

Abstract Abstract
Pulmonary blood vessel structure and tone are maintained by a complex interplay between endogenous vasoactive factors and oxygen-sensing intermediaries. Under physiological conditions, these signaling networks function as an adaptive interface between the pulmonary circulation and environmental or acquired perturbations to preserve oxygenation and maintain systemic delivery of oxygen-rich hemoglobin. Chronic exposure to hypoxia, however, triggers a range of pathogenetic mechanisms that include hypoxia-inducible factor 1α (HIF-1α)–dependent upregulation of the vasoconstrictor peptide endothelin 1 in pulmonary endothelial cells. In pulmonary arterial smooth muscle cells, chronic hypoxia induces HIF-1α-mediated upregulation of canonical transient receptor potential proteins, as well as increased Rho kinase-Ca2+ signaling and pulmonary arteriole synthesis of the profibrotic hormone aldosterone. Collectively, these mechanisms contribute to a contractile or hypertrophic pulmonary vascular phenotype. Genetically inherited disorders in hemoglobin structure are also an important etiology of abnormal pulmonary vasoreactivity. In sickle cell anemia, for example, consumption of the vasodilator and antimitogenic molecule nitric oxide by cell-free hemoglobin is an important mechanism underpinning pulmonary hypertension. Contemporary genomic and transcriptomic analytic methods have also allowed for the discovery of novel risk factors relevant to sickle cell disease, including GALNT13 gene variants. In this report, we review cutting-edge observations characterizing these and other pathobiological mechanisms that contribute to pulmonary vascular and right ventricular vulnerability.
Keywords: hypoxia, pulmonary hypertension, genetics
In the autumn of 2015 at the Lost Valley Ranch in Sedalia, Colorado, the 34th Grover Conference, on pulmonary circulation in the “omics” era, convened for a 4-day symposium to discuss contemporary scientific concepts in the genetics, genomics, proteomics, and epigenetics of pulmonary vascular diseases. This biannual meeting, sponsored by the American Thoracic Society and co-led this year by Drs. C. Gregory Elliot, Wendy K. Chung, D. Hunter Best, and Eric D. Austin, commemorates the perpetual and transformative scientific contributions of Dr. Robert Grover1 to the fields of high-altitude medicine and pulmonary vascular disease. This review is part of an ongoing Pulmonary Circulation thematic series that aims to provide a synopsis of key concepts presented at this year’s Grover Conference.
Here, we summarize the discussions that constituted a section of the conference emphasizing novel genetic and molecular mechanisms underpinning “pulmonary vascular and ventricular dysfunction in the susceptible patient.” To accomplish this, three concepts are reviewed in detail: the functional relevance of chronic hypoxia (CH), discovered through genomic analyses; novel molecular mechanisms and phenotypic signatures of pulmonary vascular disease in sickle cell disease; and hormonal regulation of vascular fibrosis. Although distinct in many regards, findings presented in these works intersect by virtue of their common intent to clarify the relationship between classical risk factors for vascular injury and the development of pulmonary vascular disease. In particular, these efforts capitalize on unbiased scientific methodologies, big data sets, and/or cell-specific properties unique to the pulmonary circulation in order to elucidate the pathobiological mechanisms that link hypoxia, hemoglobinopathies, and vasoactive hormone exposure to pulmonary hypertension (PH).
Hypoxic PH
It has long been recognized that protracted exposure to alveolar hypoxia has a negative impact on the pulmonary vasculature. In the mid-twentieth century, the migration of Han Chinese from mainland China into the highlands of Tibet uncovered the deleterious effects of residence at high altitude. Han infants born at low altitude and subsequently moved to altitudes of 3,000–4,000 m suffered symptoms consistent with congestive heart failure, including dyspnea, cyanosis, enlarged right heart, and pulmonary vascular remodeling (reviewed by Anand and Wu2). Similar symptoms were described with military troop movements into the Himalayas3,4 and later in studies performed in lowland adults who were stationed at high altitude.5,6 These results were consistent with studies being performed on the other side of the world in native high-altitude populations in the Peruvian Andes, which revealed elevations in pulmonary arterial pressure (PPA) and pulmonary vascular resistance (PVR), right heart enlargement, and pulmonary vascular remodeling, including medial hypertrophy, pulmonary arterial smooth muscle cell (PASMC) hyperplasia, and muscle extension into distal arterioles.7-13
Starting in the 1960s, experiments conducted by Robert Grover and his colleagues in the high mountains of Colorado (>8,000 ft [2,428.4 m]) provided insights into the cardiac problems that develop as a result of PH.14 Studying cattle living at altitude, Will et al.14 described animals with edema surrounding the brisket, or lower chest, and enlarged, dilated, and “flabby” hearts. In subsequent studies, extensive vascular remodeling was also observed in this model, characterized by collagen deposition, thickened adventitia, increased medial thickness, extension of muscle down to precapillary arterioles, and intimal narrowing in small pulmonary arteries (PAs).15-17
It is now clear that for those not adapted to living at altitude (usually >2,500 m), ascent to and residence at high altitude results in the development of PH,8,18,19 defined as a resting PPA of ≥25 mmHg, pulmonary vascular remodeling,7,12 right ventricular hypertrophy,20 and, in some cases, right ventricular failure.21,22 What is also clear is that certain populations have genetically adapted to life at altitude, and genomic studies in these groups, coupled with laboratory studies in animal models, have provided insight into some of the mechanisms involved in the pathogenesis of hypoxic PH (HPH).
Mechanisms of HPH: lessons from animal models
The exact mechanisms underlying the development of HPH are still being unraveled. To aid in this endeavor, a number of animal models have been developed, one of the most reliable being the rodent CH model. Rats or mice placed in normobaric or hypobaric hypoxia (fraction of inspired oxygen [FIo2] ∼ 10%) develop significant increases in PPA, right ventricular mass, and PVR resulting from both active contraction and remodeling (reviewed by Stenmark et al.15). In rats, substantial vascular remodeling has been described,23-28 findings consistent with observations reported in humans residing at high attitude.12,29 Similar results have been reported in mice, pigs, sheep, and cows, although the exact amount of remodeling and levels of PPA vary with species (reviewed by Rhodes16).
Given that initial experiments detailing the hypoxia-induced structural changes in the pulmonary circulation revealed thickening of the medial layer due to increased smooth muscle and fibroblast proliferation and that acute exposure to O2, calcium channel blockers, or other vasodilators had little effect on PPA,30-33 it was widely regarded that contraction played a minimal role in sustained HPH and that “fixed” remodeling was the underlying cause of the increased PVR. This assertion was challenged, however, by striking studies demonstrating that inhibitors of Rho kinase (ROCK), an important mediator of smooth muscle cell contractility, acutely normalized PPA in chronically hypoxic rats when administered systemically34,35 or via inhalation,36 indicating that active contraction may, in fact, be the most important factor determining PPA. This hypothesis was supported by morphological studies demonstrating that with CH, the increase in smooth muscle cells extended outward and did not, in fact, cause luminal narrowing.37 Thus, while hypoxic exposure might increase muscularity of the small PAs, this remodeling likely contributes to the increase in PVR primarily by facilitating vasoconstriction.
The mechanisms underlying smooth muscle contraction and remodeling include factors that are both intrinsic and extrinsic to smooth muscle. With respect to contraction, under normal conditions, excess production of vasodilators, such as nitric oxide (NO) and prostacyclin, results in low pulmonary vasomotor tone. During hypoxia, reduced synthesis of vasodilators, coupled with increased production of vasoconstrictors, such as endothelin 1 (ET-1) and serotonin, shifts the balance to enhanced contraction (reviewed by Stenmark et al.38). In addition, increased ROCK activation enhances the Ca2+ sensitivity of the contractile apparatus,39,40 rendering the PAs more sensitive to vasoconstrictor stimuli.
Abnormal mechanisms intrinsic to PASMCs that contribute to the increased contractile nature of PASMCs include alterations in ion channels and transporters, leading to changes in membrane potential, elevated intracellular calcium concentration ([Ca2+]i), and increased responsiveness to vasoconstrictors. PASMCs derived from chronically hypoxic rats were reported to be depolarized,41 a finding later confirmed in other species (reviewed by Shimoda and Polak42). Reduced K+ channel expression and activity were subsequently identified as factors contributing to depolarization in HPH.43-46 Depolarization was initially hypothesized to drive activation of voltage-gated calcium channels (VGCCs) and calcium influx leading to elevated basal [Ca2+]i; however, later work suggested that VGCC inhibitors have little effect on basal [Ca2+]i in PASMCs from hypoxic animals,47-49 although increased expression and Ca2+ influx through VGCCs in response to stimulation has been noted in hypoxic PASMCs.50-52 Rather, elevated basal [Ca2+]i in PASMCs derived from chronically hypoxic animals appears to occur primarily via upregulation of canonical transient receptor potential (TRPC) proteins,48,49 which form nonselective cation channels (NSCCs) that are permeable to Ca2+.42 Decreasing the activity of NSCCs, either pharmacologically or by RNA silencing, reduced [Ca2+]i48,49 in PASMCs from animals with HPH.
Direct effects of hypoxia on PASMCs are also likely to play a role in vascular remodeling. For example, cells exposed to hypoxia ex vivo or derived from chronically hypoxic animals exhibit increases in both migration and proliferation,53-56 and both K+ channel activity and [Ca2+]i are likely to play a role. Agents that activate and/or increase expression of K+ channels, including the steroid hormone dehydroepiandrosterone and the metabolic modulator dichloroacetate, reduced remodeling in chronically hypoxic rats,46,57,58 likely because increased intracellular K+ confers resistance to apoptosis.59 Increased [Ca2+]i is required for PASMC growth60,61 and migration55 and has been documented in cells from hypoxic animals.42 VGCCs contribute to stimulated proliferation62 and may participate in the remodeling process in HPH, particularly in the presence of excessive growth factors. Increased [Ca2+]i activates a number of downstream signal transduction pathways and transcription factors that could be involved in PASMC proliferation (reviewed by Kuhr et al.63). In particular, Ca2+ activates nuclear factor of activated T cells (NFAT), which reduces K+ channel expression,64 providing a link between alterations in [Ca2+]i, dysregulated K+ channel expression/activity, and PASMC growth. Moreover, hypoxia causes Ca2+-dependent increases in the expression of aquaporin 1 (AQP1), a water channel, which is required for proliferation and migration of PASMCs.55 In endothelial and tumor cells, increased AQP1 levels may result in localized control of water flux across the cell membrane, possibly allowing for directed cell movement.65 However, in PASMCs, the actions of AQP1 appear to be independent of water transport56 and instead require the C-terminal tail portion of the protein, which may act to regulate the levels of β-catenin,66 a dual-function protein that controls both migration and proliferation.
The Na+/H+ exchanger (NHE) is another membrane transporter that contributes to hypoxia-induced remodeling. NHE is a major contributor to maintenance of PASMC pH homeostasis,67,68 and NHE activation mediates growth factor–induced proliferation.69 Expression of NHE isoform 1 (NHE1) is upregulated in PASMCs from chronically hypoxic animals, leading to increased NHE activity and alkaline pH.70,71 Pharmacological inhibition72 or genetic deletion73,74 of NHE1 decreased hypoxia-induced vascular remodeling and PASMC proliferation and migration.74 In PASMCs, loss of NHE1 increased p27, a cyclin-dependent kinase inhibitor, and decreased E2F1, a nuclear transcription factor that controls proliferation,74 suggesting that NHE1 activation represses a growth inhibitor pathway while stimulating proliferation. NHE1 may also facilitate cell growth and migration via regulation of cytoskeletal arrangement.75
ROCK may represent a node of convergence with channels/transporters involved in remodeling, as ROCK activation opens Ca2+ channels51,76 and stimulates the activity of77 and is activated by73 NHE1. Chronic treatment with ROCK inhibitors reduced neovascularization and remodeling in HPH models;34,78,79 ROCK activation is necessary for migration and proliferation in a variety of vascular cell types, including PASMCs;80-83 and the upstream activators of ROCK, RhoA and RhoB, mediate cytoskeletal rearrangement in PASMCs,84 a process necessary for cell movement, a key component of both migratory and proliferative processes.
All of the pathways outlined above are linked, in that they are regulated by the oxygen-sensitive transcription factor hypoxia-inducible factor (HIF). Hypoxia-inducible factor 1 (HIF-1) was originally identified as a heterodimeric transcription factor consisting of a β subunit (HIF-1β) that is constitutively expressed and an α subunit (HIF-1α) that is typically not detectable under normoxic conditions.85 Subsequent studies identified HIF-2α, which is structurally similar to HIF-1α and also binds HIF-1β, resulting in the HIF-2 transcription factor. The mystery of how HIFs are regulated by changes in oxygen was solved when it was reported that HIF-1α is hydroxylated on two proline residues (via prolyl hydroxyalse domain proteins), using molecular oxygen as a substrate, allowing binding to the von Hippel–Lindau protein, ubiquitination, and targeting for proteasomal degradation (reviewed by Semenza86 and Prabhakar and Semenza87). Thus, falling oxygen levels limit hydroxylation, allowing HIF-1α/HIF-2α accumulation. Unlike HIF-1α, which is found in all cells, HIF-2α exhibits a more restricted expression pattern.
Animal studies revealed a role for HIFs in the development of HPH. Generation of mice homozygous for a null Hif1α allele resulted in embryonic lethality,88 whereas mice heterozygous for the null allele (Hif1a+/−) were viable, with phenotypically normal appearance under normoxic conditions. When exposed to CH, Hif1a+/− mice exhibited impaired development of HPH and reduced vascular remodeling.89 Similar results were observed in mice with partial deficiency in Hif2a.90 When studied ex vivo, PASMCs from Hif1a+/− mice also exhibited reduced hypoxia-induced proliferation.91 Surprisingly, mice with a selective Hif1a homozygous deletion targeted to vascular smooth muscle cells92 or Hif2a homozygous deletion targeted to endothelial cells93 resulted in enhanced HPH, although inducible smooth muscle–specific deletion of Hif1a exhibited protection from HPH similar to global Hif1a heterozygosity.94The differences observed between targeted homozygous and inducible targeted or global heterozygous genetic modifications clearly require further investigation.
Recent studies using pharmacologic inhibitors of HIF activity demonstrated that targeting HIF reduced CH-induced vascular remodeling in rodents.95 The exact mechanisms by which HIF mediates contraction and/or remodeling during CH are still being investigated (Fig. 1) but involve both Ca2+ and pH homeostasis.71,91,96 HIF also regulates K+ channel expression as well as other factors involved in the pathogenesis of HPH, including ET-1 and vascular endothelial derived growth factor (VEGF).87 While HIF can induce ET-1 production, ET-1 also upregulates HIF-1,97,98 creating feed-forward enhancement of HIF-1 expression. PASMCs treated with exogenous ET-1 accumulated HIF-1α protein and exhibited downregulation of prolyl hydroxylase domain 2 (PHD2) expression,97 Moreover, blockade of endothelin type A (ETA) receptors prevented upregulation of HIF-1α in PASMCs exposed to moderate hypoxia. In contrast, ET-1 failed to augment HIF-1α in aortic smooth muscle cells, suggesting a feature unique to PASMCs.97 Since the primary source of ET-1 in vivo appears to be endothelial cells, these data, taken as a whole, suggest a possible model whereby enhanced HIF-2α expression in endothelial cells during CH produces ET-1, which then augments HIF-1α in PASMCs, initiating a feed-forward mechanism for upregulation of HIF-1 and, consequently, HIF target genes to promote the development of HPH.
Figure 1.

Schematic showing some of the pathways by which hypoxia-inducible factor (HIF) mediates pulmonary arterial smooth muscle changes during chronic hypoxia. ET-1: endothelin 1; TRPCs: transient potential receptors (canonical); ROCK: Rho kinase; NHE1: Na+/H+ exchanger 1; AQP1: aquaporin 1.
Mechanisms of HPH: lessons from high-altitude genomics
While experiments conducted in laboratories using animal models revealed significant effects of hypoxia on the pulmonary circulation and began to unmask some of the mechanisms involved, the molecular-biology revolution and development of techniques for genetic screening allowed for “experiments in nature” to be conducted in populations indigenous to, and therefore adapted to residence at, high altitude. Currently, it is estimated that 140 million people reside in high-altitude regions throughout the world. Three major native high-altitude populations have been studied in depth, and, not surprisingly, those indigenous populations who have adapted to survive in the harsh environments of high-altitude regions have done so differently. In Tibetans, who have been documented to live at high altitude for 30,000 years, resting PPA and PVR were normal or borderline in residents at high altitude.99 Native Tibetans do not exhibit polycythemia but do exhibit higher hypoxic ventilatory drive and higher exhaled NO levels and rarely develop chronic mountain sickness. Separate genomic studies detailed variants in Tibetan highlanders in the genes EPAS1 (encoding HIF-2α) and EGLN1 (one of the prolyl hydroxylases that regulate HIF1α/2α).100-104Furthermore, a genetic mutation resulting in HIF-2α overexpression was associated with development of PH.105
Andeans have lived at high altitude for approximately 11,000 years. Although PPA was normal in the Aymara and Quenchua Indians of the Peruvian Andes,99 at elevations of 4,540 there was a mild increase in PPA.8,106 Development of PH and polycythemia is fairly common in residents of the Andean plateau,18,107 unlike Tibetans. Variants in EPAS1 have not been identified in adapted Andeans, although recent studies have identified other genes that may be associated with adaptation, including VEGFB,108EDNRB (encoding the endothelin receptor subtype B),109 and the prolyl hydroxylase PHD3.110,111
The Amarah have been documented to reside on the Ethiopian plateau for at least 5,000 years, but evidence suggests that they have lived at 2,300–2,400 ft (∼700–730 m) for ∼70,000 years. This last finding may explain why this population appears the best adapted to living at high altitude, with a lack of polycythemia or increased PPA, hemoglobin with higher O2 affinity, and higher O2 saturations. Gene variants associated with adaptation in this population include ARNT (encoding HIF-1β) and EDNRB.112
Finally, in addition to studies conducted in humans residing at high altitude, recent work has also examined the genetics associated with development of HPH in cattle. Comparison between cattle that developed PH at altitude and those that did not revealed variants in EPAS1 that likely confer gain of function and were associated with HPH susceptibility.113 Taken together, the genomic studies in both humans and cattle at high altitude independently identified variants in the HIF signaling pathway that confer protection or susceptibility to high-altitude PH.
Thus, while many “-omics” studies result in discovery science that is then tested in animal models to establish relevance, the field of HPH is one case in which the opposite occurred, with studies in animal models initially paving the way in identifying a prime role for the HIF pathway that was subsequently confirmed by data from large genomic studies in high-altitude populations. While additional downstream and parallel pathways continue to be identified, the central role of HIFs in the development of HPH appears to have been firmly established.
PH in sickle cell disease and the hypoxic response
Sickle cell disease
Sickle cell disease (SCD) is an autosomal recessive disorder affecting millions of individuals worldwide, making it one of the most common monogenetic diseases. Sickle cell anemia, the most common and severe form of SCD, occurs in patients with a single amino acid substitution of glutamic acid for valine in the β-globin gene, resulting in an abnormal hemoglobin form called hemoglobin S (HbS) within the erythrocytes. Mutant HbS has reduced solubility when deoxygenated, compared to normal hemoglobin A (HbA), leading to polymerization and aggregation of sickle erythrocytes in the microvasculature. Structural abnormalities and cumulative damage to the cellular membrane of sickled erythrocytes results in hemolysis and the development of chronic hemolytic anemia, as well as vaso-occlusion and multiorgan pathology. Compound heterozygosity of HbS with other β-globin gene mutations results in additional types of SCD, such as hemoglobin SC or hemoglobin S-β-thalassemia.114 The pathobiology of SCD is multifactorial and results from the structural and functional changes of sickled erythrocytes. Deoxygenated HbS polymerizes and aggregates inside erythrocytes, forming rigid, sickled cells. The abundance of polymerized HbS is strongly influenced by both the level of deoxygenation and the concentration of intracellular HbS115 and determines the severity of SCD.116 Sickled erythrocytes navigating the microcirculation are continually exposed to mechanical and oxidant injury, damaging the cell membrane and leading to a shortened erythrocyte life span, increased hemolysis, and the development of chronic hemolytic anemia in patients. Importantly, intravascular hemolysis depletes the bioavailability of NO levels through the abundant release of both cell-free Hb, an NO scavenger, and arginase, which catabolizes arginine, a substrate necessary for NO synthesis.117 Reduction in NO disrupts vascular homeostasis, leading to endothelial dysfunction, proliferation of cells within the vascular wall, thrombosis, and inflammatory stress.118-121 In addition, pathologic sickled erythrocytes have increased adherence to the vascular endothelium, leading to microvasculature occlusion and subsequent multiorgan damage. Ischemia-reperfusion injury at sites of vessel occlusion can also promote inflammation and oxidative stress.122-125 Finally, the low pressure and oxygen tension within the pulmonary arterial bed can promote HbS polymerization and erythrocyte sickling.126
PH in SCD
In spite of significant improvements in the life expectancy of patients with SCD, estimates of the median age at death range from 42 to 53 years for men and from 48 to 58.5 years for women.127,128 Cardiopulmonary disorders are a major cause of death in patients with SCD.118,127,129-131 Among these complications, PH occurs in 6%–10% of adults with SCD, and the presence of PH is associated with significant increase in the risk of death in these patients.132-135 Although the etiology of PH in patients with SCD is multifactorial, including pulmonary arterial hypertension (PAH), pulmonary venous hypertension, and chronic thromboembolic disease, data suggest that mortality in adults with SCD and PH is proportional to the severity of precapillary PH.136
Transcriptome analysis, PH, and the hypoxic response in SCD
Despite SCD’s Mendelian inheritance, the clinical course of SCD patients is highly variable. There is significant phenotypic heterogeneity in the rate of development of acute and chronic complications, including PH, suggesting that other genetic and genomic modifiers could play a role in these complications. Therefore, we explored the usefulness of peripheral blood mononuclear cell (PBMC)–derived gene signatures as biomarkers for PH in SCD.137 Genome-wide gene and micro-RNA (miRNA) expression profiles were correlated against estimated right ventricular systolic pressure (RVSP), yielding 631 transcripts and 12 miRNAs. Biological-pathway analysis of these 631 genes revealed Wnt signaling, calcium signaling, vascular smooth muscle contraction, and cancer pathways as significantly represented in patients with elevated RVSP. These pathways have been associated with pulmonary vascular remodeling in other studies and suggest biological relevance to these findings. In order to identify a more succinct gene list that could be capable of identifying patients with an elevated RVSP, we conducted support vector machine analysis and identified a 10-gene signature including GALNT13 (encoding polypeptide N-acetylgalactosaminyltransferase 13, a glycosyltransferase enzyme responsible for the synthesis of O-glycan138) that distinguishes patients with and without increased RVSP with 100% accuracy. This finding was then validated in a separate cohort of patients who had SCD without (n = 10) or with (n = 10) PH, with an overall accuracy of 90%. Increased RVSP-related miRNAs revealed strong in-silico binding predictions of miR-301a to GALNT13, corroborated by microarray analyses demonstrating an inverse correlation between their expression. A genetic-association study comparing patients with an elevated (n = 49) and those with a normal (n = 63) RVSP revealed five significant single-nucleotide polymorphisms within GALNT13 associated with an elevated RVSP. These studies suggest the potential clinical usefulness of genomic signatures as biomarkers of PH in SCD (Fig. 2). Given that aberrant glycosylation patterns are a hallmark of the tumor phenotype influencing proliferation, invasion, angiogenesis, and metastasis potentially involved in pulmonary vascular remodeling, GALNT13 may be speculated as a potential candidate gene in SCD-associated PH.
Figure 2.
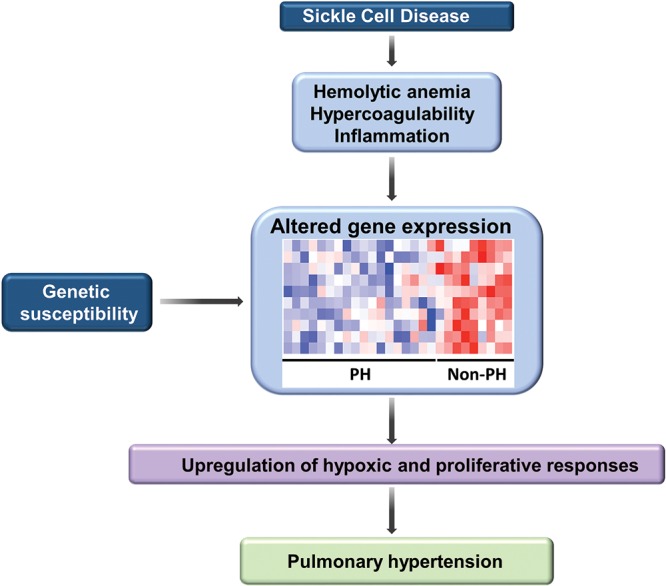
Schematic diagramming mechanisms involved in the development of pulmonary hypertension in sickle cell disease. Abnormalities characteristically observed in sickle cell disease, including hemolytic anemia, hypercoagulability, and inflammation, coupled with individual genetic susceptibility, result in altered expression of a wide panel of genes. Pathways associated with vascular remodeling, including Wnt signaling, calcium signaling, vascular smooth muscle contraction, and cancer pathways, were significantly represented in patients with elevated right systolic pressures, suggesting that these pathways are likely to play a contributory role in the development of pulmonary hypertension (PH).
As discussed above, CH is recognized to be an important cause of PH. In addition, normoxic activation of HIF-1α is involved in the pathobiology of PH and pulmonary vascular remodeling.139 SCD is characterized by steady-state high circulating erythropoietin concentrations,140 suggesting that this chronic hemolytic anemia is accompanied by chronic upregulation of the hypoxic response. In addition to the anemic state, increases in placental growth factor, a mediator that activates HIF-1α in normoxia, have been associated with elevated systolic PA pressures in SCD.141On the basis of these observations, we hypothesized that the upregulated hypoxic response in SCD might contribute to altered gene expression and the development of PH. To test this hypothesis, we compared clinical data and PBMC genomic profiles of subjects with SCD to those of subjects with Chuvash polycythemia, a monogenic hematologic disorder characterized by an upregulated hypoxic response and elevated systolic PA pressures.142,143 In Chuvash polycythemia, homozygosity for VHLR200W leads to posttranslational stabilization of the alpha subunits of HIF at normoxia via decreased binding of these subunits to the mutant VHL (von Hippel–Lindau) protein, leading to increased levels of HIF-1 and HIF-2 and altered transcription of several genes.142,144 We observed a strong correlation between gene expression profiles in hemoglobin SS subjects and VHLR200W homozygote polycythemic subjects, whereas a control analysis did not show correlations between differential gene expression in hemoglobin SS and Down syndrome patients.145 Taken together, these data suggest that a substantial proportion of PBMC gene expression variation in hemoglobin SS patients may be related to the hypoxic response. Specifically, at a 5% false-discovery rate, 1,040 genes exhibited a more than 1.15-fold change in both conditions; 297 were upregulated and 743 downregulated, including MAPK8, which encodes a mitogen-activated protein kinase important for apoptosis, T-cell differentiation, and inflammatory responses.146 Expression of MAPK8 was downregulated in SCD and Chuvash polycythemia, where the hypoxic response is upregulated. Association mapping with a focus on local regulatory polymorphisms in 61 patients with SCD identified expression quantitative trait loci (eQTLs) for 103 of the hypoxia-response genes. Among these findings, an eQTL of MAPK8 (rs10857560) was associated with precapillary PH in SCD (odds ratio: 13.8, P = 0.00036). The A allele of rs10857560 was associated with a further decrease in expression in SCD. Homozygosity for the A allele was present in all 14 of the patients with precapillary PH that we examined, suggesting that the dosage effect of the A allele on MAPK8 gene expression might contribute to the pathogenic mechanism of precapillary PH in SCD.145
Hypoxia, endothelial aldosterone synthesis, and pulmonary vascular fibrosis
The functional consequences of CH or cell-free Hb on circulatory performance differ between the pulmonary and systemic vascular circuits. This observation is in agreement with a wide body of literature indicating cell-specific properties that delineate pulmonary endothelial and vascular smooth muscle cells from corresponding cell types in resistance blood vessels. Along these lines, a unique relationship between hypoxia, the vasoactive hormone aldosterone, and remodeling proteins in pulmonary endothelial cells was explored recently.
Vascular fibrosis is a cornerstone histopathologic feature of remodeled pulmonary arterioles that is observed across the spectrum of pulmonary vascular disease, including hypoxic lung disease.147 Overactivation of hypoxia-dependent signaling in pulmonary vascular cells results in vascular and perivascular collagen deposition that decreases pulmonary circulatory compliance to promote PH and a predilection for right heart failure.148 However, effective treatments to abrogate vascular fibrosis in patients with PH due to chronic hypoxic lung disease are not currently available. Thus, further understanding of the basic mechanisms by which hypoxia regulates vascular remodeling, particularly fibrosis, is required to identify novel treatments that improve on elevated rates of morbidity and mortality in at-risk patients.149-151
Endothelial-mesenchymal transition (EndMT) is a principal mechanism implicated in the development of perivascular myofibroblast proliferation that evinces collagen deposition in PAs.152 Under hypoxic conditions or in the setting of hypoxia-independent upregulation of HIF signaling, as is the case in PAH,153 EndMT induces a complex sequence of molecular events characterized by an acquired loss of cell-cell contact membrane proteins, such as vascular endothelial cadherin and PECAM-1 (platelet endothelial cell adhesion molecule), which permits dissociation of pulmonary artery endothelial cells (PAECs) and their migration inward toward the blood vessel lumen. In PAH, endothelial cell phenotype switching to myofibroblasts occurs in parallel to the histological rearrangement of PAECs in remodeled pulmonary arterioles and is linked to dysregulated bone morphogenetic protein receptor type 2 (BMPR-2) signaling, cell proliferation, and, presumably, fibrosis.154 However, diethylaminoethyl-cellulose chromatography and other sensitive methods have confirmed detectable expression levels of (fibrillar) collagen III and the profibrotic protein connective tissue growth factor (CTGF) in PAECs under basal conditions.155,156 Importantly, however, CTGF–collagen III signaling is increased in PAECs exposed to hypoxia for 24 hours,157 which is a time point well in advance of the 3–7-day duration necessary to complete EndMT.152 Observations from coculture systems in vitro that demonstrate upregulation of fibrillar collagen synthesis in PASMCs and resident fibroblasts induced by PAECs overexpressing CTGF lend additional evidence indicating that EndMT is not required for vascular or perivascular fibrosis.
There are accumulating data to suggest that the mineralocorticoid hormone aldosterone contributes to the pathobiology of pulmonary vascular fibrosis that occurs concomitantly with chronic hypoxic lung injury. For example, in one study of 25 patients referred for right heart catheterization, pulmonary arterial plasma levels of aldosterone were increased in PAH patients compared to controls (595 ± 281 [mean ± SD] vs. 120 ± 42.3 pg/dL, P < 0.02) and correlated positively with hemodynamic measurements of pulmonary vascular remodeling, such as PVR (r = 0.72, P < 0.01) and transpulmonary gradient (r = 0.69, P < 0.02).158 Findings from other groups support these observations and suggest further that elevated circulating levels of aldosterone correlate with disease burden in PAH patients.159 In the rat monocrotaline (inflammatory), rat SU-5416/hypoxia (angioproliferative), and porcine vein banding (hypertrophic remodeling) experimental models of PAH, circulating aldosterone levels are increased, compared to those in control animals, by 4.2-fold (P < 0.05), 4.7-fold (P < 0.05),160 and 2.5-fold (P < 0.05),161 respectively.
Overactivation of the renin-angiotensin axis is a likely contributor to hyperaldosteronism in PAH, because pulmonary aldosterone levels associate inversely with cardiac output (r = −0.79, P < 0.005).158 However, extra-adrenal steroidogenesis is also suggested by the finding that, aldosterone levels are increased in plasma-free saline-perfused lung tissue homogenates harvested from PAH rats, compared to those in controls in vivo (median: 100.0 [interquartile range: 87.3–113.4] vs. 183.1 [126.1–197.4] pg/μg protein, P < 0.04).160 The possibility that local aldosterone synthesis in lung is relevant to patients is supported by data from a recent study in which hormone levels were measured across the pulmonary circulation in a cohort of controls (N = 18) and patients with mild PH and heart failure due to reduced left ventricular ejection fraction (HFrEF; N = 42) referred for invasive cardiopulmonary exercise testing. In that study, a transpulmonary (i.e., PA levels − radial artery levels) increase in aldosterone was also observed between controls and HFrEF patients at rest (7.5 [−54 to 40] vs. 61.6 [−13.6 to 165] ng/dL, P = 0.01) and during peak exercise (−20.7 [−39.6 to 79.1] vs. 25.8 [−29.2 to 109.3] ng/dL, P = 0.02), while peak volume of oxygen consumption (pVo2), which is a clinical measure of exercise reserve capacity, correlated inversely with PA (r = −0.31, P = 0.01) and radial (r = −0.32, P = 0.01) aldosterone levels.162 Taken together, these observations are in agreement with previous findings suggesting paracrine functionality of lung tissue, as a transpulmonary difference has been reported for ET-1 and the vasodilator second-messenger molecule cyclic guanosine monophosphate (cGMP), as well as other vasoactive intermediates relevant to pulmonary vascular disease.163,164
The possibility of extra-adrenal aldosterone synthesis in the lung is supported further by data indicating that lung tissue homogenates, PASMCs, and/or PAECs express key intermediaries required for aldosterone biosynthesis from cholesterol, including steroidogenic acute regulatory protein (StAR),165,166 which promotes transport of cholesterol across the mitochondrial membrane to the site of cholesterol metabolism and thus catalyzes the first and rate-limiting step of aldosterone biosynthesis, as well as CYP11β2 (aldosterone synthase),160,167 which catalyzes the final step of aldosterone biosynthesis from 18-hydroxy-corticosterone. Hypoxia as a regulator of aldosterone bioactivity in the pulmonary circulation has been suggested by findings from population studies indicating that genetic variants coding for the angiotensin II receptor 1 (T174M polymorphism; 1166C/C) and CYP11β2 (T-344C promoter region) cluster at a greater frequency in patients with PAH168 or those with a diagnosis of acute high-altitude pulmonary edema, compared to individuals tolerant of acute high-altitude exposure or high-altitude dwellers.169
There is indirect evidence that hypoxia may affect aldosterone synthesis in PAECs through ET-1 signaling. For example, PAEC exposure to FIo2 of 1% for 24 hours was associated with a 2.8-fold increase in messenger RNA transcription of the ET-1 precursor preproendothelin 1, which corresponded to an increase in ET-1 protein expression at 24 and 48 hours. Although a number of mechanisms have been proposed to account for this effect, hypoxia-induced HIF-1 binding to the ET-1 hypoxia response element has been shown to correlate with increased ET-1 activity in various vascular cell lines.170,171 In turn, ET-1, which is an established aldosterone secretogogue in adrenocortical cells, has also been shown in PAECs to induce binding of the steroidogenic transcription factors PGC-1α and steroidogenic factor 1 (SF-1) to the promoter region of CYP11β2 that increases synthesis of aldosterone.160 On the basis of observations indicating that StAR expression is increased in fibrotic pulmonary arterioles from mice exposed to CH, SU-5416/hypoxia-PAH rats, and patients with PAH, the direct relationship between hypoxia and aldosterone through StAR signaling has been assessed. Indeed, compared to normoxia, PAEC exposure to hypoxia (2% FIo2) for 24 hours increases aldosterone levels in the cell culture media significantly (187.2 ± 86.0 [mean ± SEM] vs. 376.0 ± 94.0 pg/μg protein, P < 0.02), as assessed by enzyme immunoassay and subsequently confirmed by liquid chromatography–mass spectrometry.157 Hypoxia increased expression of the steroidogenic transcription factors c-Jun and c-Fos and induced their binding to the activator protein 1 (AP-1) site of the StAR promoter, whereas pharmacological antagonism of the AP-1 site or molecular inhibition of StAR prevented endothelial synthesis of aldosterone by hypoxia, providing one possible mechanism to account for regulation of steroid biogenesis by oxygen tension in PAECs. Interestingly, stimulation of c-Fos/c-Jun-StAR signaling by hypoxia was selective to PAECs, and in that study157 it was not evident in systemic vascular cells, lung fibroblasts, or other cell types involved in pulmonary arterial fibrosis (Fig. 3). The mechanistic basis for this observation remains incompletely characterized but overall appears to be in line with expanding evidence demonstrating critical differences in the response pattern to hypoxia distinguishing pulmonary vascular (and right ventricular) cell types from their systemic (and left ventricular) cell type correlates.172
Figure 3.

Hypoxia increases aldosterone (ALDO) synthesis in pulmonary endothelial cells to promote vascular fibrosis and pulmonary hypertension. In pulmonary artery endothelial cells (PAECs), chronic hypoxia induces binding of the transcription factors c-Fos and c-Jun to the activator protein 1 site upstream of the StAR promoter. Increased StAR expression and activity induced by hypoxia increase pulmonary endothelial ALDO production, which, in turn, upregulates expression of (fibrillar) collagen III and the remodeling proteins matrix metalloproteinase 2 (MMP-2) and MMP-9. Hypoxia-aldosterone signaling in PAECs is also associated with induced collagen III synthesis in pulmonary artery smooth muscle cells (PASMCs) in vitro and fibrotic vascular remodeling of distal pulmonary arterioles and pulmonary hypertension in vivo. Fio2: fraction of inspired oxygen; StAR: steroidogenic acute regulatory protein.
The relevance of aldosterone to the vasculopathy of hypoxia appears to involve activation of remodeling and fibrotic proteins. For example, pharmacological inhibition of the mineralocorticoid receptor with spironolactone or eplerenone prevented hypoxia-induced upregulation of matrix metalloproteinase 2 (MMP-2), MMP-9, CTGF, and collagen III in PAECs in vitro. Furthermore, administration of these pharmacotherapies, using disease prevention or reversal protocols, decreased distal pulmonary arteriole collagen burden significantly to improve PPA, PVR, right ventricular mass, and cardiac index in vivo in experimental models of hypoxic pulmonary vascular disease, including PH.160,173 Preston and colleagues173 noted further that the inhibitory effects of mineralocorticoid receptor antagonism on fibrosis were extended beyond the pulmonary circulation and abrogated fully in the right ventricular interstitial compartment in HPH in vivo. These converging lines of evidence provide the rationale for several ongoing randomized clinical trials assessing the effect of mineralocorticoid receptor inhibition on clinical outcome in PAH patients.174
Conclusions
The principal determinates of pulmonary vascular and right ventricular susceptibility include inherited predispositions, such as specific variants in GALNT13 or VHLR200W homozygosity that promote pathogenic interactions between red blood cells and the pulmonary arterial blood vessel wall, as well as HIF-dependent upregulation of ROCK and intracellular Ca2+ flux due to chronic exposure to hypoxia that induces a contractile phenotype in pulmonary arterioles. CH is also linked to upregulation of StAR through activation of the oxygen-sensitive transcription factors c-Fos and c-Jun that induces pulmonary endothelial aldosterone synthesis and vascular fibrosis. Taken together, these findings illustrate complex interplay between genetic, environmental, and hormonal factors that mediate pulmonary vascular injury, but they also identify numerous potential therapeutic targets to improve on increased rates of morbidity reported in patients with PH due to hypoxia, hemoglobinopathies, and other etiologies sharing related pathobiology.
Acknowledgments
We acknowledge the American Thoracic Society and Drs. Eric Austin and Gregory Eliot for the opportunity to contribute to the 2015 Grover Conference.
Source of Support: This work was supported by National Institutes of Health grants HL126514 (LAS), HL73859 (LAS), 1K08HL11207-01A1 (BAM), R01HL127342 (RFM), and R01HL111656 (RFM) and awards from the American Heart Association (AHA 15GRNT25080016), the Pulmonary Hypertension Association, the Cardiovascular Medical Research and Education Fund (CMREF), and the Klarman Foundation at Brigham and Women’s Hospital to BAM.
Conflict of Interest: BAM reports investigator-initiated research supported by Gilead Sciences. The other authors declare no conflicts of interest.
References
- 1.Grover RF. The paradox of hypoxic pulmonary hypertension (2013 Grover Conference series). Pulm Circ 2014;4(2):151–157. [DOI] [PMC free article] [PubMed]
- 2.Anand IS, Wu T. Syndromes of subacute mountain sickness. High Alt Med Biol 2004;5(2):156–170. [DOI] [PubMed]
- 3.Singh I, Kapila CC, Khanna PK, Nanda RB, Rao BD. High-altitude pulmonary oedema. Lancet 1965;285(7379):229–234. [DOI] [PubMed]
- 4.Menon ND. High-altitude pulmonary edema: a clinical study. N Engl J Med 1965;273(2):66–73. [DOI] [PubMed]
- 5.Anand IS. Hypoxia and the pulmonary circulation. Thorax 1994;49(Suppl):S19–S24. [DOI] [PMC free article] [PubMed]
- 6.Anand IS, Malhotra RM, Chandrashekhar Y, Bali HK, Chauhan SS, Jindal SK, Bhandari RK, Wahi PL. Adult subacute mountain sickness—a syndrome of congestive heart failure in man at very high altitude. Lancet 1990;335(8689):561–565. [DOI] [PubMed]
- 7.Peñaloza D, Arias-Stella J, Sime F, Recavarren S, Marticorena E. The heart and pulmonary circulation in children at high altitudes: physiological, anatomical, and clinical observations. Pediatrics 1964;34(4):568–582. [PubMed]
- 8.Rotta A, Cánepa A, Hurtado A, Velásquez T, Chávez R. Pulmonary circulation at sea level and at high altitudes. J Appl Physiol 1956;9(3):328–336. [DOI] [PubMed]
- 9.Pryor R, Weaver WF, Blount SG Jr. Electrocardiographic observation of 493 residents living at high altitude (10,150 feet). Am J Cardiol 1965;16(4):494–499. [DOI] [PubMed]
- 10.Naeye RL. Hypoxemia and pulmonary hypertension. A study of the pulmonary vasculature. Arch Pathol 1961;71(Apr):447–452. [PubMed]
- 11.Naeye RL. Children at high altitude: pulmonary and renal abnormalities. Circ Res 1965;16(1):33–38. [DOI] [PubMed]
- 12.Arias-Stella J, Saldaña M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 1963;28(5):915–925. [DOI] [PubMed]
- 13.Heath D, Smith P, Rios Dalenz J, Williams D, Harris P. Small pulmonary arteries in some natives of La Paz, Bolivia. Thorax 1981;36(8):599–604. [DOI] [PMC free article] [PubMed]
- 14.Will DH, Alexander AF, Reeves JT, Grover RF. High altitude–induced pulmonary hypertension in normal cattle. Circ Res 1962;10(2):172–177. [DOI] [PubMed]
- 15.Stenmark KR, Meyrick B, Galiè N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 2009;297(6):L1013–L1032. [DOI] [PubMed]
- 16.Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol 2005;98(3):1092–1100. [DOI] [PubMed]
- 17.Stenmark KR, Fasules J, Hyde DM, Voelkel NF, Henson J, Tucker A, Wilson H, Reeves JT. Severe pulmonary hypertension and arterial adventitial changes in newborn calves at 4,300 m. J Appl Physiol 1987;62(2):821–830. [DOI] [PubMed]
- 18.Peñaloza D, Banchero N, Sime F, Gamboa R. The heart in chronic hypoxia. Biochem Clin 1963;2:283–298. [PubMed]
- 19.Hultgren HN, Kelly J, Miller H. Effect of oxygen upon pulmonary circulation in acclimatized man at high altitude. J Appl Physiol 1965;20(2):239–243.
- 20.Peñaloza D, Gamboa R, Marticorena E, Echevarría M, Dyer J, Gutierrez E. The influence of high altitudes on the electrical activity of the heart: electrocardiographic and vactorcardiographic observations in adolescence and adulthood. Am Heart J 1961;61(1):101–115. [DOI] [PubMed]
- 21.Aldashev AA, Sarybaev AS, Sydykov AS, Kalmyrzaev BB, Kim EV, Mamanova LB, Maripov R, et al. Characterization of high-altitude pulmonary hypertension in the Kyrgyz: association with angiotensin-converting enzyme genotype. Am J Respir Crit Care Med 2002;166(10):1396–1402. [DOI] [PubMed]
- 22.Peñaloza D, Sime F. Chronic cor pulmonale due to loss of altitude acclimatization (chronic mountain sickness). Am J Med 1971;50(6):728–743. [DOI] [PubMed]
- 23.Meyrick B, Reid L. Normal postnatal development of the media of the rat hilar pulmonary artery and its remodeling by chronic hypoxia. Lab Invest 1982;46(5):505–514. [PubMed]
- 24.Hales CA, Kradin RL, Brandstetter RD, Zhu YJ. Impairment of hypoxic pulmonary artery remodeling by heparin in mice. Am Rev Respir Dis 1983;128(4):747–751. [DOI] [PubMed]
- 25.Davies P, Maddalo F, Reid L. Effects of chronic hypoxia on structure and reactivity of rat lung microvessels. J Appl Physiol 1985;58(3):795–801. [DOI] [PubMed]
- 26.Janssens SP, Thompson BT, Spence CR, Hales CA. Polycythemia and vascular remodeling in chronic hypoxic pulmonary hypertension in guinea pigs. J Appl Physiol 1991;71(6):2218–2223. [DOI] [PubMed]
- 27.Rabinovitch M, Konstam MA, Gamble WJ, Papanicolaou N, Aronovitz MJ, Treves S, Reid L. Changes in pulmonary blood flow affect vascular response to chronic hypoxia in rats. Circ Res 1983;52(4):432–441. [DOI] [PubMed]
- 28.Sobin SS, Chen PC. Ultrastructural changes in the pulmonary arterioles in acute hypoxic pulmonary hypertension in the rat. High Alt Med Biol 2000;1(4):311–322. [DOI] [PubMed]
- 29.Sui GJ, Liu YH, Cheng XS, Anand IS, Harris E, Harris P, Heath D. Subacute infantile mountain sickness. J Pathol 1988;155(2):161–170. [DOI] [PubMed]
- 30.Brown SE, Linden GS, King RR, Blair GP, Stansbury DW, Light RW. Effects of verapamil on pulmonary haemodynamics during hypoxaemia, at rest, and during exercise in patients with chronic obstructive pulmonary disease. Thorax 1983;38(11):840–844. [DOI] [PMC free article] [PubMed]
- 31.Oka M, Morris KG, McMurtry IF. NIP-121 is more effective than nifedipine in acutely reversing chronic pulmonary hypertension. J Appl Physiol 1993;75(3):1075–1080. [DOI] [PubMed]
- 32.Moinard J, Manier G, Pillet O, Castaing Y. Effect of inhaled nitric oxide on hemodynamics and VA/Q inequalities in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1994;149(6):1482–1487. [DOI] [PubMed]
- 33.Barberà JA, Roger N, Roca J, Rovira I, Higenbottam TW, Rodriguez-Roisin R. Worsening of pulmonary gas exchange with nitric oxide inhalation in chronic obstructive pulmonary disease. Lancet 1996;347(8999):436–440. [DOI] [PubMed]
- 34.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 2004;287(4):L656–L664. [DOI] [PubMed]
- 35.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2004;287(4):L665–L672. [DOI] [PubMed]
- 36.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 2005;171(5):494–499. [DOI] [PubMed]
- 37.Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 2003;547(1):133–145. [DOI] [PMC free article] [PubMed]
- 38.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006;99(7):675–691. [DOI] [PubMed]
- 39.Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2006;290(2):L284–L290. [DOI] [PubMed]
- 40.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2008;295(3):L515–L529. [DOI] [PMC free article] [PubMed]
- 41.Suzuki H, Twarog BM. Membrane properties of smooth muscle cells in pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol 1982;242(5):H907–H915. [DOI] [PubMed]
- 42.Shimoda LA, Polak J. Hypoxia. 4. Hypoxia and ion channel function. Am J Physiol Cell Physiol 2011;300(5):C951–C969. [DOI] [PMC free article] [PubMed]
- 43.Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel α subunits in pulmonary artery smooth muscle cells. J Clin Invest 1997;100(9):2347–2353. [DOI] [PMC free article] [PubMed]
- 44.Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol 2005;288(6):L1049–L1058. [DOI] [PubMed]
- 45.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 2001;281(1):L202–L208. [DOI] [PubMed]
- 46.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 2002;105(2):244–250. [DOI] [PubMed]
- 47.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 2000;279(5):L884–L894. [DOI] [PubMed]
- 48.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 2006;98(12):1528–1537. [DOI] [PubMed]
- 49.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 2004;95(5):496–505. [DOI] [PubMed]
- 50.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, et al. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 2013;305(2):L154–L164. [DOI] [PMC free article] [PubMed]
- 51.Luke T, Maylor J, Undem C, Sylvester JT, Shimoda LA. Kinase dependent activation of voltage-gated Ca2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2012;302(10):L1128–L1139. [DOI] [PMC free article] [PubMed]
- 52.Hirenallur-S DK, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2008;295(5):L915–L924. [DOI] [PMC free article] [PubMed]
- 53.Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 2010;299(6):L861–L871. [DOI] [PMC free article] [PubMed]
- 54.Walker J, Undem C, Yun X, Lade J, Jiang H, Shimoda LA. Role of Rho kinase and Na+/H+ exchange in hypoxia-induced pulmonary arterial smooth muscle cell proliferation and migration. Physiol Rep 2016;4(6):e12702. doi:10.14814/phy2.12702. [DOI] [PMC free article] [PubMed]
- 55.Leggett K, Maylor J, Undem C, Lai N, Lu W, Schweitzer KS, King LS, et al. Hypoxia-induced migration in pulmonary arterial smooth muscle cells requires calcium-dependent upregulation of aquaporin 1. Am J Physiol Lung Cell Mol Physiol 2012;303(4):L343–L353. [DOI] [PMC free article] [PubMed]
- 56.Lai N, Lade J, Leggett K, Yun X, Baksh S, Chau E, Crow MT, Sidhaye V, Wang J, Shimoda LA. The aquaporin 1 C-terminal tail is required for migration and growth of pulmonary arterial myocytes. Am J Respir Cell Mol Biol 2014;50(6):1010–1020. [DOI] [PMC free article] [PubMed]
- 57.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA 2003;100(16):9488–9493. [DOI] [PMC free article] [PubMed]
- 58.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004;95(8):830–840. [DOI] [PubMed]
- 59.Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Brit J Pharmacol 2008;153(Suppl 1):S99–S111. [DOI] [PMC free article] [PubMed]
- 60.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 2001;280(2):H746–H755. [DOI] [PubMed]
- 61.Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 2004;19(2):44–50. [DOI] [PubMed]
- 62.Rodman DM, Reese K, Harral J, Fouty B, Wu S, West J, Hoedt-Miller M, et al. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ Res 2005;96(8):864–872. [DOI] [PubMed]
- 63.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 2012;302(8):H1546–H1562. [DOI] [PMC free article] [PubMed]
- 64.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA 2007;104(27):11418–11423. [DOI] [PMC free article] [PubMed]
- 65.Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005;434(7034):786–792. [DOI] [PubMed]
- 66.Yun X, Jiang H, Lai N, Shimoda LA. Aquaporin 1 (AQP1) enhances pulmonary arterial smooth muscle cell (PASMC) migration and proliferation via regulation of β-catenin [abstract]. FASB J 2015;29(1 suppl):662.13.
- 67.Madden JA, Ray DE, Keller PA, Kleinman JG. Ion exchange activity in pulmonary artery smooth muscle cells: the response to hypoxia. Am J Physiol Lung Cell Mol Physiol 2001;280(2):L264–L271. [DOI] [PubMed]
- 68.Quinn DA, Honeyman TW, Joseph PM, Thompson BT, Hales CA, Scheid CR. Contribution of Na+/H+ exchange to pH regulation in pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 1991;5(6):586–591. [DOI] [PubMed]
- 69.Quinn DA, Dahlberg CG, Bonventre JP, Scheid CR, Honeyman T, Joseph PM, Thompson BT, Hales CA. The role of Na+/H+ exchange and growth factors in pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 1996;14(2):139–145. [DOI] [PubMed]
- 70.Rios EJ, Fallon M, Wang J, Shimoda LA. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2005;289(5):L867–L874. [DOI] [PubMed]
- 71.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2006;291(5):L941–L949. [DOI] [PubMed]
- 72.Quinn DA, Du HK, Thompson BT, Hales CA. Amiloride analogs inhibit chronic hypoxic pulmonary hypertension. Am J Respir Crit Care Med 1998;157(4):1263–1268. [DOI] [PubMed]
- 73.Yu L, Quinn DA, Garg HG, Hales CA. Deficiency of the NHE1 gene prevents hypoxia-induced pulmonary hypertension and vascular remodeling. Am J Respir Crit Care Med 2008;177(11):1276–1284. [DOI] [PMC free article] [PubMed]
- 74.Yu L, Hales CA. Silencing of sodium-hydrogen exchanger 1 attenuates proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells via E2F1. Am J Respir Cell Mol Biol 2011;45(5):923–930. [DOI] [PMC free article] [PubMed]
- 75.Denker SP, Huang DC, Orlowski J, Furthmayr H, Barber DL. Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol Cell 2000;6(6):1425–1436. [DOI] [PubMed]
- 76.Wang J, Weigand L, Foxson J, Shimoda LA, Sylvester JT. Ca2+ signaling in hypoxic pulmonary vasoconstriction: effects of myosin light chain and Rho kinase antagonists. Am J Physiol Lung Cell Mol Physiol 2007;293(3):L674–L685. [DOI] [PubMed]
- 77.Undem C, Rios EJ, Maylor J, Shimoda LA. Endothelin-1 augments Na+/H+ exchange activity in murine pulmonary arterial smooth muscle cells via Rho kinase. PLoS ONE 2012;7(9):e46303. doi:10.1371/journal.pone.0046303. [DOI] [PMC free article] [PubMed]
- 78.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 2005;97(2):185–191. [DOI] [PubMed]
- 79.Abe K, Tawara S, Oi K, Hizume T, Uwatoku T, Fukumoto Y, Kaibuchi K, Shimokawa H. Long-term inhibition of Rho-kinase ameliorates hypoxia-induced pulmonary hypertension in mice. J Cardiovasc Pharmacol 2006;48(6):280–285. [DOI] [PubMed]
- 80.Yang X, Huang HC, Yin H, Alpern RJ, Preisig PA. RhoA required for acid-induced stress fiber formation and trafficking and activation of NHE3. Am J Physiol Renal Physiol 2007;293(4):F1054–F1064. [DOI] [PubMed]
- 81.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res 2007;100(5):607–621. [DOI] [PubMed]
- 82.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol 2008;155(4):444–454. [DOI] [PMC free article] [PubMed]
- 83.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase–induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 2004;95(6):579–586. [DOI] [PubMed]
- 84.Wojciak-Stothard B, Leiper J. Rho GTPases and hypoxia in pulmonary vascular endothelial cells. Methods Enzymol 2008;439:267–283. [DOI] [PubMed]
- 85.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012;148(3):399–408. [DOI] [PMC free article] [PubMed]
- 86.Semenza GL. Pulmonary vascular responses to chronic hypoxia mediated by hypoxia-inducible factor 1. Proc Am Thorac Soc 2005;2:68–70. [DOI] [PubMed]
- 87.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 2012;92(3):967–1003. [DOI] [PMC free article] [PubMed]
- 88.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 1998;12(2):149–162. [DOI] [PMC free article] [PubMed]
- 89.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 1999;103(5):691–696. [DOI] [PMC free article] [PubMed]
- 90.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003;111(10):1519–1527. [DOI] [PMC free article] [PubMed]
- 91.Shimoda LA. 55th Bowditch Lecture: effects of chronic hypoxia on the pulmonary circulation: role of HIF-1. J Appl Physiol 2012;113(9):1343–1352. [DOI] [PMC free article] [PubMed]
- 92.Kim YM, Barnes EA, Alvira CM, Ying L, Reddy S, Cornfield DN. Hypoxia-inducible factor-1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ Res 2013;112(9):1230–1233. [DOI] [PMC free article] [PubMed]
- 93.Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, Iruela-Arispe L, Simon MC, Keith B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009;114(2):469–477. [DOI] [PMC free article] [PubMed]
- 94.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 2014;189(3):314–324. [DOI] [PMC free article] [PubMed]
- 95.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 2012;109(4):1239–1244. [DOI] [PMC free article] [PubMed]
- 96.Wang J, Semenza G, Sylvester JT, Shimoda LA. HIF-1 regulates hypoxic-induction of canonical transient receptor potential (TRPC) channels in pulmonary arterial smooth muscle cells. FASEB J 2005;19:A1278.
- 97.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 2013;304(8):L549–L561. [DOI] [PMC free article] [PubMed]
- 98.Li M, Liu Y, Jin F, Sun X, Li Z, Liu Y, Fang P, Shi H, Jiang X. Endothelin-1 induces hypoxia inducible factor 1α expression in pulmonary artery smooth muscle cells. FEBS Lett 2012;586(21):3888–3893. [DOI] [PubMed]
- 99.Groves BM, Droma T, Sutton JR, McCullough RG, McCullough RE, Zhuang J, Rapmund G, Sun S, Janes C, Moore LG. Minimal hypoxic pulmonary hypertension in normal Tibetans at 3,658 m. J Appl Physiol 1993;74(1):312–318. [DOI] [PubMed]
- 100.Simonson TS, McClain DA, Jorde LB, Prchal JT. Genetic determinants of Tibetan high-altitude adaptation. Hum Genet 2012;131(4):527–533. [DOI] [PubMed]
- 101.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, et al. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 2010;107(25):11459–11464. [DOI] [PMC free article] [PubMed]
- 102.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 2010;329(5987):75–78. [DOI] [PMC free article] [PubMed]
- 103.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010;329(5987):72–75. [DOI] [PubMed]
- 104.Lorenzo FR, Huff C, Myllymäki M, Olenchock B, Swierczek S, Tashi T, Gordeuk V, et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet 2014;46(9):951–956. [DOI] [PMC free article] [PubMed]
- 105.Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood 2008;112(3):919–921. [DOI] [PubMed]
- 106.Peñazola D, Sime F, Banchero N, Gamboa R, Cruz J, Marticorena E. Pulmonary hypertension in healthy men born and living at high altitudes. Am J Cardiol 1963;11(2):150–157. [DOI] [PubMed]
- 107.Antezana AM, Antezana G, Aparicio O, Noriega I, Velarde FL, Richalet JP. Pulmonary hypertension in high-altitude chronic hypoxia: response to nifedipine. Eur Respir J 1998;12(5):1181–1185. [DOI] [PubMed]
- 108.Eichstaedt CA, Antão T, Pagani L, Cardona A, Kivisild T, Mormina M. The Andean adaptive toolkit to counteract high altitude maladaptation: genome-wide and phenotypic analysis of the Collas. PLoS ONE 2014;9(3):e93314. doi:10.1371/journal.pone.0093314. [DOI] [PMC free article] [PubMed]
- 109.Stobdan T, Zhou D, Ao-Ieong E, Ortiz D, Ronen R, Hartley I, Gan Z, et al. Endothelin receptor B, a candidate gene from human studies at high altitude, improves cardiac tolerance to hypoxia in genetically engineered heterozygote mice. Proc Natl Acad Sci USA 2015;112(33):10425–10430. [DOI] [PMC free article] [PubMed]
- 110.Mejia OM, Prchal JT, León-Velarde F, Hurtado A, Stockton DW. Genetic association analysis of chronic mountain sickness in an Andean high-altitude population. Haematologica 2005;90(1):13–19. [PubMed]
- 111.Moore LG, Shriver M, Bemis L, Hickler B, Wilson M, Brutsaert T, Parra E, Vargas E. Maternal adaptation to high-altitude pregnancy: an experiment of nature—a review. Placenta 2004;25(Suppl):S60–S71. [DOI] [PubMed]
- 112.Scheinfeldt LB, Soi S, Thompson S, Ranciaro A, Woldemeskel D, Beggs W, Lambert C, et al. Genetic adaptation to high altitude in the Ethiopian highlands. Genome Biol 2012;13(1):R1. doi:10.1186/gb-2012-13-1-r1. [DOI] [PMC free article] [PubMed]
- 113.Newman JH, Holt TN, Cogan JD, Womack B, Phillips JA III, Li C, Kendall Z, et al. Increased prevalence of EPAS1 variant in cattle with high-altitude pulmonary hypertension. Nat Commun 2015;6:6863. doi:10.1038/ncomms7863. [DOI] [PMC free article] [PubMed]
- 114.Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol 2000;151(9):839–845. [DOI] [PubMed]
- 115.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337(11):762–769. [DOI] [PubMed]
- 116.Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65(1):183–189. [PubMed]
- 117.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 2005;293(13):1653–1662. [DOI] [PubMed]
- 118.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350(9):886–895. [DOI] [PubMed]
- 119.Frei AC, Guo Y, Jones DW, Pritchard KA Jr., Fagan KA, Hogg N, Wandersee NJ. Vascular dysfunction in a murine model of severe hemolysis. Blood 2008;112(2):398–405. [DOI] [PMC free article] [PubMed]
- 120.Hsu L, McDermott T, Brown L, Aguayo SM. Transgenic HbS mouse neutrophils in increased susceptibility to acute lung injury: implications for sickle acute chest syndrome. Chest 1999;116(suppl 1):92S. [DOI] [PubMed]
- 121.Gladwin MT. Role of the red blood cell in nitric oxide homeostasis and hypoxic vasodilation. Adv Exp Med Biol 2006;588:189–205. [DOI] [PubMed]
- 122.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest 2000;106(3):337–338. [DOI] [PMC free article] [PubMed]
- 123.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest 2000;106(3):411–420. [DOI] [PMC free article] [PubMed]
- 124.Klings ES, Farber HW. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir Res 2001;2:280. doi:10.1186/rr70. [DOI] [PMC free article] [PubMed]
- 125.Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol/Oncol Clin North Am 2014;28(2):181–198. [DOI] [PubMed]
- 126.Powars D, Weidman JA, Odom-Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine 1988;67(1):66–76. [PubMed]
- 127.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med 1994;330(23):1639–1644. [DOI] [PubMed]
- 128.Wierenga KJ, Hambleton IR, Lewis NA. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. Lancet 2001;357(9257):680–683. [DOI] [PubMed]
- 129.Minter KR, Gladwin MT. Pulmonary complications of sickle cell anemia: a need for increased recognition, treatment, and research. Am J Respir Crit Care Med 2001;164(11):2016–2019. [DOI] [PubMed]
- 130.Siddiqui AK, Ahmed S. Pulmonary manifestations of sickle cell disease. Postgrad Med J 2003;79(933):384–390. [DOI] [PMC free article] [PubMed]
- 131.Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol 2010;85(6):403–408. [DOI] [PMC free article] [PubMed]
- 132.Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012;307(12):1254–1256. [DOI] [PMC free article] [PubMed]
- 133.Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012;39(1):112–118. [DOI] [PubMed]
- 134.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, Habibi A, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011;365(1):44–53. [DOI] [PubMed]
- 135.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood 2003;101(4):1257–1261. [DOI] [PubMed]
- 136.Mehari A, Alam S, Tan X, Cuttica MJ, Barnett CF, Miles G, Xu D, et al. Hemodynamic predictors of mortality in adults with sickle cell disease. Am J Respir Crit Care Med 2013;187(8):840–847. [DOI] [PMC free article] [PubMed]
- 137.Desai AA, Zhou T, Ahmad H, Zhang W, Mu W, Trevino S, Wade MS, et al. A novel molecular signature for elevated tricuspid regurgitation velocity in sickle cell disease. Am J Respir Crit Care Med 2012;186(4):359–368. [DOI] [PMC free article] [PubMed]
- 138.Berois N, Blanc E, Ripoche H, Mergui X, Trajtenberg F, Cantais S, Barrois M, et al. ppGalNAc-T13: a new molecular marker of bone marrow involvement in neuroblastoma. Clin Chem 2006;52(9):1701–1712. [DOI] [PubMed]
- 139.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121(18):2045–2066. [DOI] [PMC free article] [PubMed]
- 140.Sachdev V, Kato GJ, Gibbs JS, Barst RJ, Machado RF, Nouraie M, Hassell KL, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 2011;124(13):1452–1460. [DOI] [PMC free article] [PubMed]
- 141.Sundaram N, Tailor A, Mendelsohn L, Wansapura J, Wang X, Higashimoto T, Pauciulo MW, et al. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood 2010;116(1):109–112. [DOI] [PMC free article] [PubMed]
- 142.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 2002;32(4):614–621. [DOI] [PubMed]
- 143.Sable CA, Aliyu ZY, Dham N, Nouraie M, Sachdev V, Sidenko S, Miasnikova GY, et al. Pulmonary artery pressure and iron deficiency in patients with upregulation of hypoxia sensing due to homozygous VHLR200W mutation (Chuvash polycythemia). Haematologica 2012;97(2):193–200. [DOI] [PMC free article] [PubMed]
- 144.Hickey MM, Lam JC, Bezman NA, Rathmell WK, Simon MC. Von Hippel–Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2α signaling and splenic erythropoiesis. J Clin Invest 2007;117(12):3879–3889. [DOI] [PMC free article] [PubMed]
- 145.Zhang X, Zhang W, Ma SF, Desai AA, Saraf S, Miasniakova G, Sergueeva A, et al. Hypoxic response contributes to altered gene expression and precapillary pulmonary hypertension in patients with sickle cell disease. Circulation 2014;129(16):1650–1658. [DOI] [PMC free article] [PubMed]
- 146.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene 2008;27(48):6245–6251. [DOI] [PMC free article] [PubMed]
- 147.Rich S, Rabinovitch M. Diagnosis and treatment of secondary (non-category 1) pulmonary hypertension. Circulation 2008;118(21):2190–2199. [DOI] [PubMed]
- 148.Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187(8):865–878. [DOI] [PMC free article] [PubMed]
- 149.Wells JM, Washko GR, Han MK, Abbas N, Nath H, Mamary AJ, Regan E, et al. Pulmonary arterial enlargement and acute exacerbations of COPD. N Engl J Med 2012;367(10):913–921. [DOI] [PMC free article] [PubMed]
- 150.Hurdman J, Condliffe R, Elliot CA, Swift A, Rajaram S, Davies C, Hill C, et al. Pulmonary hypertension in COPD: results from the ASPIRE registry. Eur Respir J 2013;41(6):1292–1301. [DOI] [PubMed]
- 151.Blanco I, Santos S, Gea J, Güell R, Torres F, Gimeno-Santos E, Rodriguez DA, et al. Sildenafil to improve respiratory rehabilitation outcomes in COPD: a controlled trial. Eur Respir J 2013;42(4):982–992. [DOI] [PubMed]
- 152.Slukvin II, Vodyanik M. Endothelial origin of mesenchymal stem cells. Cell Cycle 2011;10(9):1370–1373. [DOI] [PMC free article] [PubMed]
- 153.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012;110(11):1484–1497. [DOI] [PMC free article] [PubMed]
- 154.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015;131(11):1006–1018. [DOI] [PubMed]
- 155.Macarak EJ. Collagen synthesis by cloned pulmonary artery endothelial cells. J Cell Physiol 1984;119(2):175–182. [DOI] [PubMed]
- 156.Mendoza FA, Piera-Velazquez S, Farber JL, Feghali-Bostwick C, Jiménez SA. Endothelial cells expressing endothelial and mesenchymal cell gene products in lung tissue from patients with systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol 2016;68(1):210–217. [DOI] [PMC free article] [PubMed]
- 157.Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 2014;130(2):168–179. [DOI] [PMC free article] [PubMed]
- 158.Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail 2013;15(3):277–283. [DOI] [PMC free article] [PubMed]
- 159.Calvier L, Legchenko E, Grimm L, Sallmon H, Hatch A, Plouffe BD, Schroeder C, Bauersachs J, Murthy SK, Hansmann G. Galectin-3 and aldosterone as potential tandem biomarkers in pulmonary arterial hypertension. Heart 2016;102(5):390–396. [DOI] [PubMed]
- 160.Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 2012;126(8):963–974. [DOI] [PMC free article] [PubMed]
- 161.Aguero J, Ishikawa K, Hadri L, Santos-Gallego C, Fish K, Hammoudi N, Chaanine A, et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am J Physiol Heart Circ Physiol 2014;307(8):H1204–H1215. [DOI] [PMC free article] [PubMed]
- 162.Maron BA, Stephens TE, Farrell LA, Oldham WM, Loscalzo J, Leopold JA, Lewis GD. Elevated pulmonary arterial and systemic plasma aldosterone levels associate with impaired cardiac reserve capacity during exercise in left ventricular systolic heart failure patients: a pilot study. J Heart Lung Transplant 2016;35(3):342–351. [DOI] [PMC free article] [PubMed]
- 163.Ooi H, Colucci WS, Givertz MM. Endothelin mediates increased pulmonary vascular tone in patients with heart failure: demonstration by direct intrapulmonary infusion of sitaxsentan. Circulation 2002;106(13):1618–1621. [DOI] [PubMed]
- 164.Melenovsky V, Al-Hiti H, Kazdova L, Jabor A, Syrovatka P, Malek I, Kettner J, Kautzner J. Transpulmonary B-type natriuretic peptide uptake and cyclic guanosine monophosphate release in heart failure and pulmonary hypertension: the effects of sildenafil. J Am Coll Cardiol 2009;54(7):595–600. [DOI] [PubMed]
- 165.Pezzi V, Mathis JM, Rainey WE, Carr BR. Profiling transcript levels for steroidogenic enzymes in fetal tissues. J Steroid Biochem Mol Biol 2003;87(2–3):181–189. [DOI] [PubMed]
- 166.Provost PR, Boucher E, Tremblay Y. Glucocorticoid metabolism in the developing lung: adrenal-like synthesis pathway. J Steroid Biochem Mol Biol 2013;138:72–80. [DOI] [PubMed]
- 167.Hatakeyama H, Miyamori I, Fujita T, Takeda Y, Takeda R, Yamamoto H. Vascular aldosterone: biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem 1994;269(39):24316–24320. [PubMed]
- 168.Chung WK, Deng L, Carroll JS, Mallory N, Diamond B, Rosenzweig EB, Barst RJ, Morse JH. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J Heart Lung Transplant 2009;28(4):373–379. [DOI] [PMC free article] [PubMed]
- 169.Srivastava S, Bhagi S, Kumari B, Chandra K, Sarkar S, Ashraf MZ. Association of polymorphisms in angiotensin and aldosterone synthase genes of the renin-angiotensin-aldosterone system with high-altitude pulmonary edema. J Renin Angiotensin Aldosterone Syst 2012;13(1):155–160. [DOI] [PubMed]
- 170.Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest 1991;88(3):1054–1057. [DOI] [PMC free article] [PubMed]
- 171.Minchenko A, Caro J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Mol Cell Biochem 2000;208(1–2):53–62. [DOI] [PubMed]
- 172.Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol 2015;119(10):1164–1172. [DOI] [PMC free article] [PubMed]
- 173.Preston IR, Sagliani KD, Warburton RR, Hill NS, Fanburg BL, Jaffe IZ. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2013;304(10):L678–L688. [DOI] [PMC free article] [PubMed]
- 174.Elinoff JM, Rame JE, Forfia PR, Hall MK, Sun J, Gharib AM, Abd-Elmoniem K, et al. A pilot study of the effect of spironolactone therapy on exercise capacity and endothelial dysfunction in pulmonary arterial hypertension: study protocol for a randomized controlled trial. Trials 2013;14:91. doi:10.1186/1745-6215-14-91. [DOI] [PMC free article] [PubMed]