

Abstract
Background
Despite important progress in the field of innate immunity, our understanding of host immune responses to parasitic nematode infections lags behind that of responses to microbes. A limiting factor has been the obligate requirement for a vertebrate host which has hindered investigation of the parasitic nematode infective process. The nematode parasite Heterorhabditis bacteriophora offers great potential as a model to genetically dissect the process of infection. With its mutualistic Photorhabdus luminescens bacteria, H. bacteriophora invades multiple species of insects, which it kills and exploits as a food source for the development of several nematode generations. The ability to culture the life cycle of H. bacteriophora on plates growing the bacterial symbiont makes it a very exciting model of parasitic infection that can be used to unlock the molecular events occurring during infection of a host that are inaccessible using vertebrate hosts.
Results
To profile the transcriptional response of an infective nematode during the early stage of infection, we performed next generation RNA sequencing on H. bacteriophora IJs incubated in Manduca sexta hemolymph plasma for 9 h. A subset of up-regulated and down-regulated genes were validated using qRT-PCR. Comparative analysis of the transcriptome with untreated controls found a number of differentially expressed genes (DEGs) which cover a number of different functional categories. A subset of DEGs is conserved across Clade V parasitic nematodes revealing an array of candidate parasitic genes.
Conclusions
Our analysis reveals transcriptional changes in the regulation of a large number of genes, most of which have not been shown previously to play a role in the process of infection. A significant proportion of these genes are unique to parasitic nematodes, suggesting the identification of a group of parasitism factors within nematodes. Future studies using these candidates may provide functional insight into the process of nematode parasitism and also the molecular evolution of parasitism within nematodes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3468-6) contains supplementary material, which is available to authorized users.
Keywords: Heterorhabditis bacteriophora, RNA-seq, Hemolymph, Parasitism, Nematode
Background
Parasitic nematodes (PN) continue to place a considerable burden on human health and agricultural production. PN diseases cause a variety of unfavorable conditions ranging from lethargy to fever, diarrhea, blindness and death. An estimated 1–1.2 billion people are infected with PNs leading to an increase of up to 52.1 million disability adjusted life years with hookworm infections contributing more than 40% of the lost years alone [1]. Current control strategies of deworming, while effective in the short-term, are inadequate due to frequent reinfection and the development of drug resistant nematode populations. Furthermore, attempts at creating a vaccine have been hindered due to the lack of good animal models and effective antigens [2].
While PN infections are understood to be immunomodulatory in nature, the molecular mechanisms of infection are poorly understood [3]. While efforts have been made to study parasitism in PNs directly, the requirement of a vertebrate host for development makes in vitro cultivation currently impossible, thereby making access to parasitic life stages and the interactions between the host immune system and the parasite difficult [4]. Even though a great deal has been learned about nematode biology and development using the free-living nematode Caenorhabditis elegans, it is not a parasite and thus does not allow for relevant investigations of PN infection mechanisms. For this reason, interest in developing Heterorhabditis bacteriophora into a model organism in order to study nematode parasitism has recently grown [5–8].
Heterorhabditis bacteriophora is a member of the Eurhabditis clade, which also contains other PNs such as the vertebrate hookworms Ancylostoma ceylanicum and Necator americanus as well as the model organism C. elegans. Heterorhabditis bacteriophora is an entomopathogenic nematode (EPN) which uses host insects and the mutualistic bacteria Photorhabdus luminescens to successfully reproduce [9]. The free-living stage of H. bacteriophora, the infective juvenile (IJ), is a developmentally arrested stage analogous to the infective stage of hookworms and the dauer of C. elegans [10, 11]. The IJs seek host insects to colonize and reproduce. Once established, the IJs resume their development and progress through the complete life cycle of H. bacteriophora. After 2 to 3 generations of reproduction, the nutrition of the host’s cadaver is exhausted and juveniles begin to arrest in mass as IJs. These IJs leave the cadaver and begin to search for a new host. Unlike hookworms and most other PNs, H. bacteriophora and its bacterial symbiont P. luminescens can be manipulated and cultured in vitro. Additionally, advanced molecular tools (e.g. gene silencing by RNAi) are being developed for H. bacteriophora as well as the recent publication of its genome, making H. bacteriophora potentially an excellent alternative model for nematode parasitism [5, 8, 12–14]. Furthermore, the ability to propagate H. bacteriophora in the immunology model Drosophila melanogaster, allows the study of host responses to PN infection mechanisms [15–17].
While the basic tools to develop H. bacteriophora as a model organism have been or are in the process of being developed [8, 13], genes directly involved in parasitism are still poorly understood. Studies examining the transcriptome of H. bacteriophora have been performed, but our results described here is the first study, to our knowledge, to utilize advanced next-generation sequencing technologies and the published H. bacteriophora genome to examine the transcriptional program during host invasion [18, 19]. A better understanding of this crucial transition period could help define parasitism genes and possibly allow the establishment of new interventions that prevent infection of hosts. To begin teasing apart the molecular biology of the early infection, we performed next generation RNA sequencing on H. bacteriophora IJs incubated in Manduca sexta hemolymph plasma for 9 h. Comparative analysis of the transcriptome with untreated controls found a number of differentially expressed genes (DEGs) which cover a number of different functional categories. Furthermore, a subset of the DEGs is conserved across Clade V parasitic nematodes. This subset of genes may serve as potential targets for future studies investigating nematode parasitism.
Results
Illumina sequencing
RNA-sequencing using the Illumina HiSeq4000 platform was performed on H. bacteriophora (TT01 strain) IJs soaked for 9 h in M. sexta hemolymph plasma in order to identify nematode genes that are expressed during the initial stages of insect infection. Total RNA was collected from six independent samples (three 9 h hemolymph plasma treated and three untreated 0 h controls) to characterize the transcriptome and measure differential expression of genes. An outline of the pipeline used to analyze RNA-seq data is shown in Additional file 1: Figure S1. After quality checks and trimming, RNA-sequencing yielded an average of ~41.8 million reads per sample, with an average of 89.1% of control and 81.3% of hemolymph-treated reads mapping to the H. bacteriophora genome. There was an average of 4,084,703 and 8,600,584 unmapped reads for control and treated IJ respectively, making the total number of mapped reads 33,554,746 and 37,399,072. Further details of the read mapping are shown in Additional file 1: Table S1. Of the 20,964 genes contained within the H. bacteriophora genome, 1641 were significantly (p < 0.05) differentially expressed after a 9-h exposure to hemolymph plasma. 881 of these DEGs were upregulated (fold change ≥ 2) and 760 were downregulated (fold change ≤ −2) relative to the 0 h control.
Validation of RNA-seq by qRT-PCR
In order to confirm changes in expression observed by differential expression analysis of RNA-seq data, qRT-PCR was performed on selected genes. Genes with the largest fold changes, either positive or negative and with significant differential expression (p < 0.05), were used for validation. Genes used to test upregulation were Hba_07265 (logFC = 5.08), Hba_11987 (logFC = 3.02), Hba_15382 (logFC = 7.97), Hba_15540 (logFC = 6.47), and Hba_20350 (logFC = 3.75). Hba_05395 (logFC = −3.17), Hba_05947 (logFC = −3.33), Hba_15875 (logFC = −4.44), Hba_17909 (logFC = −3.31), and Hba_18611 (logFC = −2.81) were used as representatives of downregulated genes. In order to confirm differential regulation, qRT-PCR was also performed on untreated control and hemolymph plasma soaked IJs made independently of the samples used for RNA-seq. Using rpl-32 as an expression control, the directionality of the calculated ΔΔCT values for the treated IJ used for RNA-seq agreed with the changes observed in the RNA-seq differential analysis (Fig. 1). The changes observed by RNA-seq were also present in the samples made exclusively for qRT-PCR.
Fig. 1.
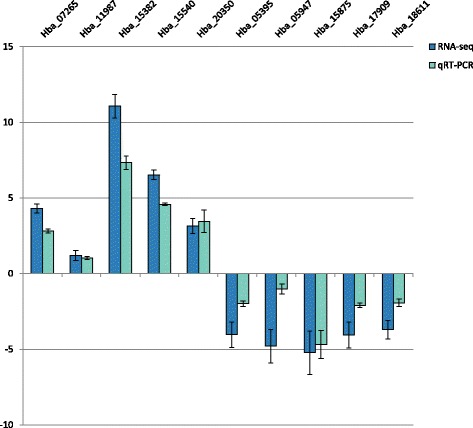
qRT-PCR of genes identified as differentially expressed by RNA-seq. Quantitative RT-PCR using rpl-32 as an expression control was performed on samples used for RNA-seq to confirm changes in expression. Additionally, qRT-PCR was also performed on a set of samples prepared independently from the samples used for RNA-seq to further validate expression changes. ΔΔCT values are relative to matched control samples. Similar changes in expression were seen in both the RNA-seq and independent samples. Error bars represent SEM
GO analysis of RNA-seq data
Gene Ontology (GO) annotations of differentially expressed genes were collected from the WormBase ParaSite Biomart [20]. In order to better understand the functional distribution of the genes at a global level, WEGO software was used to classify the terms into high level categories. The 1641 DEGs were annotated to 40 functional groups (Fig. 2). 10 groups were contained within the Cellular Component root category, 10 in the Molecular Function root category, and 20 in the Biological Process root. Within Cellular Component, the groups containing the most genes were cell (GO:0005623), organelle (GO:0043226) and macromolecular complex (GO:0032991). Within Molecular Function, a large proportion of genes were categorized to catalytic activity (GO:0003824), binding (GO:0005488), structural molecule activity (GO:0005198) and transporter activity (GO:0005215). The categories of metabolic process (GO:0008152), cellular process (GO:0009987), reproduction (GO:0000003), developmental process (GO:0032502) and growth (GO:0040007) contained a large number of genes within the root level Biological Process category.
Fig. 2.
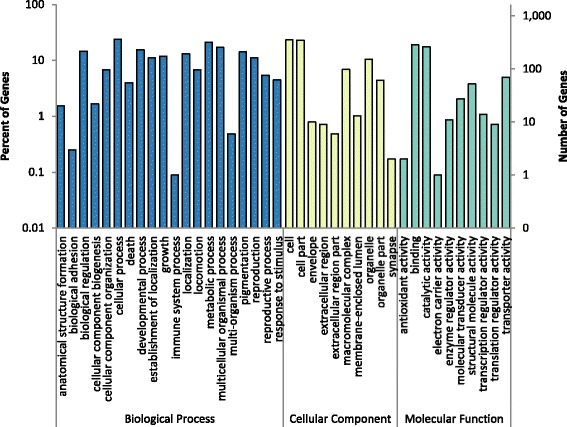
Common GO annotations of DEGs. GO terms were condensed into higher order functional categories using WEGO, in order to more easily understand global changes of expression. Changes were seen in a number of different functional categories with the most occurring in the Biological Process GO domain
To further explore which, if any, GO terms were overrepresented, gene set enrichment analysis was performed using the topGO R package [21]. Categories of significance were found for each of the root GO categories (Table 1). A total of 14 GO groups were found to be enriched at an unadjusted p-value < 0.05. The groups include metabolic process, cell cycle, body morphogenesis, carbohydrate metabolic process, cytoplasm, endoplasmic reticulum, translation elongation factor activity and acid phosphatase activity. The most significantly enriched groups (unadjusted p < 0.001) were proteolysis (GO:0006508) and structural constituents of the ribosome (GO:0003735).
Table 1.
Significantly enriched GO terms in DEGs
| GO Domain | Go Accession | GO Term | Number of Genes (Significant/Total Annotated) |
p-value (Fisher’s Exact Test) |
|---|---|---|---|---|
| Molecular Function | GO:0003735 | structural constituent of ribosome | 39/52 | <1E-20 |
| Biological Process | GO:0006508 | proteolysis | 26/103 | 1.07E-05 |
| Molecular Function | GO:0003746 | translation elongation factor activity | 3/3 | 0.00167 |
| Molecular Function | GO:0003993 | acid phosphatase activity | 5/10 | 0.00351 |
| Biological Process | GO:0010171 | body morphogenesis | 3/4 | 0.00744 |
| Biological Process | GO:0008152 | metabolic process | 105/673 | 0.01048 |
| Molecular Function | GO:0016788 | hydrolase activity, acting on ester bonds | 10/35 | 0.02239 |
| Molecular Function | GO:0004129 | cytochrome-c oxidase activity | 3/6 | 0.02535 |
| Molecular Function | GO:0016810 | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds | 4/8 | 0.03872 |
| Molecular Function | GO:0004359 | glutaminase activity | 2/3 | 0.039 |
| Molecular Function | GO:0008137 | NADH dehydrogenase (ubiquinone) activity | 2/3 | 0.039 |
| Cellular Component | GO:0005737 | cytoplasm | 19/97 | 0.0408 |
| Biological Process | GO:0005975 | carbohydrate metabolic process | 14/66 | 0.04955 |
| Cellular Component | GO:0005789 | endoplasmic reticulum | 2/3 | 0.0496 |
KEGG annotations
In addition to annotating DEGs with GO terms, Kyoto Encyclopedia of Genes and Genomes KEGG functional categories and pathways were also found. KEGG functional categories covered enzymes (Enzymes, Protein phosphatases and associated proteins, Peptidases, Chaperones and folding catalysts) and nucleic acid machinery (Ribosome, Chromosome, Ribosome biogenesis, Messenger RNA biogenesis and DNA replication proteins). Similarly, KEGG pathway annotation identified pathways involved in ribosome biogenesis and protein processing pathways (Fig. 3).
Fig. 3.
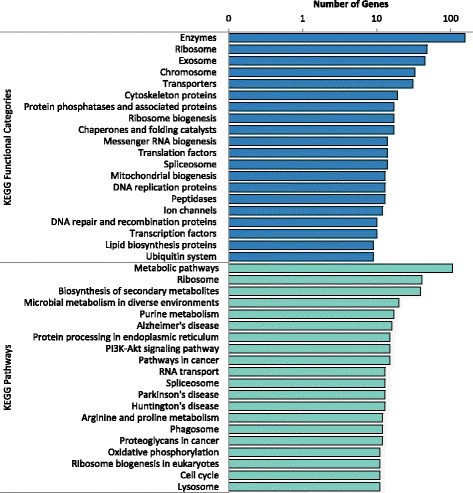
KEGG functional and pathway annotations for DEGs. Using the peptide sequences of DEGs, functional and pathway annotations were found using the KEGG Automatic Annotation Server and eggNOG, respectively. Annotations covered a variety of different pathway and functional annotations. The 20 most represented categories by KAAS and eggNOG annotation are shown
Comparison of DEGs with different nematode clades and representative species
In order to identify a set of common PN genes expressed during the early stages of infection, a series of data filters were used on genes identified as significantly changed (FC ≥ 2 or FC ≤ −2, p < 0.05) during differential expression analysis. Protein sequences of DEGs were blasted against all sequenced nematode genomes in each clade (H. bacteriophora was excluded from the Clade V blast). Alignments were considered matches if the percent identity was greater than or equal to 60 and the e-value was less than 0.00005. The greatest number of shared genes (802) was between H. bacteriophora and Clade V. A total of 130 genes were found to be common between H. bacteriophora and the nematodes of Clades I, III, IV and V (Fig. 4). GO annotation was performed on the genes within the common pool. The genes were primarily categorized to groups involving reproduction, development and growth.
Fig. 4.
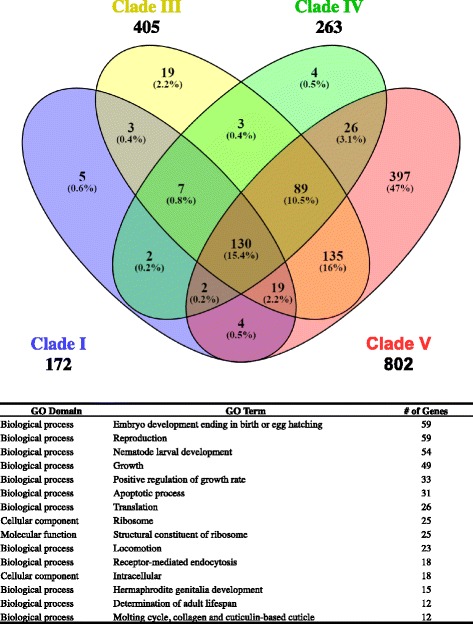
Distribution of DEGs across nematode clades. Overlaps between alignments between H. bacteriophora and different nematode clades were found using Venny 2.1. The majority of alignments were exclusive to Clade V (excluding H. bacteriophora). However a number of genes were shared between all Clades and single genes were annotated to multiple GO categories. The top fifteen GO categories for the 130 genes shared by all clades are shown in the table below the diagram
Due to the DEGs predominantly aligning with Clade V nematodes, further comparisons between parasites and H. bacteriophora were made using only Clade V nematodes. A second blast of the 1641 genes was performed against the genomes of C. elegans, Ancylostoma ceylanicum, Necator americanus and Haemonchus contortus. The same criteria used for the blast against nematode clades were used to determine if alignments were matches with the above nematodes. After analysis, 551 genes were hits for C. elegans genes, 790 genes matched A. ceylanicum, 706 genes matched N. americanus, and 363 genes matched H. contortus. 226 of the genes were held in common by the four nematode species and H. bacteriophora, while 75 genes were shared between the parasitic nematodes A. ceylanicum, H. contortus and N. americanus (Fig. 5). Of the 75 genes, only 45 had annotation data available in the ParaSite Biomart database. The GO terms, InterPro ID and other protein information for the 45 matches are shown in Table 2.
Fig. 5.
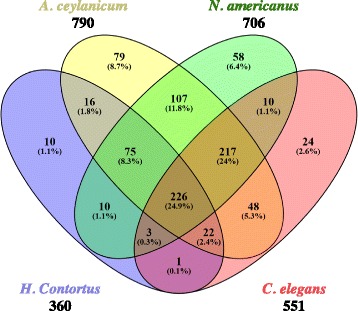
Distribution of DEGs across clade V parasitic nematodes and C. elegans. Overlaps between alignments between H. bacteriophora and different Clade V nematodes were found using Venny 2.1. While 226 genes were found to be common between C. elegans and the parasitic nematodes surveyed, a subset of 75 genes was shared among parasitic nematodes
Table 2.
Common genes of clade V parasitic nematodes
| Gene stable ID | GO term accession | GO term name | InterPro ID | InterPro short description | Signal Present | Coiled coils | Transmembrane domain |
|---|---|---|---|---|---|---|---|
| Hba_00415 | GO:0016021 | integral component of membrane | IPR010761 | Clc_prot-like | - | - | TMhelix |
| Hba_03156 | GO:0050660 | flavin adenine dinucleotide binding | IPR004113 | FAD-linked_oxidase_C | - | - | TMhelix |
| Hba_05467 | GO:0006810 | transport | IPR003439 | ABC_transporter-like | - | - | TMhelix |
| Hba_06397 | GO:0005975 | carbohydrate metabolic process | IPR001223 | Glyco_hydro18cat | - | - | - |
| Hba_06467 | GO:0007017 | microtubule-based process | IPR001372 | Dynein_light_chain_typ-1/2 | - | - | - |
| Hba_06770 | GO:0005975 | carbohydrate metabolic process | IPR001360 | Glyco_hydro_1 | - | - | - |
| Hba_06875 | GO:0005921 | gap junction | IPR000990 | Innexin | - | - | TMhelix |
| Hba_06876 | GO:0005921 | gap junction | IPR000990 | Innexin | - | - | TMhelix |
| Hba_06877 | GO:0005921 | gap junction | IPR000990 | Innexin | - | - | - |
| Hba_07556 | GO:0006810 | transport | IPR002259 | Eqnu_transpt | - | - | TMhelix |
| Hba_07557 | GO:0006810 | transport | IPR002259 | Eqnu_transpt | - | - | TMhelix |
| Hba_08573 | GO:0016020 | membrane | IPR006029 | Neurotrans-gated_channel_TM | - | - | TMhelix |
| Hba_08939 | GO:0004871 | signal transducer activity | IPR015898 | G-protein_gamma-like_dom | - | - | - |
| Hba_09140 | GO:0016020 | membrane | IPR007265 | COG_su3 | - | - | TMhelix |
| Hba_09704 | GO:0005515 | protein binding | IPR001611 | Leu-rich_rpt | - | Coil | TMhelix |
| Hba_09919 | GO:0006334 | nucleosome assembly | IPR002164 | NAP_family | - | - | - |
| Hba_10371 | GO:0005524 | ATP binding | IPR004523 | Asp-tRNA_synthase | - | - | - |
| Hba_10698 | GO:0040010 | positive regulation of growth rate | IPR026847 | VPS13 | - | - | TMhelix |
| Hba_11850 | GO:0003677 | DNA binding | IPR000536 | Nucl_hrmn_rcpt_lig-bd_core | - | - | - |
| Hba_12267 | GO:0000138 | Golgi trans cisterna | IPR007258 | Vps52 | - | - | TMhelix |
| Hba_12451 | GO:0005515 | protein binding | IPR000626 | Ubiquitin_dom | - | Coil | - |
| Hba_13253 | GO:0005515 | protein binding | IPR000210 | BTB/POZ-like | - | - | - |
| Hba_13349 | GO:0016021 | integral component of membrane | IPR011701 | MFS | - | - | TMhelix |
| Hba_13917 | GO:0016020 | membrane | IPR005027 | Glyco_trans_43 | - | - | - |
| Hba_14055 | GO:0016020 | membrane | IPR002159 | CD36 | - | - | TMhelix |
| Hba_14609 | GO:0009792 | embryo development ending in birth or egg hatching | - | - | - | - | - |
| Hba_14679 | GO:0005515 | protein binding | IPR001611 | Leu-rich_rpt | SignalP-noTM | - | - |
| Hba_14742 | GO:0001104 | RNA polymerase II transcription cofactor activity | IPR019145 | Mediator_Med10 | - | Coil | - |
| Hba_14927 | GO:0006189 | ‘de novo’ IMP biosynthetic process | IPR000031 | PurE_dom | - | - | - |
| Hba_15073 | GO:0006355 | regulation of transcription, DNA-templated | IPR000536 | Nucl_hrmn_rcpt_lig-bd_core | - | - | - |
| Hba_15994 | GO:0005634 | nucleus | IPR002999 | Tudor | - | - | - |
| Hba_17350 | GO:0006270 | DNA replication initiation | IPR003874 | CDC45 | - | - | - |
| Hba_17394 | GO:0006508 | proteolysis | IPR001969 | Aspartic_peptidase_AS | SignalP-noTM | - | - |
| Hba_17412 | GO:0016020 | membrane | IPR000731 | SSD | - | - | TMhelix |
| Hba_18346 | GO:0006457 | protein folding | IPR002777 | PFD_beta-like | - | Coil | - |
| Hba_18755 | GO:0008146 | sulfotransferase activity | IPR000863 | Sulfotransferase_dom | - | - | - |
| Hba_19983 | GO:0003777 | microtubule motor activity | IPR013594 | Dynein_heavy_dom-1 | - | Coil | - |
| Hba_20009 | GO:0009792 | embryo development ending in birth or egg hatching | IPR007051 | CHORD | - | - | - |
| Hba_20121 | GO:0009792 | embryo development ending in birth or egg hatching | - | - | - | - | - |
| Hba_20127 | GO:0055114 | oxidation-reduction process | IPR008972 | Cupredoxin | - | - | - |
| Hba_20282 | GO:0016021 | integral component of membrane | IPR008855 | TRAP-delta | SignalP-noTM | - | - |
| Hba_20782 | GO:0005515 | protein binding | IPR003172 | ML_dom | - | - | - |
| Hba_20870 | GO:0016020 | membrane | IPR003492 | Battenin_disease_Cln3 | - | - | TMhelix |
| Hba_21214 | GO:0004568 | chitinase activity | IPR000726 | Glyco_hydro_19_cat | SignalP-noTM | - | - |
| Hba_21297 | GO:0003723 | RNA binding | IPR001040 | TIF_eIF_4E | - | - | - |
Identification of genes activated during the initial stages of host infection
Upregulated DEGs were further analyzed to determine if any genes could be identified as highly conserved PN genes. Out of the 239 differentially expressed up regulated genes (p < 0.05, FDR < 0.05, FC ≥ 2), 52 genes (21.8%) were found to contain a classic or non-classical signal peptide, and to be non-orthologous to C. elegans but orthologous to A. ceylanicum (Bioproject PRJNA231479). Of these 52 H. bacteriophora genes, 17 were determined to be initial activation candidates due to either the identification of proteins and/or molecular mechanisms believed to be involved in parasitism (by GO grouping and InterPro ID) or the lack of any identifiable protein motifs (Table 3). Of the 17 genes, 16 were identified as having enzymatic motifs, including both hydrolases and kinases. The remaining gene, Hba_13349, was identified as being involved in membrane transport.
Table 3.
Highly conserved clade V parasitic nematode genes upregulated in H. bacteriophora incubated in hemolymph plasma
| Gene ID | GO Accession | GO Name | InterPro ID | InterPro Description |
|---|---|---|---|---|
| Hba_05422 | GO:0005515 | Protein binding | IPR011105 | Cell_wall_hydrolase_SleB |
| Hba_06426 | GO:0004190 | Aspartic-type endopeptidase activity | IPR021109 | Peptidase_aspartic_dom |
| Hba_07292 | GO:0016758 | Transferase activity, transferring hexosyl groups | IPR002213 | UDP_glucos_trans |
| Hba_07973 | GO:0004181 | Metallocarboxypeptidase activity | IPR000834 | Peptidase_M14 |
| Hba_08473 | GO:0008483 | Transaminase activity | IPR005814 | Aminotrans_3 |
| Hba_11636 | GO:0004185 | Serine-type carboxypeptidase activity | IPR001563 | Peptidase_S10 |
| Hba_11637 | GO:0004185 | Serine-type carboxypeptidase activity | IPR001563 | Peptidase_S10 |
| Hba_13000 | GO:0008236 | Serine-type peptidase activity | IPR008758 | Peptidase_S28 |
| Hba_13072 | GO:0008378 | Galactosyltransferase activity | IPR002659 | Glyco_trans_31 |
| Hba_13349 | GO:0055085 | Transmembrane transport | IPR020846 | MFS_dom |
| Hba_13477 | GO:0004672 | Protein kinase activity | IPR011009 | Kinase-like_dom |
| Hba_14122 | GO:0003993 | Acid phosphatase activity | IPR029033 | His_PPase_superfam |
| Hba_15308 | GO:0008234 | Cysteine-type peptidase activity | IPR013128 | Peptidase_C1A |
| Hba_17215 | GO:0003824 | Catalytic activity | IPR031319 | A-amylase_C |
| Hba_19909 | GO:0003796 | Lysozyme activity | IPR008597 | Destabilase |
| Hba_20878 | GO:0055085 | Transmembrane transport | IPR020846 | MFS_dom |
| Hba_20939 | GO:0003993 | Acid phosphatase activity | IPR029033 | His_PPase_superfam |
Discussion
Parasitic nematode infections continue to pose a considerable burden to human health. However, the obligate need for a vertebrate host has made efforts to study the molecular mechanisms of parasitic infection intractable and highlights the need for an analogous model nematode. Due to its life cycle and recent studies, the possibility of developing and using H. bacteriophora as a model for parasitic infections has grown. Basic advances in the development of a molecular toolbox and the publication of its genome provides a way for manipulating and identifying the function of parasitic genes [5, 8, 13, 19]. In order to create a smaller list of genes involved in infection for future investigation, we performed RNA-seq on H. bacteriophora strain TT01 IJs incubated in hemolymph plasma for 9 h. After differential expression analysis, a total of 1641 genes were identified as being differentially expressed after a 9 h exposure to hemolymph plasma. Hemolymph plasma from Manduca sexta was selected as an activation media because M. sexta is a natural host, and its large size facilitates extraction of large quantities of hemolymph. Previous studies identified hemolymph soaking as a valid method for inducing synchronous activation of entomopathogenic IJs, while also allowing for the mass activation of IJs necessary to produce enough high quality RNA for RNA-seq [19, 22–24]. Additionally, our pilot studies used to determine the maximum concentration of IJs per milliliter hemolymph plasma showed IJs develop into J4s within 48 h of soaking and continue to adults by 72 h (data not shown). After 1 week of soaking in hemolymph plasma, multiple generations of nematodes were present.
Assignment of GO terms to DEGs categorized 769 of the genes to probable functions. Some of the most represented functional categories included reproduction, reproductive process, developmental process, binding and catalytic activity. By checking for overrepresented GO terms, we found a number of genes involved in proteolysis, acid phosphatase activity, NADH dehydrogenase (ubiquinone) activity and hydrolases, as well as sequences associated with ribosomal constituents and translation elongation factors. Also, a number of overrepresented terms were also found in categories related to development such as body morphogenesis, carbohydrate metabolism and metabolic processes, consistent with the resumption of development by the IJs.
KEGG functional categories and pathways were also assigned to DEGs. KEGG assignments showed similar categories as the overrepresented GO terms. A large number of sequences were categorically assigned ribosome, ribosome biogenesis, messenger RNA biogenesis, cytoskeleton proteins and chaperones and folding catalysts suggesting upregulation in protein production is necessary to exit the developmentally arrested dauer stage. Also seen in the KEGG functional categories again are protein phosphatases and associated proteins, ubiquitin system and peptidases. While the KEGG and GO analysis does not provide enough resolution to identify parasitic genes directly, the recurrence and overrepresentation of proteolytic enzymes suggests some of these genes may be involved in the production of proteins/catalysts necessary for parasitism. This idea is further supported by an increase in the transporters category since gene products important for PN-host interactions are likely secreted into host tissues. The KEGG pathway assignments mirror the KEGG functional categories by also showing a number of genes involved in biosynthesis of secondary metabolites, protein processing in endoplasmic reticulum, ribosome biogenesis and lysosome.
In an attempt to identify common parasitic genes, a series of protein blasts were performed against each nematode clade and also against representative nematodes of Clade V. Blasting the protein sequences of the H. bacteriophora DEGs against all nematodes included in Clades I, III, IV and V (not including H. bacteriophora) returned 172, 405, 263 and 802 gene hits respectively. As expected, the majority of hits (397 genes) were found only in Clade V due to H. bacterophora’s close phylogenetic relationship with its members. Interestingly, while there is overlap with Clades III and IV, there are 405 hits with Clade III and only 263 with Clade IV. Given their phylogenetic relationship to Clade V, hits would be expected to decrease as the distance between clades increases. The blast searches by clade also suggest a subset of “pan-nematode” genes (130) which are shared by all four of the clades surveyed. GO term assignment identifies these genes as being involved in embryo development, reproduction, nematode larval development, life cycle development and growth. Given the apparent conservation of genes related to reproduction and development, it is possible these genes represent the basic genetic needs for the reproduction and development of nematodes.
Given the relatively large number of hits with Clade V nematodes, another protein blast was performed in order to further investigate the presence of a set of PN genes. For this reason, this Clade V blast was limited to A. ceylanicum, H. contortus, N. americanus and C. elegans. While C. elegans is not a parasitic nematode, given the deep understanding and robust annotation of its genome, it was included in order to further separate Clade V nematode genes from the highly conserved genes of Clade V PNs. While a number of DEGs were found to hit each nematode surveyed, 75 genes were found to be shared by A. ceylanicum, H. contortus and N. americanus. Of these 75 genes, 30 have no GO, InterPro ID or protein motifs assigned in the ParaSite Biomart database. The 45 remaining genes contain a variety of different functional categories, but are predominantly represented by categories involved in growth and development. However, some proteins of possible parasitic interest are present. Most notably, 4 genes (Hba_14679, Hba_17394, Hba_20282 and Hba_21214) contain signal peptides with no transmembrane region. Hba_14679 contains a protein motif for leucine-rich repeats, which serve as structural supports for protein-protein interactions [25]. While a wide variety of proteins make use of these supports, given the presence of a signaling peptide, the product of Hba_14679 may play a role in host-parasite interactions external to the worm. Hba_20282 contains a motif for the delta subunit precursor of a translocon. While the exact function of this protein is unknown, translocons are known to transport peptides across membranes. Within eukaryotes translocons are commonly used to transfer polypeptides into the endoplasmic reticulum [26]. However, in prokaryotes, translocons can be assembled to export virulence factors outside of the cell [27, 28] While this protein may serve no other purpose than shuttling molecules into the endoplasmic reticulum, its conservation between 4 different parasites suggests that it may be needed for the transport of parasitism factors.
Hba_17394 and Hba_21214 are both enzymes which may be externally secreted since they both contain signal peptides. Hba_17394 is identified to have an aspartic peptidase motif. While further functional investigation is necessary for an exact identity, aspartic peptidases are known to have a range of functions from the digestion of peptides to the production of active proteins from precursor proteins [29, 30]. Within parasites, such a peptidase could be secreted for use as a virulence factor. Hba_21214 contains a protein motif for a glycoside hydrolase, family 19. Glycoside hydrolases are chitinases which break down glycosidic bonds between carbohydrates [31]. Chitinases are most commonly used as defense mechanisms to break down the cell walls of fungal and insect pathogens [32–34]. Within H. bacteriophora, the presence of a chitinase seems obligate since the digestion of chitin would be necessary upon infection of a host insect. However, given its presence across several parasitic nematodes, it is possible a secreted chitinase may serve a more general role in parasitism [35].
Given that any likely parasitism genes would potentially be upregulated upon exposure to a host, further analyses was carried out to identify any upregulated DEGs conserved within parasitic nematodes by comparison to A. ceylanicum (Bioproject PRJNA231479). Out of 861 upregulated DEGs, 239 had a false discovery rate less than 0.05. Of these 239 genes, 17 genes were identified as being potential conserved parasitism genes due to being orthologous to A. ceylanicum and containing a signal peptide without a transmembrane region and lacking orthology with free-living nematodes. The majority of these genes (16) were identified to have enzymatic protein motifs by InterPro ID. These enzymes include peptidases (7), phosphatases (2) and a kinase. The non-catalytic peptide is identified as having a transmembrane transport motif. While individually, the peptidases listed in Table 2 could be present for a number of different biological processes, their inclusion as secreted peptides orthologous to A. ceylanicum and non-orthologous with C. elegans suggests they are conserved parasitism genes. Furthermore, it is possible these genes are involved in the mechanisms of parasitism either as virulence factors or modulators of the host immune system.
Conclusions
Taken together, our RNA-Seq analysis reveals transcriptional changes in the regulation of a large number of genes, most of which have not been shown previously to play a role in parasitic responses. A significant proportion of these genes are conserved amongst closely related parasitic nematodes, suggesting the identification of a group of parasitism factors within nematodes. These genes provide ideal candidates for functional characterization using recently developed tools in H. bacteriophora to dissect the contribution of these genes in infection. While our selection criteria potentially misses some genes of interest, future studies using these candidates, in addition to further data-mining of our sequencing data, will not only provide functional insight into the process of nematode parasitism but may also shed light on the evolution of parasitism within nematodes as our data also reveals the existence of conserved genes amongst PN that are upregulated during early infection.
Methods
Culturing of Heterorhabditis bacteriophora TT01
Heterorhabditis bacteriophora strain TT01 was maintained by infecting Galleria mellonella larvae. Briefly, 20 G. mellonella, approximately 5th to 6th instar, were placed onto 8.5 cm filter papers held in 9 cm petri dishes. Approximately 100 IJs per larva were added to the filter paper (~1 mL total volume) and placed in a resealable bag in the dark at room temperature for incubation. After 10 days, infected insects turned brick red in color and were transferred to a White trap [36] containing distilled water with 0.01% Tween 20. Thirteen to fifteen days post-infection the IJs began to emerge from the carcasses and entered the liquid. After 7 days on the trap, IJs were collected and transferred to sterile culture flasks until use. IJs were used within 3 weeks of collection.
Hemolymph plasma extraction from Manduca sexta
Five 5th instar Manduca sexta larvae were placed on ice for 15 min. The posterior end of the insect was sterilized with an alcohol wipe and using aseptic technique, an incision was made at the distal end of the insect’s horn. The insect was gently exsanguinated by squeezing, and the hemolymph was collected in a sterile 2 mL microfuge tube on ice. To inhibit melanization, a solution of 20 mM phenylthiocarbamide in phosphate buffered saline was immediately added to the hemolymph at a final concentration of 0.33 mM. In order to further inhibit melanization of the hemolymph during incubation, the hemolymph was centrifuged at 4 °C for 5 min at 4000 × g to precipitate the hemocytes. The resulting hemolymph plasma was diluted 1:1 with Ringer’s solution (100 mM NaCl, 1.8 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 6.9) and filtered through a 0.45 μm syringe filter. Hemolymph plasma samples were stored at −80 °C until needed.
Infective juvenile incubation in hemolymph plasma solution
Approximately 50,000 H. bacteriophora TT01 infective juveniles were surface-sterilized with 10 mL of 3% commercial bleach in Ringer’s solution (hypochlorite final concentration of 0.26%) for 5 min. The nematodes were centrifuged at 500 × g for 2 min at room temperature. The resulting pellet was washed three times with 10 mL sterile Ringer’s solution and centrifuged. The nematode pellet was either suspended in 2 mL of hemolymph plasma solution and transferred to a 12.5 cm2 tissue flask or reserved as a 0 h control. Tissue flasks were incubated for 9 h in the dark at 27 °C with shaking at 300 RPM. The flask contents were transferred to a sterile 15 mL conical tube and centrifuged at 1100 × g at room temperature for 2 min. The pelleted nematodes were washed a total of three times with 10 mL sterile Ringer’s solution with centrifugation at 1100 × g between washes. The pellet was suspended in 500 μL ice-cold Trizol reagent (Thermo Fisher) and transferred to a 1.5 mL microfuge tube containing 1.4 g of 0.5 mm zirconium oxide beads. Tubes were placed in a Bullet Blender Blue (NextAdvance, Averill Park, NY) and lysed (3 cycles of 5 min at speed 10, with a 2 min rest on ice between each cycle). The resulting homogenate was transferred to a clean microfuge tube and an additional 500 μL of Trizol was added. RNA purification was carried out using the Trizol RNA Plus Purification Kit according to the manufacturer’s protocol and the PureLink DNase protocol was used to remove contaminating DNA. RNA from 0 h IJs was isolated as above, with the exception that RNA extraction began immediately after washing the surface sterilized IJs. RNA samples were analyzed for quantity and quality using an Agilent 2100 Bioanalyzer with the RNA 6000 Nano Kit. Samples with a RIN greater than 9.0 were sent to the Institute for Genome Sciences (University of Maryland School of Medicine) for RNA-Seq using the Illumina HiSeq4000 platform with 150 bp, paired-end sequencing. RNA-seq libraries were prepared using the TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA). Double-stranded cDNA was ligated to seven indexed nucleotide adapters and purified between enzymatic reactions. Library size selection was performed using SPRIselect beads (Beckman Coulter Genomics, Danvers, MA).
RNA-Seq analysis using subread and edgeR
Illumina adapter sequences, leading and trailing bases and low quality base reads (Phred-64 score < 15) were removed from RNA-Seq data using Trimmomatic (version 0.33) [37]. After trimming, only reads greater than or equal to 25 bases were retained. Reads were then mapped to the H. bacteriophora genome (Bioproject PRJNA13977) [5] using subread-aligner (version 1.5.0-p1) and the H. bacteriophora genome [38]. The aligned reads were counted using featureCounts and differential expression analysis was performed using the edgeR package’s quasi-likelihood F test [39, 40]. Genes were considered differentially expressed if they had a p-value < 0.05 and a fold change ≥ 2 or a fold change ≤ −2.
qRT-PCR validation of RNA-seq
Selected DEGs were validated by qRT-PCR using the Brilliant II SYBR Green QRT-PCR 1-step Master Mix (Agilent Technologies) and CFX96 Real-Time System (Bio-Rad Laboratories) with Hba-rpl-32 as an expression control. Primer efficiencies for primers targeting the selected DEGs were determined using serial dilutions (0.001 to 100 ng total RNA) of RNA extracted from untreated IJs, while the rpl-32 primers used were previously published [8]. PCR reactions were carried out using the manufacturer’s suggested two-step protocol with an annealing temperature of 55 °C and a dissociation curve performed at the end of the run. In addition to validating expression in samples used for RNA-seq, changes in expression were also verified in an additional set of samples prepared by incubating IJs in hemolymph plasma, as described above. Differential expression was measured using the ΔΔCT method, using matched, untreated IJs as relative controls.
Annotation and analysis of differentially expressed genes
DEGs were annotated and further analyzed using a variety of bioinformatic tools and databases. Peptide sequences, C. elegans orthologues, GO terms, InterProScan IDs, transmembrane regions, coiled coils and classical secretory sequences were gathered from the WormBase ParaSite Biomart database for H. bacteriophora [41]. Non-classical secretory peptides were found using SecretomeP v1.0f [42]. In order to better understand global gene ontology classifications, terms were condensed into higher functional categories using WEGO [43]. To determine if any GO terms were significantly over-represented, an enrichment analysis was performed with the topGO R package using Fisher’s exact test and the default hybrid classic/elim algorithm [21]. Annotation of KEGG pathways was performed using the peptide sequences of the DEGs and eggNOG verion 4.5 [44]. KEGG functional annotations were also gathered using the same peptide sequences and the KEGG Automatic Annotation Server [45]. Orthology between DEGs and other nematodes was examined using blastp and the genomes available for Clade I, Clade III, Clade IV and Clade V (excluding H. bacteriophora) nematodes. Blast results were considered hits if the aligned sequence had percent identity greater than 60% and an e-value less than 0.00005. Overlaps between alignments from different search targets (e.g. Clades I, III, IV and V) were found and graphed using Venny 2.1 [46].
Acknowledgements
The authors thank Dr. David Clarke of University College Cork, Ireland for providing the TT01 strain of H. bacteriophora, Caitlyn Leasure, Olivia Granger and Marcie Gsteiger-Cox for technical assistance, and the Institute of Genome Sciences (University of Maryland School of Medicine) for providing RNA sequencing services.
Funding
The project was supported by grant 5R21AI109517 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
All the data generated from this research are freely available to download at the following webpage: http://ohalloranlab.net/RNA_seq.html. To further facilitate data exploration using our research, we developed a frontend interface to our data. Users can query our database by accession number, keyword, protein sequence, Interpro ID, Panther ID, Prosite ID, Pfam ID or Gene3 ID. The user can also select or deselect specific tables within the database to query, order the data or limit the number of matches returned. The backend of our stack is served using a LAMP (Linux, Apache, MySQL, PHP/Perl) bundle; the database from our research is organized into a series of tables within a MySQL database and custom Perl scripts convert frontend requests into MySQL queries.
Author’s contributions
JV participated in the study design, prepared the IJs and isolated the RNA, analyzed the data, maintained the parasites and drafted the manuscript. RR participated in the study design. EK contributed to gene validation. MK assisted with parasite husbandry. JMH, IE and DO conceived of, designed and coordinated the study, and edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Abbreviations
- DEG
Differentially expressed gene
- EPN
Entomopathogenic nematode
- GO
Gene Ontogeny
- IJ
Infective juvenile
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PN
Parasitic nematode
- qRT-PCR
Quantitative reverse transcriptase-polymerase chain reaction
Additional file
Summary of Illumina sequencing reads. Figure S1 RNA-seq Analysis Pipeline. Pipeline used to collect, trim and analyze RNA-seq data. RNA sequencing reactions were performed by the Institute of Genome Sciences (University of Maryland School of Medicine) using high quality total RNA obtained from IJs incubated in hemolymph plasma (9 h) or Ringer’s solution (0 h). The resulting reads were trimmed, screened for quality and mapped to the H. bacteriophora reference genome. Expression analysis for DEGs was performed using the edgeR package (PDF 634 kb)
Contributor Information
Ioannis Eleftherianos, Email: ioannise@gwu.edu.
Damien O’Halloran, Email: damienoh@gwu.edu.
John M. Hawdon, Email: jhawdon@gwu.edu
References
- 1.Hotez PJ, Fenwick A, Savioli L, Molyneux DH. Rescuing the bottom billion through control of neglected tropical diseases. Lancet. 2009;373(9674):1570–1575. doi: 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- 2.John M. Hawdon: Controlling Soil-Transmitted Helminths: Time to Think Inside the Box? J Parasitol. 2014;100(2):166–188. doi: 10.1645/13-412.1. [DOI] [PubMed] [Google Scholar]
- 3.Erb KJ. Can helminths or helminth-derived products be used in humans to prevent or treat allergic diseases? Trends Immunol. 2009;30(2):75–82. doi: 10.1016/j.it.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Castillo JC, Reynolds SE, Eleftherianos I. Insect immune responses to nematode parasites. Trends Parasitol. 2011;27(12):537–547. doi: 10.1016/j.pt.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Bai X, Adams BJ, Ciche TA, Clifton S, Gaugler R, Kim K, Spieth J, Sternberg PW, Wilson RK, Grewal PS. A lover and a fighter: the genome sequence of an entomopathogenic nematode Heterorhabditis bacteriophora. PLoS One. 2013;8(7):e69618. doi: 10.1371/journal.pone.0069618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciche T. The biology and genome of Heterorhabditis bacteriophora (February 20, 2007), WormBook, ed. The C. elegans Research Community, WormBook. doi:10.1895/wormbook.1.135.1. [DOI] [PMC free article] [PubMed]
- 7.Hallem EA, Rengarajan M, Ciche T, Sternberg PW. Nematodes, Bacteria, and Flies: A Tripartite Model for Nematode Parasitism. Curr Biol. 2007;17(10):898–904. doi: 10.1016/j.cub.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 8.Ratnappan R, Vadnal J, Keaney M, Eleftherianos I, O’Halloran D, Hawdon JM. RNAi-mediated gene knockdown by microinjection in the model entomopathogenic nematode Heterorhabditis bacteriophora. Parasit Vectors. 2016;9:160. doi: 10.1186/s13071-016-1442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciche TA, Darby C, Ehlers R, Forst S, Goodrich-Blair H. Dangerous liaisons: The symbiosis of entomopathogenic nematodes and bacteria. Biol Control. 2006;38(1):22–46. doi: 10.1016/j.biocontrol.2005.11.016. [DOI] [Google Scholar]
- 10.Hotez P, Hawdon J, Schad GA. Hookworm larval infectivity, arrest and amphiparatenesis: the Caenorhabditis elegans daf-c paradigm. Parasitol Today. 1993;9(1):23–26. doi: 10.1016/0169-4758(93)90159-D. [DOI] [PubMed] [Google Scholar]
- 11.Crook M. The dauer hypothesis and the evolution of parasitism: 20 years on and still going strong. Int J Parasitol. 2014;44(1):1–8. doi: 10.1016/j.ijpara.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashmi S, Abu Hatab MA, Gaugler RR. GFP : green fluorescent protein a versatile gene marker for entomopathogenic nematodes. Fundam Appl Nematol. 1997;20(4):323–327. [Google Scholar]
- 13.Ciche TA, Sternberg PW. Postembryonic RNAi in Heterorhabditis bacteriophora: a nematode insect parasite and host for insect pathogenic symbionts. BMC Dev Biol. 2007;7(1):101. doi: 10.1186/1471-213X-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashmi S, Hashmi G, Gaugler R. Genetic Transformation of an Entomopathogenic Nematode by Microinjection. J Invertebr Pathol. 1995;66(3):293–296. doi: 10.1006/jipa.1995.1103. [DOI] [PubMed] [Google Scholar]
- 15.Castillo JC, Shokal U, Eleftherianos I. A novel method for infecting Drosophila adult flies with insect pathogenic nematodes. Virulence. 2012;3(3):339–347. doi: 10.4161/viru.20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eleftherianos I, Ffrench-Constant RH, Clarke DJ, Dowling AJ, Reynolds SE. Dissecting the immune response to the entomopathogen Photorhabdus. Trends Microbiol. 2010;18(12):552–560. doi: 10.1016/j.tim.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Eleftherianos I, Joyce S, Ffrench-Constant RH, Clarke DJ, Reynolds SE. Probing the tri-trophic interaction between insects, nematodes and Photorhabdus. Parasitology. 2010;137(11):1695–1706. doi: 10.1017/S0031182010000508. [DOI] [PubMed] [Google Scholar]
- 18.Bai X, Adams BJ, Ciche TA, Clifton S, Gaugler R, Hogenhout SA, Spieth J, Sternberg PW, Wilson RK, Grewal PS. Transcriptomic analysis of the entomopathogenic nematode Heterorhabditis bacteriophora TTO1. BMC Genomics. 2009;10(1):205. doi: 10.1186/1471-2164-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moshayov A, Koltai H, Glazer I. Molecular characterisation of the recovery process in the entomopathogenic nematode Heterorhabditis bacteriophora. Int J Parasitol. 2013;43(10):843–852. doi: 10.1016/j.ijpara.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Howe KL, Bolt BJ, Cain S, Chan J, Chen WJ, Davis P, Done J, Down T, Gao S, Grove C, Harris TW, Kishore R, Lee R, Lomax J, Li Y, Muller H, Nakamura C, Nuin P, Paulini M, Raciti D, Schindelman G, Stanley E, Tuli MA, Van Auken K, Wang D, Wang X, Williams G, Wright A, Yook K, Berriman M, Kersey P, Schedl T, Stein L, Sternberg PW. WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 2016;44(D1):D774. doi: 10.1093/nar/gkv1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexa A, Rahnenfuhrer J. topGO: Enrichment Analysis for Gene Ontology. 2016. [Google Scholar]
- 22.Hao Y, Montiel R, Nascimento G, Toubarro D, Simoes N. Identification, characterization of functional candidate genes for host–parasite interactions in entomopathogenetic nematode Steinernema carpocapsae by suppressive subtractive hybridization. Parasitol Res. 2008;103(3):671–683. doi: 10.1007/s00436-008-1030-4. [DOI] [PubMed] [Google Scholar]
- 23.Ciche TA, Ensign JC. For the Insect Pathogen Photorhabdus luminescens, Which End of a Nematode Is Out? Appl Environ Microbiol. 2003;69(4):1890–1897. doi: 10.1128/AEM.69.4.1890-1897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao Y, Montiel R, Abubucker S, Mitreva M, Simões N. Transcripts analysis of the entomopathogenic nematode Steinernema carpocapsae induced in vitro with insect haemolymph. Mol Biochem Parasitol. 2010;169(2):79–86. doi: 10.1016/j.molbiopara.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kedzierski L, Montgomery J, Bullen D, Curtis J, Gardiner E, Jimenez-Ruiz A, Handman E. A leucine-rich repeat motif of Leishmania parasite surface antigen 2 binds to macrophages through the complement receptor 3. J Immunol. 2004;172(8):4902–4906. doi: 10.4049/jimmunol.172.8.4902. [DOI] [PubMed] [Google Scholar]
- 26.Holthuis JC, van Riel MC, Martens GJ. Translocon-associated protein TRAP delta and a novel TRAP-like protein are coordinately expressed with pro-opiomelanocortin in Xenopus intermediate pituitary. Biochem J. 1995;312(Pt 1):205–213. doi: 10.1042/bj3120205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atkinson S, Williams P. Yersinia virulence factors - a sophisticated arsenal for combating host defences. F1000Res. 2016;5:10. doi: 10.12688/f1000research.8466.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coombes BK, Finlay BB. Insertion of the bacterial type III translocon: not your average needle stick. Trends Microbiol. 2005;13(3):92–95. doi: 10.1016/j.tim.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J. 1993;290(Pt 1):205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sojka D, Hartmann D, Bartosova-Sojkova P, Dvorak J. Parasite Cathepsin D-Like Peptidases and Their Relevance as Therapeutic Targets. Trends Parasitol. 2016;32(9):708–723. doi: 10.1016/j.pt.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3(9):853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 32.Hamid R, Khan MA, Ahmad M, Ahmad MM, Abdin MZ, Musarrat J, Javed S. Chitinases: An update. J Pharm Bioallied Sci. 2013;5(1):21–29. doi: 10.4103/0975-7406.106559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Punja ZK, Zhang YY. Plant chitinases and their roles in resistance to fungal diseases. J Nematol. 1993;25(4):526–540. [PMC free article] [PubMed] [Google Scholar]
- 34.Bordon Y. Parasite immunity: chitinase-like proteins smoke out worms. Nat Rev Immunol. 2014;14(12):775. doi: 10.1038/nri3770. [DOI] [PubMed] [Google Scholar]
- 35.Tachu B, Pillai S, Lucius R, Pogonka T. Essential Role of Chitinase in the Development of the Filarial Nematode Acanthocheilonema viteae. Infect Immun. 2008;76(1):221–228. doi: 10.1128/IAI.00701-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindegren JE, Valero KA, Mackey BE. Simple In Vivo Production and Storage Methods for Steinernema carpocapsae Infective Juveniles. J Nematol. 1993;25(2):193–197. [PMC free article] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41(10):e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 40.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howe KL, Bolt BJ, Shafie M, Kersey P, Berriman M. WormBase ParaSite - a comprehensive resource for helminth genomics. Mol Biochem Parasitol. 2016;(16)30160–8. [DOI] [PMC free article] [PubMed]
- 42.Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17(4):349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 43.Ye J, Lin F, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(2):W293. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, von Mering C, Bork P. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44(D1):D286. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliveros JC. Venny. An interactive tool for comparing lists with Venn’s diagrams. 2007-2015. http://bioinfogp.cnb.csic.es/tools/venny/index.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data generated from this research are freely available to download at the following webpage: http://ohalloranlab.net/RNA_seq.html. To further facilitate data exploration using our research, we developed a frontend interface to our data. Users can query our database by accession number, keyword, protein sequence, Interpro ID, Panther ID, Prosite ID, Pfam ID or Gene3 ID. The user can also select or deselect specific tables within the database to query, order the data or limit the number of matches returned. The backend of our stack is served using a LAMP (Linux, Apache, MySQL, PHP/Perl) bundle; the database from our research is organized into a series of tables within a MySQL database and custom Perl scripts convert frontend requests into MySQL queries.