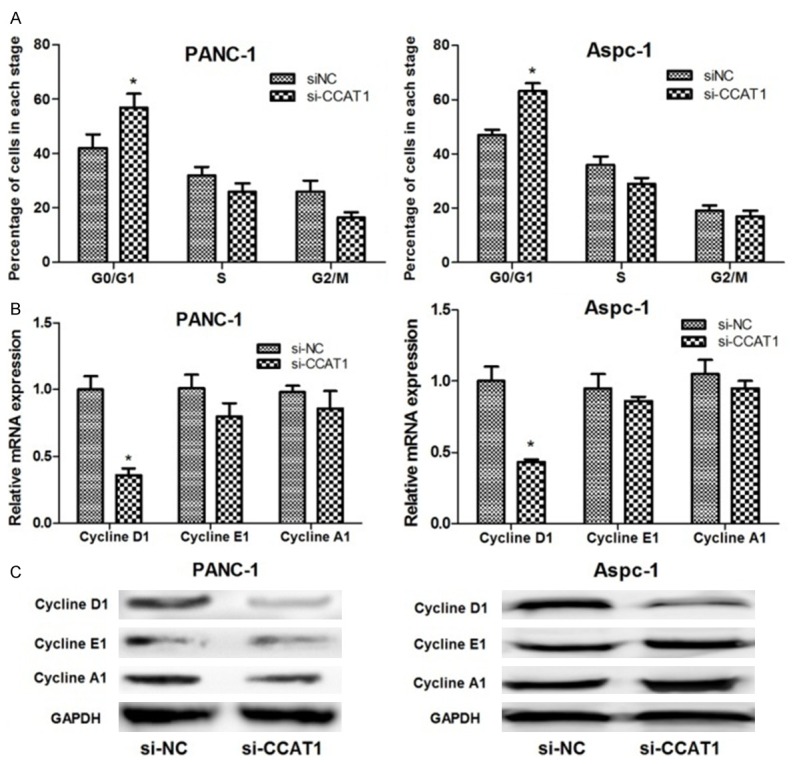
The role of lncRNA CCAT1 suppression on pancreatic cancer cell cycle. (A) Flow cytometry showed that lncRNA CCAT1 suppression significantly increased the percentage of the two cancer cell lines in G0/G1 stage; (B and C) qRT-PCR (B) and western blot analysis (C) showed that lncRNA CCAT1 suppression significantly decreased cyclin D1 expression, but had no significant effect on cyclin E1 and A1 expressions in the two pancreatic cancer cell lines. *: P<0.05, **: P<0.01.
To determine whether depletion of CCAT1 arrested the cell cycle in the G0/G1 stage, mRNA and protein expression levels of cell cycle-related proteins, including cyclin D1, cyclin E1, and cyclin A1 were analyzed. As presented in Figure 3B and 3C, the expression of cyclin D1 drastically decreased in the siRNA-CCAT1 group compared with that in the control group (P<0.05). However, the expression levels of cyclin A1 and E1 did not change significantly in the two groups.
siRNA-CCAT1 inhibited cell migration
In order to investigate the role of CCAT1 in regulating PC cancer cell migration, we performed wound healing assay. The results showed that suppression of CCAT1 significantly inhibited the migration of PANC-1 and Aspc-1 cells compared with controls (P<0.05) (Figure 4A and 4B).
Figure 4.
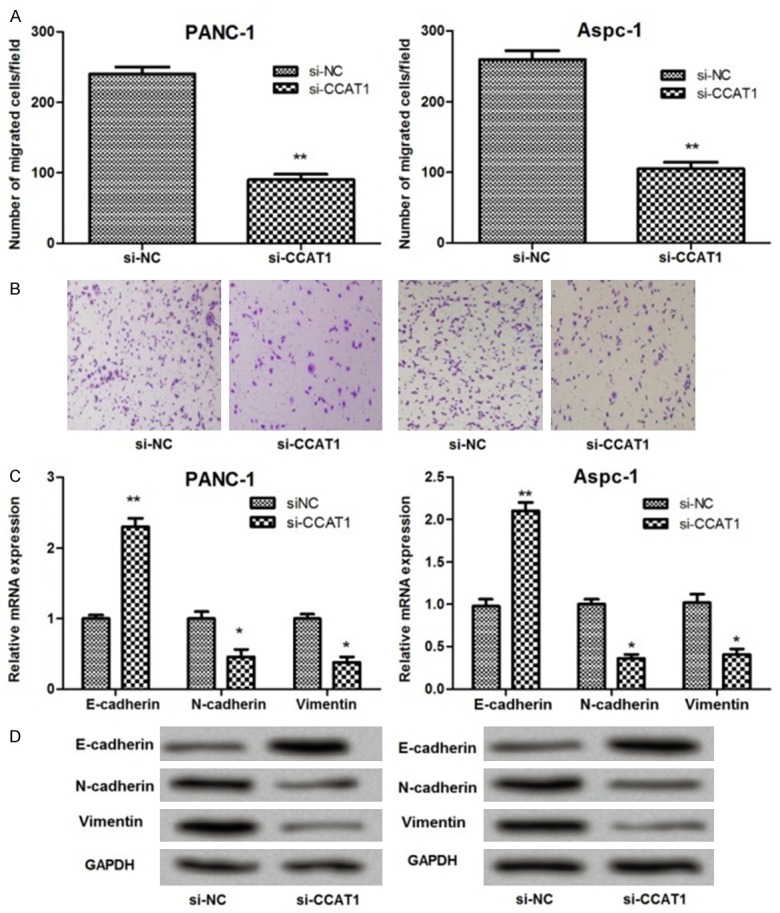
The effect of CCAT1 suppression on pancreatic cancer cell migration. (A and B) Wound healing assay showing that lncRNA CCAT1 suppression significantly decreased the number of migrated cells; (C and D) qRT-PCR (C) and western blot analysis (D) showing that lncRNA CCAT1 suppression significantly increased E-cadherin expression, while decreased N-cadherin and vimentin expressions. *: P<0.05, **: P<0.01.
Further studies demonstrated that the expression levels of epithelial-mesenchymal transition (EMT) markers, including E-cadherin, N-cadherin, and vimentin changed significantly before and after CCAT1 suppression. As shown in Figure 4C and 4D, the expression of E-cadherin increased significantly in the siRNA-CCAT1 group compared with the control group (P<0.01). On the contrary, the expression of N-cadherin and vimentin decreased significantly when cells were transfected with siRNA-CCAT1 (P<0.05) (Figure 4C and 4D).
CCAT1 is up-regulated by c-Myc through binding to E-box
It has been suggested that c-Myc is overexpressed in PC [19,20]. In this study, the mRNA and protein expression levels of c-Myc in PC tissues were found to be significantly higher than that in the adjacent normal pancreatic tissues (P<0.01) (Figure 5A). After PC cells were transfected with siRNA-c-Myc, mRNA and protein expressions of c-Myc decreased significantly in the two PC cell lines (P<0.01). Additionally, the expression of CCAT1 significantly decreased (P<0.01) (Figure 5B).
Figure 5.

c-Myc bound to the promoter regions of lncRNA CCAT1 and regulated its expression. A: mRNA and protein expressions of c-myc in pancreatic cancer tissues and cell lines assayed by qRT-PCR and western blot analysis; B: The effects of c-myc suppression on mRNA and protein expressions of lncRNA CCAT1; C: RNA immunoprecipitation (RIP) assay showing that endogenous E-box was highly enriched by c-myc RIP compared to IgG RIP; D: Chromatin immunoprecipitation (ChIP) assay showed that c-myc immunoprecipitates were highly enriched in promoter region of CCAT1 containing E-box element compared with that without E-box. IgG indicated the negative control of immunoprecipitation. *: P<0.05, **: P<0.01.
In order to explore further the interaction between CCAT1 and c-Myc, we conducted RIP assay in ANC-1 and Aspc-1 cells. The results showed that endogenous E-box was highly enriched by c-Myc RIP compared to IgG RIP (Figure 5C), indicating that c-Myc may bind to E-box. To determine whether c-Myc directly binds to the E-box element and contribute to the overexpression of CCAT1, we performed ChIP assay. The result showed that the c-Myc immunoprecipitates were highly enriched in the promoter region of CCAT1 containing the E-box element compared with control IgG immunoprecipitates in PANC-1 and Aspc-1 cells. The CCAT1 promoter with silenced E-box site displayed no significant enrichment (Figure 5D), which implied that c-Myc may alter the expression of CCAT1 by directly binding to E-box. The regulatory mechanism of CCAT1 in PC is shown in Figure 6.
Figure 6.
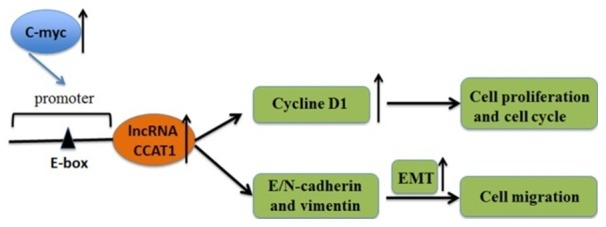
The regulatory mechanism of lncRNA CCAT1 in pancreatic cancer.
Discussion
Recently, large-scale gene expression studies have revealed that lncRNAs were dysregulated in many human cancers [21,22]. Many studies showed that lncRNA CCAT1 may serve as a possible candidate for exploring the mechanisms in cancers. However, only a few studies have reported the potential role of CCAT1 in PC [16,17]. The data of the present study showed that CCAT1 was significantly up-regulated in PC tissues and cancer cells. CCAT1 suppression significantly inhibited cell proliferation and migration in two PC cell lines. Besides, CCAT1 suppression arrested the cell cycle at the G0/G1 stage and significantly decreased the expression of cyclin D1 in PC cells. Moreover, c-Myc was also found to be overexpressed in PC tissues and cells, and silencing of c-Myc altered CCAT1 expression by targeting its promoter at E-box. However, all of these results need to be further discussed.
In previous studies, overexpression of CCAT1 has been shown to be associated with the development of several cancers, such as colon, gastric, and gallbladder cancers [16,17,23]. In accordance with the above findings, we also observed that CCAT1 was significantly up-regulated in PC tissues, as well as in the two PC cell lines. Therefore, we speculate that the overexpression of CCAT1 may be correlated with the pathogenesis of PC.
We then analyzed the effects of CCAT1 expression on PC cell proliferation using siRNA-mediated gene silencing. Yang et al. [24] suggested that the silencing of CCAT1 inhibits cell proliferation in gastric cancer. In agreement with the above result, our study showed that PC cell proliferation was significantly suppressed after siRNA-mediated depletion of CCAT1, indicating that CCAT1 could promote PC cell proliferation. Additionally, we analyzed the effects of CCAT1 expression on PC cell cycle and cell cycle-associated protein expressions to explore further the mechanism of CCAT1 suppression regulating PC cell proliferation. We found that PC cell cycle was arrested at the G0/G1 stage after CCAT1 suppression, and the expression of cyclin D1 decreased significantly. Increasing the number of evidence support a central role of cyclin D1 in promoting cancer cell proliferation [25]. Importantly, cyclin D1 has been considered as a “pivotal element” of malignant transformation in human cancer, including PC [26]. Taken together, our results corroborated the role of CCAT1 in regulating PC cell proliferation.
Accumulating evidence has demonstrated that epithelial-mesenchymal transition (EMT) is an important biological process contributing to the progression of primary tumors towards metastasis [27]. The expression of E-cadherin, one of EMT markers, was found to be lower in PC tissues than in non-tumor tissues [28]. Two other EMT markers, namely N-cadherin and vimentin were found to be overexpressed in PC [29,30]. In this study, we found that E-cadherin expression increased significantly, whereas the expressions of N-cadherin and vimentin decreased significantly when PANC-1 and Aspc-1 cells were transfected with siRNA-CCAT1, suggesting the role of CCAT1 in modulating tumor metastasis of PC.
c-Myc is a transcription factor that is generally recognized as an important regulator of cell cycle, proliferation, differentiation, and apoptosis [31,32]. It has been found to be overexpressed in PC [19,20] and to promote CCAT1 transcription and regulate CCAT1 expression in colon cancer cells [33]. Importantly, Yang et al. [17] revealed that c-Myc regulates the expression of CCAT1 by directly binding to the E-box. Interestingly, the current study also found that c-Myc could activate CCAT1 expression through interaction with the E-box in the CCAT1 promoter region. Besides, c-Myc knockdown decreased the expression of CCAT1. Thus, the current study further supported the regulatory effects of c-Myc on CCAT1 expression.
In conclusion, our study indicates that up-regulated lncRNA CCAT1 is a tumor promoter, which may promote the progression of PC. Moreover, CCAT1 may be activated by transcription factor c-Myc by directly binding to its promoter region (E-box). These findings implicate that lncRNA CCAT1 may serve as an important target in PC therapy. However, further studies are still required to confirm these findings and reveal the potential molecular mechanism for CCAT1 in regulating PC.
Acknowledgements
This study was supported by the Zhejiang Provincial Natural Science Foundation of China (Grant No: Y2080096) and the Medical and Health Science Research Fund of Zhejiang Provincial Health Department (Grant No: 2007A172).
Disclosure of conflict of interest
None.
References
- 1.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 2.Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, Goldstein D, Padbury R, Moore MJ, Gallinger S, Mariette C. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304:1073–1081. doi: 10.1001/jama.2010.1275. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang G, He P, Tan H, Budhu A, Gaedcke J, Ghadimi BM, Ried T, Yfantis HG, Lee DH, Maitra A. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983–4993. doi: 10.1158/1078-0432.CCR-13-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 6.Gutschner T, Hämmerle M, Eißmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Groß M. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davalos V, Esteller M. Non-coding RNAs and Cancer. Springer; 2014. Unraveling the complex network of interactions between noncoding RNAs and epigenetics in cancer; pp. 125–148. [Google Scholar]
- 8.Li A, Yu J, Kim H, Wolfgang CL, Canto MI, Hruban RH, Goggins M. MicroRNA array analysis finds elevated serum miR-1290 accurately distinguishes patients with low-stage pancreatic cancer from healthy and disease controls. Clin Cancer Res. 2013;19:3600–3610. doi: 10.1158/1078-0432.CCR-12-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arora S, Swaminathan SK, Kirtane A, Srivastava SK, Bhardwaj A, Singh S, Panyam J, Singh AP. Synthesis, characterization, and evaluation of poly (D, L-lactide-co-glycolide)-based nanoformulation of miRNA-150: potential implications for pancreatic cancer therapy. Int J Nanomedicine. 2014;9:2933. doi: 10.2147/IJN.S61949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheetham S, Gruhl F, Mattick J, Dinger M. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108:2419–2425. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Guo Z. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013;23:1446–1461. doi: 10.1101/gr.152942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan M, Li X, Jiang W, Huang Y, Li J, Wang Z. A long non-coding RNA, PTCSC3, as a tumor suppressor and a target of miRNAs in thyroid cancer cells. Exp Ther Med. 2013;5:1143–1146. doi: 10.3892/etm.2013.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Liao LM, Liu AW, Wu JB, Cheng XL, Lin JX, Zheng M. Overexpression of long noncoding RNA HOTAIR predicts a poor prognosis in patients with cervical cancer. Arch Gynecol Obstet. 2014;290:717–723. doi: 10.1007/s00404-014-3236-2. [DOI] [PubMed] [Google Scholar]
- 14.Pang EJ, Yang R, Fu XB, Liu YF. Overexpression of long non-coding RNA MALAT1 is correlated with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2015;36:2403–2407. doi: 10.1007/s13277-014-2850-8. [DOI] [PubMed] [Google Scholar]
- 15.Peng W, Gao W, Feng J. Long noncoding RNA HULC is a novel biomarker of poor prognosis in patients with pancreatic cancer. Med Oncol. 2014;31:346. doi: 10.1007/s12032-014-0346-4. [DOI] [PubMed] [Google Scholar]
- 16.He X, Tan X, Wang X, Jin H, Liu L, Ma L, Yu H, Fan Z. C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol. 2014;35:12181–12188. doi: 10.1007/s13277-014-2526-4. [DOI] [PubMed] [Google Scholar]
- 17.Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y, Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013;139:437–445. doi: 10.1007/s00432-012-1324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franken NA, Rodermond HM, Stap J, Haveman J, Van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 19.Ding X, Zhou X, Jiang B, Zhao Q, Zhou G. Triptolide suppresses proliferation, hypoxia-inducible factor-1α and c-Myc expression in pancreatic cancer cells. Mol Med Rep. 2015;12:4508–4513. doi: 10.3892/mmr.2015.3960. [DOI] [PubMed] [Google Scholar]
- 20.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-κB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–8580. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 21.Shore AN, Herschkowitz JI, Rosen JM. Noncoding RNAs involved in mammary gland development and tumorigenesis: there’s a long way to go. J Mammary Gland Biol Neoplasia. 2012;17:43–58. doi: 10.1007/s10911-012-9247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Xu J, Hong J, Tang R, Zhang X, Fang JY. Long noncoding RNA profiles identify five distinct molecular subtypes of colorectal cancer with clinical relevance. Mol Oncol. 2014;8:1393–1403. doi: 10.1016/j.molonc.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma M, Chu B, Zhang Y, Weng M, Qin Y, Gong W, Quan Z. Long non-coding RNA CCAT1 promotes gallbladder cancer development via negative modulation of miRNA-218-5p. Cell Death Dis. 2015;6:e1583. doi: 10.1038/cddis.2014.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y, Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013;139:437–445. doi: 10.1007/s00432-012-1324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer. 2007;55:1–14. doi: 10.1016/j.lungcan.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Tian T, Lv F, Chang Y, Wang X, Zhang L, Li X, Li L, Ma W, Wu J. Six1 promotes proliferation of pancreatic cancer cells via upregulation of cyclin D1 expression. PLoS One. 2013;8:e59203. doi: 10.1371/journal.pone.0059203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biddle A, Mackenzie IC. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 2012 doi: 10.1007/s10555-012-9345-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Zhang X, Zhang Y, Zhu D, Zhang L, Li Y, Zhu Y, Li D, Zhou J. HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J Exp Clin Cancer Res. 2016;35:1. doi: 10.1186/s13046-016-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shintani Y, Johnson KR, Wheelock MJ. N-cadherin expression and epithelial-to-mesenchyme transition in pancreatic cancer. Cancer Res. 2006;66:650–650. doi: 10.1158/0008-5472.CAN-06-2322. [DOI] [PubMed] [Google Scholar]
- 30.Xu Q, Ma J, Lei J, Duan W, Sheng L, Chen X, Hu A, Wang Z, Wu Z, Wu E. α-Mangostin suppresses the viability and epithelial-mesenchymal transition of pancreatic cancer cells by downregulating the PI3K/Akt pathway. Biomed Res Int. 2014;2014:546353–546353. doi: 10.1155/2014/546353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole MD, Henriksson M. 25 years of the c-Myc oncogene. Semin Cancer Biol. 2006;16:241–241. doi: 10.1016/j.semcancer.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 33.He X, Tan X, Wang X, Jin H, Liu L, Ma L, Yu H, Fan Z. C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol. 2014;35:12181–12188. doi: 10.1007/s13277-014-2526-4. [DOI] [PubMed] [Google Scholar]