

Abstract
Background
The simian malaria parasite Plasmodium knowlesi has been reported to cause significant numbers of human infection in South East Asia. Its merozoite surface protein-3 (MSP3) is a protein that belongs to a multi-gene family of proteins first found in Plasmodium falciparum. Several studies have evaluated the potential of P. falciparum MSP3 as a potential vaccine candidate. However, to date no detailed studies have been carried out on P. knowlesi MSP3 gene (pkmsp3). The present study investigates the genetic diversity, and haplotypes groups of pkmsp3 in P. knowlesi clinical samples from Peninsular Malaysia.
Methods
Blood samples were collected from P. knowlesi malaria patients within a period of 4 years (2008–2012). The pkmsp3 gene of the isolates was amplified via PCR, and subsequently cloned and sequenced. The full length pkmsp3 sequence was divided into Domain A and Domain B. Natural selection, genetic diversity, and haplotypes of pkmsp3 were analysed using MEGA6 and DnaSP ver. 5.10.00 programmes.
Results
From 23 samples, 48 pkmsp3 sequences were successfully obtained. At the nucleotide level, 101 synonymous and 238 non-synonymous mutations were observed. Tests of neutrality were not significant for the full length, Domain A or Domain B sequences. However, the dN/dS ratio of Domain B indicates purifying selection for this domain. Analysis of the deduced amino acid sequences revealed 42 different haplotypes. Neighbour Joining phylogenetic tree and haplotype network analyses revealed that the haplotypes clustered into two distinct groups.
Conclusions
A moderate level of genetic diversity was observed in the pkmsp3 and only the C-terminal region (Domain B) appeared to be under purifying selection. The separation of the pkmsp3 into two haplotype groups provides further evidence of the existence of two distinct P. knowlesi types or lineages. Future studies should investigate the diversity of pkmsp3 among P. knowlesi isolates in North Borneo, where large numbers of human knowlesi malaria infection still occur.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-016-1935-1) contains supplementary material, which is available to authorized users.
Keywords: Plasmodium knowlesi, Merozoite surface protein-3, Genetic diversity, Natural selection, Haplotypes
Background
Malaria is a disease caused by the infection of blood protozoa belonging to the genus Plasmodium. Molecular evidence suggests that the simian malaria agent Plasmosium knowlesi evolved from a group which included Plasmodium cynomolgi and P. vivax some 30.5 million years ago [1]. The first report of natural transmission of P. knowlesi to humans was reported in 1965 when a US Army surveyor acquired the infection while working in Peninsular Malaysia [2]. It was observed that the parasite could be transmitted to humans through blood inoculation and thus the authors designated it the human strain or strain H. A second case was reported in southern Peninsular Malaysia 5 years later [3]. A large number of human knowlesi malaria was reported in Malaysian Borneo in 2004 [4], and reports have also been published on this infection in several neighbouring Asian countries such as Singapore [5], the Philippines [6] and Thailand [7]. However, the majority of the infections have been recorded in Malaysia. More than 300 human cases have been detected in Peninsular Malaysia since 2005 [8–10]. Recently, a study reported that more than half of the malaria cases in Malaysia were caused by P. knowlesi [11]. The highest proportion of P. knowlesi cases was found to be in the Malaysian Borneo as well as in the Peninsular Malaysia states of Kelantan, Pahang, Terengganu and Johor [11].
Malaria parasites invade the red blood cells (RBC) of many vertebrate hosts including humans and simians. The proteins involved in the invasion process have been studied to gain deeper insights of the invasion mechanism, and also to identify potential vaccine candidates against malaria [12]. One of these proteins, the merozoite surface protein-3 (MSP3), was identified in P. falciparum in 1994 [13, 14]. Subsequently, a novel surface antigen was discovered in P. vivax and was named MSP3α, due to its putative similarity to the MSP3 of P. falciparum [15]. Two paralogs of the P. vivax MSP3 protein were further identified, designated as PvMSP3β and PvMSP3γ [16]. Due to the presence of more than one such protein in a species, the P. vivax MSP3 proteins were grouped into a multi-gene family [17]. Full genome analysis on P. vivax (Salvador I strain) revealed 12 msp3 paralogs which cluster on chromosome 10 [18]. Surprisingly, these paralogs have limited similarity to the P. knowlesi MSP3 and the four P. falciparum MSP3 proteins. Although a number of studies have suggested that the msp3 genes in P. vivax and P. falciparum are related, a closer comparison between the domain organizations on chromosome 10 as well as the syntenic loci of pvmsp3, pfmsp3 and P. knowlesi putative msp3 genes suggest that these genes are not homologues [19].
Structurally the protein is characterized by a putative signal peptide and lacks a transmembrane domain or a GPI-lipid modification to anchor it to the outer membrane of the parasite. Another characteristic of the protein family is the presence of an alanine-rich central domain containing a series of heptad coiled-coil repeats [15, 20]. Recent studies have predicted that the MSP-3 proteins in P. vivax form oligometric and elongated molecules suggesting the protein may mediate interactions between host proteins and other merozoite surface proteins [21].
Genetic diversity in a natural population is usually generated by the introduction of new alleles through the process of migration, mutation, or recombination [22]. The frequency of these alleles on the other hand is governed by the actions of selection and natural drift [23]. For pathogens that infect humans, the host’s immune responses as well as modes of treatment administered are major components of selection, thus, genetic diversity can be an important indicator of how a pathogen responds to modes of intervention such as vaccines or drugs [24]. In this instance, directional selection leads towards fixing beneficial alleles in the population, resulting in reduced diversity [25]. Conversely, naturally acquired host immunity can exert balancing selection which tends to preserve or increase the allelic diversity of antigen genes. This of course occurs within the functional constraints of the encoded protein to prevent the protein from losing its native ability and function [26, 27]. The modelling of neutral processes in a population with a constant size allows for the prediction of expected frequencies of a particular allele. Thus, departures from this neutrality can thus be utilised to identify or pinpoint alleles that are targets for directional or balancing selection [28–31].
Several studies have been carried out on MSP3 proteins of P. falciparum and P. vivax; however, studies on P. knowlesi MSP3 lag far behind. In this study, the genetic diversity, natural selection and haplotype groups of pkmsp3 gene of P. knowlesi clinical isolates from Peninsular Malaysia were studied. Evidence of purifying selection in the C-terminal domain and haplotype grouping of P. knowlesi MSP3 was found. These data will be useful in understanding the genetic variation and natural selection forces acting on this gene and may indicate the gene’s potential as a vaccine candidate.
Methods
Blood sample collection
Twenty-three blood samples from knowlesi malaria patients were obtained from the University of Malaya Medical Centre (UMMC), Kuala Lumpur as well as from private clinics in Peninsular Malaysia between July 2008 and July 2012. Each blood sample was assigned a reference code for laboratory record. Knowlesi malaria infection was re-confirmed using several tests including microscopic examination of Giemsa-stained thick and thin blood smears, BinaxNOW® malaria rapid diagnostic test (Inverness Medical International, Stockport, United Kingdom) and polymerase chain reaction (PCR) based on the Plasmodium small subunit ribosomal RNA gene [4].
Genomic DNA extraction
Genomic DNA was extracted from the blood samples using a commercial blood extraction kit (QIAGEN, Hilden, Germany). One hundred μl of blood were used per extraction and the DNA was eluted into 50 μl of TE Buffer.
PCR, cloning and sequencing of pkmsp3
The pkmsp3 gene was amplified by nested PCR. For the initial primary PCR, oligonucleotide primers MSP3N1F: 5′-CCT CTT CAA CCA CAC ACA CA-3′ and MSP3N1R: 5′-GTT CAT TCT GGC GGA TAA GG-3′ were used [19]. Oligonucleotide primers MSP3N2F: 5′-CCC GTG AAA TAA CAC CCA-3′ and MSP3BN2R: 5′-CCA CCA TCT TAC GTT CAG-3′ [19] were used for the secondary PCR. Approximately 0.5 μg of genomic DNA was used in a final volume of 20 μl which also contained 0.2 mM of dNTP, 0.4 μM of forward and reverse primers, 2 mM MgCl2 and 1 unit of Taq DNA polymerase in buffer provided by the commercial kit (Promega, Madison, WI, USA). The PCR thermal profile was as follows, an initial denaturation of one cycle at 95 °C for 5 min followed by 30 cycles of 1 min at 94 °C, 1 min at 50 °C for annealing and 1 min 30 s at 72 °C for nest 1. Cycling for nest 2 consisted of a 5 min initial denaturation at 95 °C and 30 cycles of 1 min at 94 °C, 1 min at 48 °C, 1 min 30 s at 72 °C, and a final extension step at 72 °C for 10 min. PCR products were analysed by gel electrophoresis on a 1.5% agarose gel stained with SYBR® Safe DNA gel stain (Invitrogen, Eugene, USA).
Purification of PCR products and DNA cloning
PCR products were purified using QIAquick PCR purification kit (Qiagen, Hilden, Germany) per the manufacturer’s instructions. The purified PCR products were then ligated into the pGEM-T® TA cloning vector (Promega, Madison, Wisconsin, USA) and transformed into Escherichia coli TOP10F’ competent cells; colonies were then screened for the presence of recombinant plasmids harbouring the pkmsp3 fragment. These plasmids were then sequenced in a commercial laboratory (MyTACG Bioscience Enterprise, Malaysia). Between 3 and 5 recombinant plasmids were sent for sequencing per isolate. DNA for isolates showing clonal sequence variations (singletons or rare substitutions) was re-amplified and re-sequenced in order to confirm that the variations were genuine, and not the result of incorporation errors of the Taq DNA polymerase.
Analysis of pkmsp3 sequences
Editing and alignment of the pkmsp3 nucleotide sequences (including the sequence of reference P. knowlesi strain H, GenBank: XM_002259752) were performed using the BioEdit sequence alignment editor ver. 7.2.0. Gene Runner ver. 4.0.9.2 was used to deduce the respective amino acid sequences. The Neighbour Joining method described in MEGA6 was used to construct a phylogenetic tree [32] with bootstrap replicates of 1000. The Median-Joining method in NETWORK v4.6.1.2 program [33] was used to establish the genetic relationship among pkmsp3 haplotypes and construct the haplotype network. All newly-generated sequences were deposited in the GenBank database (KT900798–KT900845).
Sequence polymorphism analysis of pkmsp3
The programme DnaSP ver. 5.10.01 [34] was used to determine pkmsp3 genetic polymorphism by calculating the number of nucleotide differences per site (π), singleton sites (S), segregating sites (Ss), haplotypes (H), parsimony-informative sites (Ps), and haplotype diversity (Hd) [35].
The neutral model of molecular evolution acting on the pkmsp3 was tested according to nucleotide polymorphisms and haplotype distribution in the Fu and Li’s D* and F* tests [36]. The Tajima’s D test [22] was calculated to test the hypothesis that all mutations are selectively neutral. Tajima’s D test is based on the difference between Ss and π where positively significant values indicate balancing selection and negatively significant values indicate directional or purifying selection. In all tests carried out, sites that had gaps were excluded from the analysis. In tests requiring an outgroup, Plasmodium cynomolgi MSP3 was used (GenBank: KC907504). The FST fixation index [37] in DnaSP 5.10.00 was used to measure the genetic differentiation between the different clustering groups observed in the pkmsp3 phylogenetic tree and haplotype network.
The effect of natural selection was evaluated by the codon based Z-test, which determines whether it is negative or positive selection. Probability (P) values of less than 0.05 were considered significant. The variance of the differences was computed using the bootstrap method with 1000 replicates. The ratio between the average number of non-synonymous substitutions per non-synonymous site (dN) and the average number of synonymous substitutions per synonymous site (dS) using the Nei-Gojobori method with Jukes and Cantor correction [38] was also calculated. MEGA6 was used to calculate the Z-test and dN/dS ratio [32].
The Interpro programme (http://www.ebi.ac.uk/interpro) predicted the P. knowlesi MSP3 to have a large coiled-coil region. Genetic diversity and selection analyses were also performed separately on the coiled-coil region (Domain A) and the C-terminal (Domain B) of the protein (Fig. 1). This was carried out to investigate domain specific selective pressure.
Fig. 1.

Domain structures in pkmsp3. Organisation of the pkmsp3 gene showing the positions of coiled-coil region identified as Domain A (yellow), the C-terminal region as Domain B (blue) and the signal peptide (green)
Results
Genetic diversity at the nucleotide level
Successful PCR amplification produced DNA fragments of 1077 bp. This fragment contained a region coding a protein sequence of 338 amino acids. A total of 48 sequences were obtained for analysis.
Table 1 gives the estimates of genetic diversity for the full length pkmsp3 sequence, Domain A and Domain B. In the full length sequence, 384 segregating sites were observed; of these, 320 were parsimony-informative and 64 were singleton sites. When separated into Domain A and B, however, Domain B contained more segregating sites as compared to Domain A (273 vs 104). As for diversity, the full length sequence had haplotype diversity (Hd) of 0.997 ± 0.005. Both Domains A and B had similar Hd of 0.989 ± 0.007.
Table 1.
Estimates of DNA diversity, selection, and neutrality tests of full length, Domain A and Domain B of pkmsp3 gene
| Pkmsp3 | Sitesa | Ss | S | Ps | Hd ± SD | π ± SD | dN ± SE | dS ± SE | dN/dS | Z-test P-values | Tajima’s D |
Fu & Li’s | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dN = dS | dN > dS | dN < dS | D* | F* | |||||||||||
| Full length | 1,077 | 384 | 64 | 320 | 0.997 ± 0.005 | 0.046 ± 0.011 | 0.052 ± 0.004 | 0.058 ± 0.008 | 1.1 | 0.45 | 1.00 | 0.24 | -1.440 | -0.077 | 1.046 |
| Domain A | 534 | 104 | 44 | 60 | 0.989 ± 0.007 | 0.039 ± 0.002 | 0.039 ± 0.002 | 0.030 ± 0.008 | 1.3 | 0.34 | 0.10 | 1.00 | -0.723 | -1.852 | -1.670 |
| Domain B | 483 | 273 | 16 | 257 | 0.989 ± 0.007 | 0.067 ± 0.025 | 0.025 ± 0.007 | 0.042 ± 0.002 | 0.6 | 0.19 | 1.00 | 0.09 | -1.918 | -1.579 | -1.711 |
Abbreviations: d N non-synonymous polymorphism, d S synonymous polymorphism, d N /d S ratio of dN to dS, Hd haplotype diversity, Ps number of informative-parsimonious sites, π nucleotide diversity, S number of singleton sites, SD standard deviation, Ss number of segregating sites
aTotal number of sites analysed excluding gaps
*Modified Fu & Li’s D and F tests
Nucleotide diversity (π: 0.046 ± 0.011) for the full length sequence was found to be several times higher compared to other P. knowlesi functional genes such as PkDBPαII (π: 0.012) [39], PkAMA-1 (π: 0.00501) [40] and PkRAP-1 (π: 0.01298) [41]. Diversity for Domain B (π: 0.067 ± 0.025) was found to be higher than that for Domain A (π: 0.039 ± 0.002). A sliding window plot with a window length of 100 bp and a step size of 25 bp provided a detailed analysis of the full length sequence, with π ranging from 0.012 to 0.087 (Fig. 2). The highest peak diversity was within nucleotide positions 801–975 in Domain B, whereas in Domain A, the most conserved region was within nucleotide positions 51–150.
Fig. 2.
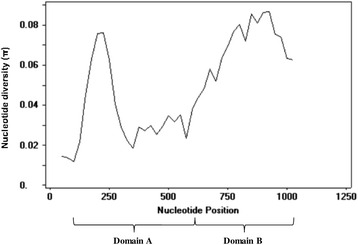
Nucleotide polymorphism in the pkmsp3. Sliding window plot of the nucleotide diversity (π) along the pkmsp3, generated with a window length of 100 bp and step size of 25 bp
Genetic diversity at the amino acid level
Comparisons and analysis with P. knowlesi strain H as a reference sequence showed mutations at 339 positions. Of these positions, 101 were synonymous changes and 238 were non-synonymous. When translated into deduced amino acids, high level polymorphism was observed (Fig. 3 and Additional file 1: Table S1). Among the 119 polymorphic sites, 100 were monomorphic mutations with a change into one amino acid type, and 19 showed dimorphic mutations with change in two amino acid types (K33R/N, T38I/S, N59E/G, L62E/Q, N66T/Y, N68D/G, T72A/M, A78K/E, V82M/A, K118N/R, K155E/R, E158Q/R, H173N/Y, Y197W/C, N228H/K, A281V/T, E307G/A, E317D/G and H319Y/P). The amino acid sequences could be categorised into 42 haplotypes (H1-42) (Fig. 3) with haplotype 11 having the highest frequency. Fifteen of the 23 patient samples had mixed haplotype infections (Table 2).
Fig. 3.
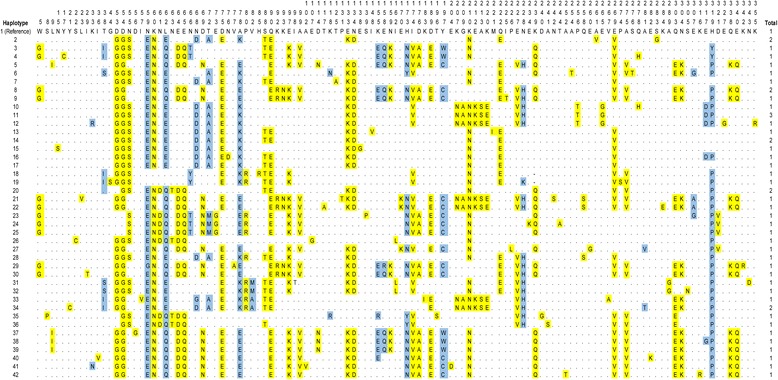
Amino acid sequence polymorphism in pkmsp3. Polymorphic amino acid residues are listed for each haplotype. Monomorphic and dimorphic amino acid changes are marked in yellow and blue, respectively. Total number of sequences for each haplotype is listed in the panel on the right
Table 2.
Haplotypes of pkmsp3 detected in human blood samples. Each blood sample was assigned a reference code (alphabetical or numerical)
| Blood sample code | Haplotype detected |
|---|---|
| ANU | H2 |
| AZI | H3, H4 |
| CHO | H5 |
| GAN | H6 |
| MAH | H7 |
| NGO | H8, H9 |
| OTH | H10, H11 |
| RAU | H11, H12 |
| SAM | H13, H14 |
| SYA | H15, H16, H17 |
| UM0001 | H18, H19 |
| UM0004 | H20 |
| UM0006 | H21, H22 |
| UM0009 | H23, H24, H25 |
| UM0014 | H26 |
| UM0015 | H27 |
| UM0016 | H28 |
| UM0018 | H29, H30 |
| UM0020 | H31, H32 |
| UM0029 | H33, H34 |
| UM0032 | H35, H36 |
| UM0047 | H37, H38, H39 |
| UM0050 | H40, H41, H42 |
| P. knowlesi strain H | H1 |
Phylogenetic analysis of pkmsp3
Analysis of the phylogenetic tree (Fig. 4) and haplotype network (Fig. 5) revealed that the haplotypes are clustered into two main groups (Group 1 and Group 2), which contained almost equal number of haplotypes. Furthermore, mixed haplotypes from the same blood sample were found to cluster into the same group in both the phylogenetic tree (Fig. 4) and haplotype network and (Fig. 5).
Fig. 4.
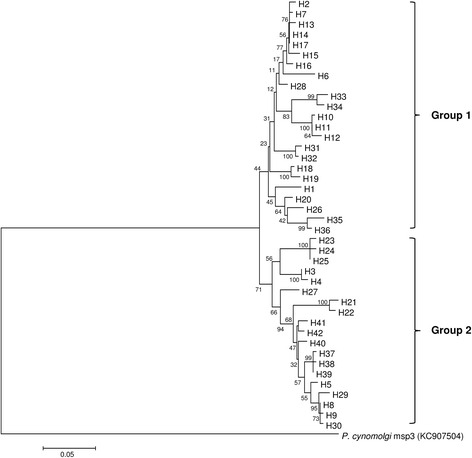
Phylogenetic tree of pkmsp3 haplotypes. The neighbour joining method was used to construct the tree, which contains 42 haplotypes. Numbers at the nodes indicate percentage support of 1000 bootstrap replicates
Fig. 5.
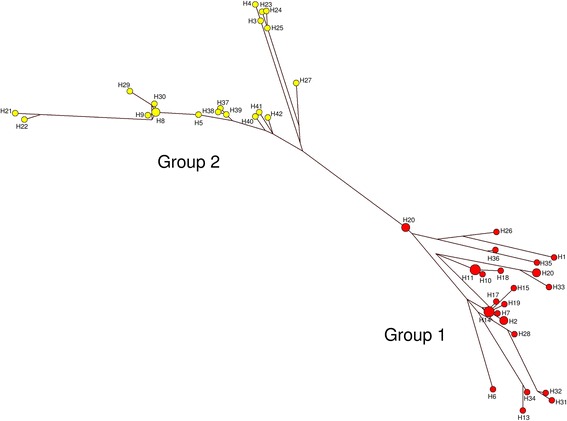
Network analysis of pkmsp3 haplotypes. The NETWORK program v4.6.1.2 was used to construct the haplotype network, which contains 42 haplotypes. Nodes in red indicate Group 1 haplotype members and nodes in yellow indicate Group 2 haplotype members
Further analysis was carried out to determine if Domain A or Domain B contributed to the haplotype clustering. A Neighbour Joining tree was constructed for both the domains (Fig. 6) and it was observed that polymorphisms in Domain A contributed to the haplotype clustering, as the clustering observed in this domain mirrored the tree constructed using the full length pkmps3 sequences.
Fig. 6.
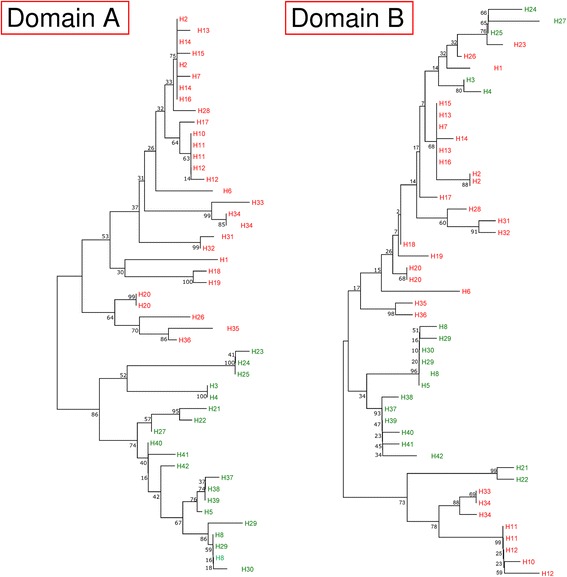
Phylogenetic trees of Domains A and B of pkmsp3. Neighbour joining method was used to construct the tree. In both trees, taxa indicated in red represent haplotypes of Group 1, whereas the taxa indicated in green are members of Group 2. The Domain A tree shows clustering similar to the tree of full length pkmsp3 (Fig. 4). Numbers at the nodes indicate percentage support of 1000 bootstrap replicates
Analysis on the diversity parameters and natural selection of members in Groups 1 and 2 was also carried out (Table 3). Haplotype diversity (Group 1: 0.993; Group 2: 0.995) and nucleotide diversity (Group 1: 0.02276; Group 2: 0.02418) of both groups were quite similar, as was the average number of nucleotide differences (Group 1: 24.31; Group 2: 25.97). The FST value between the groups was 0.402, indicating high genetic differentiation between these two groups. However, analysis of the phylogenetic tree did not indicate any temporal distribution between the two groups.
Table 3.
Estimates of DNA diversity and selection for Group 1 and Group 2, which are the major clusters obtained in the phylogenetic analysis
| Pkmsp3 | H | Hd ± SD | π ± SD | K | Tajima’s D |
|---|---|---|---|---|---|
| Group 1 | 26 | 0.993 ± 0.011 | 0.02276 ± 0.00167 | 24.31 | −0.81373 |
| Group 2 | 19 | 0.995 ± 0.018 | 0.02418 ± 0.00226 | 25.37 | −0.46858 |
Abbreviations: H number of haplotypes, Hd haplotype diversity, K average number of nucleotide differences, π nucleotide diversity, SD standard deviation
Tests of selection for pkmsp3
Tests were carried out to determine if the diversity in pkmsp3 was due to natural selection. The Tajima’s D, Fu & Li’s D* and F* tests showed no significant departure from neutrality in the full length pkmsp3, Domain A or Domain B (Table 1), thus suggesting neutral selection may be acting on these regions. Similarly, Tajima’s D test carried out on Group 1 and 2 showed no significant departure from neutrality (Table 3). This was reinforced by estimation of the dN/dS ratio, where, the dN/dS ratio for the full length sequence as well as Domain A were just slightly above 1, indicating neutral selection. However, the dN/dS ratio for Domain B was 0.6, suggesting that this domain may be under purifying selection.
Discussion
Vaccine development against malaria parasites is not a straightforward procedure. Multistage vaccines have recently been proposed because unique antigens are produced during the different stages of the parasite’s life-cycle. The merozoite has been identified as an important vaccine target due to its mobile and invasive nature, which exposes this stage to the host’s immune responses [42]. Many of the merozoite surface proteins contain polymorphic domains that signify diversifying selection, and conserved domains which indicate functional constraints of the protein. Furthermore, different strains within a Plasmodium species have been found to co-exist [43], thus vaccine candidates would need to be strain-transcending as one particular antibody generated against the protein from one strain may be ineffective against another. Antigenic diversity in vaccine candidates is one of the hurdles to design effective malaria vaccine. In vaccine development, it is prerequisite to survey genetic polymorphism of the candidate antigens, particularly the polymorphism from a wide range of field isolates. Furthermore, genetic polymorphism is also an important epidemiological tool. Plasmodium knowlesi has emerged in south-east Asia within the recent decade, and molecular epidemiological investigation may explain reasons of this recent emergence.
Although the biological functions of P. vivax and P. knowlesi MSP3 are not fully understood at this juncture, the alanine-rich central core in both proteins is predicted to form a coiled-coil tertiary structure [18]. Being located on the surface of the merozoites, the P. vivax MSP3 has been suggested to interact with other merozoite surface proteins, possibly mediated through protein-protein interactions involving the coiled-coil structure [18, 19] which is similar to what is observed in P. falciparum MSP3 [44]. In the present study, the coiled-coil region of P. knowlesi MSP3 was observed to be conserved. Therefore, similar to P. falciparum and P. vivax MSP3, the P. knowlesi MSP3 coiled-coil region may also utilise protein-protein interaction type bonds to interact with other merozoite surface proteins.
The nucleotide diversity (π: 0.046 ± 0.011) was found to be high when compared to other P. knowlesi functional genes [39–41], considering that most of the haplotypes discovered in this study were unique. A similar observation has also been reported for other merozoite surface antigens such as eba175, and this suggests that even where functional constraints exist, a range of haplotypes can still occur [45]. The low nucleotide diversity in Domain A as compared to that of the full length sequence, suggests limited polymorphism in the domain due to the presence of the coiled-coil region. Sliding window plot analysis (Fig. 2) showed high nucleotide diversity in the C-terminal, a finding also reported in pvmsp3β [20]. Temporal distribution of the haplotypes was not detected and this may be due to the fact that the P. knowlesi isolates were recent and collected within a 4-year period (2008–2012). The possibility of temporal distribution happening within such a short time is unlikely.
The pkmsp3 gene shares significant homology with the P. vivax pvmsp3 [46]. A study on pvmsp3 of P. vivax isolates from Korea revealed nucleotide diversity of 0.0727 ± 0.002 and 0.0304 ± 0.001 at the N- and C-terminal domains respectively [47], which contrast the nucleotide diversity of pkmsp3 domains (N-terminal π: 0.039 ± 0.002; C-terminal π: 0.067 ± 0.025). However, similar to pkmsp3, the C-terminal of pvmsp3 had ratio of dN/dS < 1, indicating purifying selection in that region. A study on pvmsp3 of P. vivax isolates from Thailand found nucleotide diversity of 0.0877 ± 0.005 [48], which is comparatively higher than the nucleotide diversity of pkmsp3 (π: 0.046 ± 0.011). Like pkmsp3, the C-terminal of pvmsp3 of the Thailand isolates also showed purifying selection (dN/dS < 1).
Phylogenetic and haplotype network analyses revealed that the P. knowlesi MSP3 haplotypes were clustered into two main groups. The Domain A in particular contributed to this clustering (Fig. 6). To gain a clearer picture of selection, the Z-test and Tajima’s D test for all three sets of sequences were analysed. In this instance, results for both the Z-test and Tajima’s D were not significant for the full length gene, Domain A or Domain B, indicating neutral selection. The dN/dS ratio is widely used to evaluate the effect of natural selection on genes where a lack of dN relative to dS (dN/dS < 1) suggests negative or purifying selection. Conversely, a higher value of dN compared to dS (dN/dS > 1) is indicative of positive selection. The dN/dS ratio for the full length gene as well as Domain A marginally exceeded 1, indicating neutral selection. Domain B, however, had a ratio of 0.6, indicating purifying selection on this part of the gene. Thus, it could be postulated that the P. knowlesi MSP3 has a functionally restricted Domain A which is protected from immune responses by an exposed and polymorphic Domain B.
In the present study, the phylogenetic tree showed separation of the P. knowlesi MSP3 haplotypes into two groups. Studies on P. knowlesi proteins such as the Duffy binding protein (PkDBPαII) [39], Pknbpxa [49] and PkAMA-1 domain I [50] have also reported bifurcation of haplotypes, indicating dimorphism of the genes. These findings provide support to the notion that two distinct P. knowlesi types or lineages exist in south-east Asia [51]. Microsatellite DNA analysis revealed two divergent P. knowlesi populations which have been associated with different macaque reservoir host species [52]. Recently, a whole-genome population study highlighted two major subgroups of P. knowlesi clinical isolates [53].
Conclusions
To the best of our knowledge, the present study is the first to investigate genetic diversity of the pkmsp3 gene as well as the natural selection acting on it. A moderate level of genetic diversity was observed in the pkmsp3 and only the C-terminal region (Domain B) appeared to be under purifying selection. The separation of the pkmsp3 into two groups of haplotypes provides further evidence of the existence of two distinct P. knowlesi types or lineages. Future studies should investigate the diversity of pkmsp3 among P. knowlesi isolates in North Borneo, a region with reports of the highest number of human knowlesi malaria cases to date.
Acknowledgments
We thank the Department of Parasitology Diagnostic Laboratory, Faculty of Medicine, University of Malaya and University of Malaya Medical Centre for providing patient blood samples.
Funding
This research project was supported by the University Malaya Postgraduate Research Grant (PG054-2016A) awarded to JRDS.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its Additional file 1. The nucleotide sequences of the pkmsp3 gene generated in this study are available in the GenBank database under accession numbers KT900798–KT900845.
Authors’ contributions
MYF and YLL designed the study and supervised the study process. JRDS performed all the experiments and analyzed the sequence data. JRDS and MYF performed sequence and phylogenetic analyses. JRDS, MYF and YLL wrote the manuscript. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Ethical clearance for this study was obtained from University of Malaya Medical Ethics Committee (Ref No. 817.18). Consent was obtained from patients prior to collection and they were informed of the use of these samples for research. This consent procedure was approved by the ethics committee.
Additional file
Multiple alignment of full amino acid sequences of pkmsp3. The yellow columns are the variable amino acid positions. The region highlighted in red at the top of the alignment indicates Domain A, and the region in green indicates Domain B. (XLS 389 kb)
Contributor Information
Jeremy Ryan De Silva, Email: jeremy8811@gmail.com.
Yee Ling Lau, Email: lauyeeling@um.edu.my.
Mun Yik Fong, Email: fongmy@um.edu.my.
References
- 1.Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995;12:616–26. doi: 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- 2.Chin W, Contacos PG, Coatney GR, Kimball HR. A naturally acquited quotidian-type malaria in man transferable to monkeys. Science. 1965;149:865. doi: 10.1126/science.149.3686.865. [DOI] [PubMed] [Google Scholar]
- 3.Fong YL, Cadigan FC, Coatney GR. A presumptive case of naturally occurring Plasmodium knowlesi malaria in man in Malaysia. Trans R Soc Trop Med Hyg. 1971;65:839–40. doi: 10.1016/0035-9203(71)90103-9. [DOI] [PubMed] [Google Scholar]
- 4.Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 5.Ng OT, Ooi EE, Lee CC, Lee PJ, Ng LC, Pei SW, et al. Naturally acquired human Plasmodium knowlesi infection, Singapore. Emerg Infect Dis. 2008;14:814–6. doi: 10.3201/eid1405.070863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luchavez J, Espino F, Curameng P, Espina R, Bell D, Chiodini P, et al. Human Infections with Plasmodium knowlesi, the Philippines. Emerg Infect Dis. 2008;14:811–3. doi: 10.3201/eid1405.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jongwutiwes S, Putaporntip C, Iwasaki T, Sata T, Kanbara H. Naturally acquired Plasmodium knowlesi malaria in human, Thailand. Emerg Infect Dis. 2004;10:2211–3. doi: 10.3201/eid1012.040293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vythilingam I, Noorazian YM, Huat TC, Jiram AI, Yusri YM, Azahari AH, et al. Plasmodium knowlesi in humans, macaques and mosquitoes in peninsular Malaysia. Parasit Vectors. 2008;1:26. doi: 10.1186/1756-3305-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau YL, Tan LH, Chin LC, Fong MY, Noraishah MA, Rohela M. Plasmodium knowlesi reinfection in human. Emerg Infect Dis. 2011;17:1314–5. doi: 10.3201/eid1707.101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee WC, Chin PW, Lau YL, Chin LC, Fong MY, Yap CJ, et al. Hyperparasitaemic human Plasmodium knowlesi infection with atypical morphology in peninsular Malaysia. Malar J. 2013;12:88. doi: 10.1186/1475-2875-12-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yusof R, Lau YL, Mahmud R, Fong MY, Jelip J, Ngian HU, et al. High proportion of knowlesi malaria in recent malaria cases in Malaysia. Malar J. 2014;13:168. doi: 10.1186/1475-2875-13-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conway DJ. Molecular epidemiology of malaria. Clin Microbiol Rev. 2007;20:188–204. doi: 10.1128/CMR.00021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McColl DJ, Silva A, Foley M, Kun JF, Favaloro JM, Thompson JK, et al. Molecular variation in a novel polymorphic antigen associated with Plasmodium falciparum merozoites. Mol Biochem Parasitol. 1994;68:53–67. doi: 10.1016/0166-6851(94)00149-9. [DOI] [PubMed] [Google Scholar]
- 14.Oeuvray C, Bouharoun-Tayoun H, Grass-Masse H, Lepers JP, Ralamboranto L, Tartar A, et al. A novel merozoite surface antigen of Plasmodium falciparum (MSP-3) identified by cellular-antibody cooperative mechanism antigenicity and biological activity of antibodies. Mem Inst Oswaldo Cruz. 1994;89(Suppl 2):77–80. doi: 10.1590/S0074-02761994000600018. [DOI] [PubMed] [Google Scholar]
- 15.Galinski MR, Corredor-Medina C, Povoa M, Crosby J, Ingravallo P, Barnwell JW. Plasmodium vivax merozoite surface protein-3 contains coiled-coil motifs in an alanine-rich central domain. Mol Biochem Parasitol. 1999;101:131–47. doi: 10.1016/S0166-6851(99)00063-8. [DOI] [PubMed] [Google Scholar]
- 16.Galinski MR, Ingravallo P, Corredor-Medina C, Al-Khedery B, Povoa M, Barnwell JW. Plasmodium vivax merozoite surface proteins-3beta and-3gamma share structural similarities with P. vivax merozoite surface protein-3alpha and define a new gene family. Mol Biochem Parasitol. 2001;115:41–53. doi: 10.1016/S0166-6851(01)00267-5. [DOI] [PubMed] [Google Scholar]
- 17.Jiang J, Barnwell JW, Meyer EV, Galinski MR. Plasmodium vivax merozoite surface protein-3 (PvMSP3): expression of an 11 member multigene family in blood-stage parasites. PLoS ONE. 2013;8:e63888. doi: 10.1371/journal.pone.0063888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–63. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice BL, Acosta MM, Pacheco MA, Carlton JM, Barnwell JW, Escalante AA. The origin and diversification of the merozoite surface protein 3 (msp3) multi-gene family in Plasmodium vivax and related parasites. Mol Phylogenet Evol. 2014;78:172–84. [DOI] [PMC free article] [PubMed]
- 20.Rayner JC, Huber CS, Feldman D, Ingravallo P, Galinski MR, Barnwell JW. Plasmodium vivax merozoite surface protein PvMSP-3 beta is radically polymorphic through mutation and large insertions and deletions. Infect Genet Evol. 2004;4:309–19. doi: 10.1016/j.meegid.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Jimenez MC, Ramos CH, Barbosa JA, Galinski MR, Barnwell JW, Rodrigues MM, et al. Biophysical characterization of the recombinant merozoite surface protein-3 of Plasmodium vivax. Biochim Biophys Acta. 2008;1780:983–8. doi: 10.1016/j.bbagen.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hahn MW, Rausher MD, Cunningham CW. Distinguishing between selection and population expansion in an experimental lineage of bacteriophage T7. Genetics. 2002;161:11–20. doi: 10.1093/genetics/161.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark AG. Population genetics: malaria variorum. Nature. 2002;418:283–5. doi: 10.1038/418283a. [DOI] [PubMed] [Google Scholar]
- 25.Paul RE, Day KP. Mating patterns of Plasmodium falciparum. Parasitol Today. 1998;14:197–202. doi: 10.1016/S0169-4758(98)01226-5. [DOI] [PubMed] [Google Scholar]
- 26.Kimura M. The neutral theory of molecular evolution: a review of recent evidence. Jpn J Genet. 1991;66:367–86. doi: 10.1266/jjg.66.367. [DOI] [PubMed] [Google Scholar]
- 27.Brunham RC, Plummer FA, Stephens RS. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infect Immun. 1993;61:2273–6. doi: 10.1128/iai.61.6.2273-2276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conway DJ. Natural selection on polymorphic malaria antigens and the search for a vaccine. Parasitol Today. 1997;13:26–9. doi: 10.1016/S0169-4758(96)10077-6. [DOI] [PubMed] [Google Scholar]
- 29.Conway DJ, Cavanagh DR, Tanabe K, Roper C, Mikes ZS, Sakihama N, et al. A principal target of human immunity to malaria identified by molecular population genetic and immunological analyses. Nat Med. 2000;6:689–92. doi: 10.1038/76272. [DOI] [PubMed] [Google Scholar]
- 30.Conway DJ, Polley SD. Measuring immune selection. Parasitology. 2002;125(Suppl):S3–16. doi: 10.1017/s0031182002002214. [DOI] [PubMed] [Google Scholar]
- 31.Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics. 2001;158:1505–12. doi: 10.1093/genetics/158.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.NETWORK v4.6.1.3, a programme for haplotype analysis downloaded from http://www.fluxus-engineering.com. Accessed 1 Dec 2016.
- 34.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Depaulis F, Veuille M. Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol Biol Evol. 1998;15:1788–90. doi: 10.1093/oxfordjournals.molbev.a025905. [DOI] [PubMed] [Google Scholar]
- 36.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132:583–9. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 39.Fong MY, Lau YL, Chang PY, Anthony CN. Genetic diversity, haplotypes and allele groups of Duffy binding protein (PkDBPαII) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasit Vectors. 2014;7:161. doi: 10.1186/1756-3305-7-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faber BW, Abdul Kadir K, Rodriguez-Garcia R, Remarque EJ, Saul FA, Vulliez-Le Normand B, et al. Low levels of polymorphisms and no evidence for diversifying selection on the Plasmodium knowlesi Apical Membrane Antigen 1 gene. PLoS ONE. 2015;10:e0124400. doi: 10.1371/journal.pone.0124400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rawa MS, Fong MY, Lau YL. Genetic diversity and natural selection in the rhoptry-associated protein 1 (RAP-1) of recent Plasmodium knowlesi clinical isolates from Malaysia. Malar J. 2016;15:62. doi: 10.1186/s12936-016-1127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escalante AA, Lal AA, Ayala FJ. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. 1998;149:189–202. doi: 10.1093/genetics/149.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snounou G, White NJ. The co-existence of Plasmodium: sidelights from falciparum and vivax malaria in Thailand. Trends Parasitol. 2004;20:333–9. doi: 10.1016/j.pt.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 44.McColl DJ, Anders RF. Conservation of structural motifs and antigenic diversity in the Plasmodium falciparum merozoite surface protein-3 (MSP-3) Mol Biochem Parasitol. 1997;90:21–31. doi: 10.1016/S0166-6851(97)00130-8. [DOI] [PubMed] [Google Scholar]
- 45.Schultz L, Wapling J, Mueller I, Ntsuke PO, Senn N, Nale J, et al. Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar J. 2010;9:336. doi: 10.1186/1475-2875-9-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA. Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004;20:388–95. doi: 10.1016/j.pt.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Kang JM, Ju HL, Cho PY, Moon SU, Ahn SK, Sohn WM, et al. Polymorphic patterns of the merozoite surface protein-3β in Korean isolates of Plasmodium vivax. Malar J. 2014;13:104. doi: 10.1186/1475-2875-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Putaporntip C, Miao J, Kuamsab N, Sattabongkot J, Sirichaisinthop J, Jongwutiwes S, et al. The Plasmodium vivax merozoite surface protein 3β sequence reveals contrasting parasite populations in southern and northwestern Thailand. PLoS Negl Trop Dis. 2014;8:e3336. doi: 10.1371/journal.pntd.0003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinheiro MM, Ahmed MA, Millar SB, Sanderson T, Otto TD, Lu WC, et al. Plasmodium knowlesi genome sequences from clinical isolates reveal extensive genomic dimorphism. PLoS ONE. 2015;10:e0121303. doi: 10.1371/journal.pone.0121303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fong MY, Wong SS, De Silva JR, Lau YL. Genetic polymorphism in domain I of the apical membrane antigen-1 among Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Acta Trop. 2015;152:145–50. doi: 10.1016/j.actatropica.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, et al. Accelerated diversification of nonhuman primate malarias in southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2015;32:422–39. doi: 10.1093/molbev/msu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Divis PC, Singh B, Anderios F, Hisam S, Matusop A, Kocken CH, et al. Admixture in humans of two divergent Plasmodium knowlesi populations associated with different macaque host species. PLoS Pathog. 2015;11:e1004888. doi: 10.1371/journal.ppat.1004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Assefa S, Lim C, Preston MD, Duffy CW, Nair MB, Adroub SA, et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci U S A. 2015;112:13027–32. doi: 10.1073/pnas.1509534112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the conclusions of this article are included within the article and its Additional file 1. The nucleotide sequences of the pkmsp3 gene generated in this study are available in the GenBank database under accession numbers KT900798–KT900845.