

Abstract Abstract
Among pulmonary vascular diseases, pulmonary hypertension (PH) is the best studied and has been the focus of our work. The current classification of PH is based on a relatively simple combination of patient characteristics and hemodynamics. This leads to inherent limitations, including the inability to customize treatment and the lack of clarity from a more granular identification based on individual patient phenotypes. Accurate phenotyping of PH can be used in the clinic to select therapies and determine prognosis and in research to increase the homogeneity of study cohorts. Rapid advances in the mechanistic understanding of the disease, improved imaging methods, and innovative biomarkers now provide an opportunity to define novel PH phenotypes. We have recently shown that altered metabolism may affect nitric oxide levels and protein glycosylation, the peripheral circulation (which may provide insights into the response to therapy), and exhaled-breath analysis (which may be useful in disease evaluation). This review is based on a talk presented during the 2015 Grover Conference and highlights the relevant literature describing novel methods to phenotype pulmonary arterial hypertension patients by using approaches that involve the pulmonary and systemic (peripheral) vasculature. In particular, abnormalities in metabolism, the pulmonary and peripheral circulation, and exhaled breath in PH may help identify phenotypes that can be the basis for a precision-medicine approach to PH management. These approaches may also have a broader scope and may contribute to a better understanding of other diseases, such as asthma, diabetes, and cancer.
Keywords: phenotyping, peripheral circulation, metabolism, exhaled breath, precision medicine
Pulmonary vascular diseases (PVDs) are disorders that affect pulmonary circulation and include a category of diseases such as pulmonary embolism, chronic thromboembolic disease, pulmonary hypertension (PH), pulmonary veno-occlusive disease, arteriovenous malformations, and pulmonary edema. The frequency and pathogenesis of PVDs are not known; however, clinical reports have estimated the prevalence to be 20–25 million.1 Of the aforementioned PVDs, PH is one of the best studied, and it can be associated with other PVDs or other diseases, which increases the disease overlap, the heterogeneity of the pathobiology, and the complication of disease management. For this reason, reaching a consensus for treatment of pulmonary disease has been a daunting and slow process. In PVDs, the traditional approach to treatment strategies has been to target the group as a whole entity instead of using personalized or phenotyping-based methods. Recently, an American Thoracic Society committee of world-renowned specialists in PVD developed an official statement on PH phenotypes.2 The goal of this document was to initiate a consortium of study sites to perform accurate phenotyping and derive a plan to (1) identify subsets of PH patients with a similar molecular basis of PVD regardless of World Health Organization (WHO) clinical classification; (2) define novel, clinically relevant indices of therapeutic responsiveness to be useful as intermediate or primary outcomes in clinical trials; (3) identify biomarkers of disease risk and progression to be useful as measures for PVD detection and prevention research.2 In essence, the statement emphasized the need for novel phenotyping methods (genomics, transcriptomics, proteomics, metabolomics, etc.) to help identify disease mechanisms and biomarkers (endophenotypes) for early diagnosis, prognosis, and management in PVDs.
A model endophenotype would be identifiable, steadfast, and useful for assessing clinical outcomes and/or mechanisms of a disease phenotype. For example, endothelial dysfunction and vascular remodeling are endophenotypes shared among the different PVD categories. Other endophenotypes may be shared among different diseases, including asthma, diabetes, cancer, and lung and systemic diseases. The endophenotypes one chooses to study should be based on their specific link to the pathobiology of disease and their importance in determining prognosis and/or response to management strategies. On this basis, we must employ strategies to differentiate PVD patients into phenotypic groups according to these criteria. Such strategies include physiology, clinical parameters, biomarkers, and genotyping as well as metabolomics and proteomic analysis (Fig. 1).
Figure 1.

Phenotyping model. The model illustrates the specific tools used to collect human samples/data from patients to help better understand pulmonary vascular disease.
In pulmonary arterial hypertension (PAH), any given patient will have multiple overlapping endophenotypes, making therapeutic design complicated. In light of this, the predominant therapies in PAH are those that target nitric oxide (NO) deficiency, prostacyclin, or endothelin 1.3-6 To date, these endophenotypic targets limit us to PH disease management rather than reversing the disease process or preventing it, which is the ultimate goal. However, a new paradigm of research aimed at phenotyping large groups of PVD patients will hopefully open new avenues to reverse or prevent disease based on our existing endophenotypes as well as any newly discovered ones. For example, NO deficiency and dysregulated glucose metabolism are two endophenotypes that may benefit from phenotyping methods. Indeed, the pathology of PVDs (including PH) within the lung is the predominant target of deep-phenotyping techniques. However, the systemic nature of the disease may also provide specific clues or mechanisms for combating PVDs. This review focuses on the 2015 Grover Conference discussion and relevant literature describing phenotyping of PH patients based on novel methods using pulmonary and systemic (peripheral) approaches.
Introduction
PAH is a cardiopulmonary disease with a heterogeneous pathobiology and many presenting phenotypes. PAH is characterized by high pulmonary artery pressure and increased pulmonary vascular resistance, which often result in right ventricular (RV) failure and premature death.7-10 The disease etiology is complicated and can be associated with other pulmonary, cardiac, or systemic diseases or other factors, including drug use and toxin stimuli. The idiopathic form of PAH (IPAH) has no identifiable etiology, affects mostly young women, and if treated has an average survival of 2–3 years from the time of diagnosis.7,11-14 Currently, therapies target pulmonary vasoconstriction, and our ability to manage PAH remains poor because of our limited knowledge of the disease pathogenesis.
Multiple phenotypes (other than vasoconstriction), such as dysregulated pulmonary vascular cell proliferation and remodeling, increased angiogenesis, and inflammation, contribute to the PAH disease process.15 In addition, altered metabolism, including altered glucose uptake/metabolism, dyslipidemia, leptin dysregulation, and deficits in NO production, have been established in IPAH13,16-19 and may affect the disease processes. IPAH is remarkable because of its phenotypic and metabolic similarities to cancer.20-24 For example, it is known that highly proliferative cancer cells have extensive energy requirements and utilize excess metabolites, including glucose, glutamine, acetyl coenzyme A, and nucleotides by way of glycolysis, glutaminolysis/nitrogen metabolism, fatty acid metabolism, and nucleotide metabolism, respectively. This same phenomenon is shared in PAH, and, for the most part, the pathological study takes place in the lung. However, recognition of the underlying systemic phenotypes in PAH may increase our understanding of the disease. In fact, the overall metabolic changes may include the peripheral vascular system along with lung-localized alterations. Examining both systems, as well as their relationship to cancer, may help us uncover functional mechanisms (endophenotypes) and increase our ability to design better PAH therapies.
Metabolism
Glycosylation
Interrogation of altered glycan expression in metabolic disorders such as cancer and diabetes has become a fundamental way to determine the level of disease severity.25,26 To date, one of the most remarkable functions for glycosylation is its role in cell adhesion and migration, specifically in cancer metastasis.27-31 In addition, glycans have been documented for their role in extracellular matrix (ECM) turnover and remodeling in multiple diseases.32-34 Hence, there is a great need for high-throughput technologies to phenotype clinical diseases using glycoanalytic approaches. These approaches, in particular glycomics, could provide broad and deep glycan profile coverage and characterization in metabolic disorders and diseases with aberrant ECM remodeling. Our lab focuses on the metabolic disease IPAH. We recently showed an increase in hyaluronan (HA), an ECM glycosaminoglycan (GAG), in IPAH patient lung tissue, plasma, and pulmonary arterial smooth muscle cells (PASMCs).35,36 HA, along with other GAGs, may be functionally involved in the remodeling process as well as in metabolic abnormalities. In addition, altered glycosylation through increased serine/threonine hydroxyl (O)-linked N-acetylglucosamine (O-GlcNAc) modification has been determined.37 Specifically, the O-GlcNAc transferase was shown to regulate PASMC proliferation and associate with PAH disease worsening (n = 86 PAH patients), demonstrating a potential endophenotypic target for therapy. Both of these findings are related to the dysregulated glucose metabolism demonstrated in the disease and may contribute to augmented ECM remodeling. These recently found endophenotypes could benefit from a glycoproteomic (O-GlcNAc) or metabolomic (HA/O-GlcNAc) analysis within the different PH classes, and they may help to better classify PAH on the basis of the severity of the metabolic derangements or the remodeling process.
High-density lipoprotein cholesterol
High-density lipoprotein cholesterol (HDL-C), a major lipid carrier in the bloodstream, is critically involved in vascular disease and is associated with a lower risk of coronary heart disease.38,39 HDL-C protects against lipid oxidation, has anti-inflammatory properties, reduces endothelial dysfunction, and has anticoagulant effects. Low HDL-C levels are a prominent feature of the metabolic syndrome and insulin resistance.40-42 Recent reports suggest that low HDL-C may predispose to PVD.43,44 We have previously shown that HDL-C was lower in a PAH patient cohort (n = 69) than in control subjects (n = 229) who had more cardiovascular risk factors.45 Low HDL-C was associated with worse functional capacity and higher right atrial pressure and brain natriuretic peptide levels, as well as inflammatory markers. Importantly, low HDL-C was an independent predictor of increased mortality. This could potentially be explained by the antioxidant and anti-inflammatory properties of HDL-C, where decreases result in excessive lipid oxidation. The predictive value of HDL-C in PAH has been independently validated in a Chinese cohort43 and in a separate American cohort,46 but not in a French cohort.47 The value of HDL-C in PAH phenotyping and as a marker of PAH prognostic assessment warrants further investigation.
Interestingly, an association of oxidative stress, lipid oxidation, and peroxidation was documented in the progression of PAH and may drive or affect multiple PAH endophenotypes.48,49 Along these lines, dysfunctional HDL-C has been reported in PAH patients.50 Similar to reports on HDL-C, apolipoprotein A-I (Apo A-I), a major protein component of HDL-C, was shown to be reduced in PAH and associated with endothelial dysfunction, and the Apo A-1 mimetic peptide was shown to rescue PH in two rodent models.51 In addition, apolipoprotein E–deficient mice were shown to develop right ventricle hypertrophy, pulmonary vascular remodeling, and insulin resistance when administered a high-fat diet.52 The combined deficiency of peroxisome proliferator–activated receptor γ (a ligand-activated nuclear receptor that regulates adipogenesis and glucose metabolism) and apolipoprotein E has been linked to insulin resistance and the metabolic syndrome, and both were reduced in a similar PAH patient study.53 Collectively, these studies validate the importance of cholesterol metabolism and cholesterol components in the pathogenesis of PAH.
Leptin
Leptin, a neuroendocrine hormone that is secreted by adipose tissue, regulates fat metabolism, obesity, and appetite. Studies have shown that leptin levels are related to cardiovascular function.54 Independent of obesity, leptin levels have been shown to be inversely proportional to cardiovascular mortality in coronary artery disease and diabetes.55,56 The cardioprotectiveness of leptin is most likely due to its anti-inflammatory, proapoptotic, and antithrombotic actions. Multiple reports have shown that leptin and its receptor contribute to vascular remodeling in cardiovascular disease,57,58 acting to potentiate the proliferation and migration of vascular smooth muscle cells and vascular wall inflammatory cell infiltration.
In PAH patients, leptin was reported to be elevated in endothelium and may contribute to the pathobiology of the disease.59 Conversely, we have shown that low levels of serum leptin were associated with increased overall mortality in patients with PAH, when adjusted for age, sex, body mass index (BMI), and smoking status.60 Similarly, the ratio of leptin to BMI was shown to be inversely proportional to mortality in PAH, suggesting a protective effect, albeit independent of obesity. Further investigation is needed to determine whether leptin, when adjusted for BMI, can be used to distinguish PAH patients with a more favorable prognosis. In particular, serum leptin levels adjusted for BMI may be useful in phenotyping patients who have widespread metabolic abnormalities and may help us to better understand the association with PAH severity.
Nitric oxide
NO is a potent signaling molecule and physiological regulator that serves many biological processes. In particular, NO is a major physiologic regulator of blood vessel tone, permeability, cardiovascular homeostasis, platelet adhesion and aggregation, infiltration of inflammatory cells, and smooth muscle cell migration and proliferation.61,62 The production of NO has been well described.63 Three different nitric oxide synthases (NOSs), including neuronal NOS (nNOS; NOS I), inducible NOS (iNOS; NOS II), and endothelial NOS (eNOS; NOS III), are involved in the biosynthesis of NO and convert the amino acid substrate l-arginine to l-citruline and NO. Once produced by one of the three NOSs, NO is freely diffusible, enters pulmonary smooth muscle cells, and activates soluble guanylate cyclase (sGC) to produce 3′,5′-cyclic monophosphate, which causes relaxation and inhibition of proliferation64,65 and migration.66 Interestingly, the reduced bioavailability of NO is a mainstay in PAH patients and is one of the current therapeutic treatment targets in the disease. Indeed, NO deficiency facilitates the proliferation of vascular cells and remodeling of the pulmonary vasculature in PH. It has been shown that mice genetically deficient in eNOS spontaneously develop PH and RV failure.67 Even with the recent advances uncovering the role of NO in the pathology of the PAH, the molecular mechanisms that cause NO deficits are still not well understood. Current work is focused on exploring personalized approaches to each patient’s responsiveness to different NO-based therapies (sildenafil, riociguat, etc.), which do not work in all patients. Other methods may be useful in phenotyping patients on the basis of the level of NO deficiency determined from exhaled breath (see below). This will undoubtedly change the way PAH patients are classified and treated in the future.
Systemic circulation: laser Doppler flowmetry and iontophoresis of treprostinil
IPAH has been traditionally viewed as a disease only of the pulmonary circulation. Our recently published data,68-70 along with those of several others,71-73 have shown that this view is incomplete, because of the evidence that there may be extrapulmonary vascular involvement. We have shown that patients with PAH have sublingual vessels with reduced flow, greater heterogeneity, and more-pronounced tortuosity than age- and sex-matched healthy controls.68 Similarly, other reports found lower capillary density with nail-fold capillaroscopy in IPAH patients.74 We observed that transcutaneous oxygen (O2) levels, measured noninvasively at the level of the forearm, were reduced in patients with PAH,69 suggesting that this reduction in transcutaneous O2 may be a reflection of a reduced cutaneous capillary density.75,76
Laser Doppler flowmetry can be used to challenge and provide functional assessment of the cutaneous microcirculation. Laser Doppler flowmetry is based on the change in the wavelength of light (Doppler shift) when it hits moving blood cells.77 This method has been used to associate higher cardiovascular mortality in other diseases with severe cutaneous microcirculation dysfunction.78 In addition, laser Doppler flowmetry, combined with iontophoresis, allows for examination of the effect of certain vasoactive agents on the peripheral vascular system. For example, we examined the cutaneous microcirculation as a model for studying the prostacyclin (PGI2) pathway, which is one of the putative mechanisms responsible for the pulmonary vasculopathy of IPAH.79,80 In vivo, cutaneous treprostinil (a synthetic analog of PGI2) was administered through iontophoresis to PAH patients (n = 24), and the response was significantly reduced compared to that in age- and sex-matched controls (n = 25).70 In fact, patients with IPAH had significantly lower peak perfusion units and percentage change relative to baseline levels (Fig. 2). We found that the RV function (Fig. 3A) or the cardiac index (Fig. 3B) had only a small influence on the response to treprostinil iontophoresis. Interestingly, we have observed that higher PGI2 metabolites (e.g., prostaglandin F1α in plasma) were inversely proportional to percent change in perfusion during treprostinil iontophoresis (R = −0.43, P = 0.19, n = 11), suggesting an increased metabolism of PGI2 in IPAH. Mechanisms that are potentially responsible for the decrease in the response to treprostinil iontophoresis are shown in Figure 4. Collectively, these data highlight the relevance and potential use of laser Doppler flowmetry and iontophoresis in IPAH. The use of iontophoresis for vasoactive medication administration presents an innovative approach to explore the connection between the pulmonary and extrapulmonary circulation, since there are limited biomarkers available to help understand the pathobiology of the PAH and no mechanistic tools to guide the selection process for treatment. Furthermore, this approach may help to (1) identify novel endophenotypes of PAH, (2) uncover molecular mechanism(s) underlying PAH, and (3) phenotype patients on the basis of major pathway abnormalities.2
Figure 2.
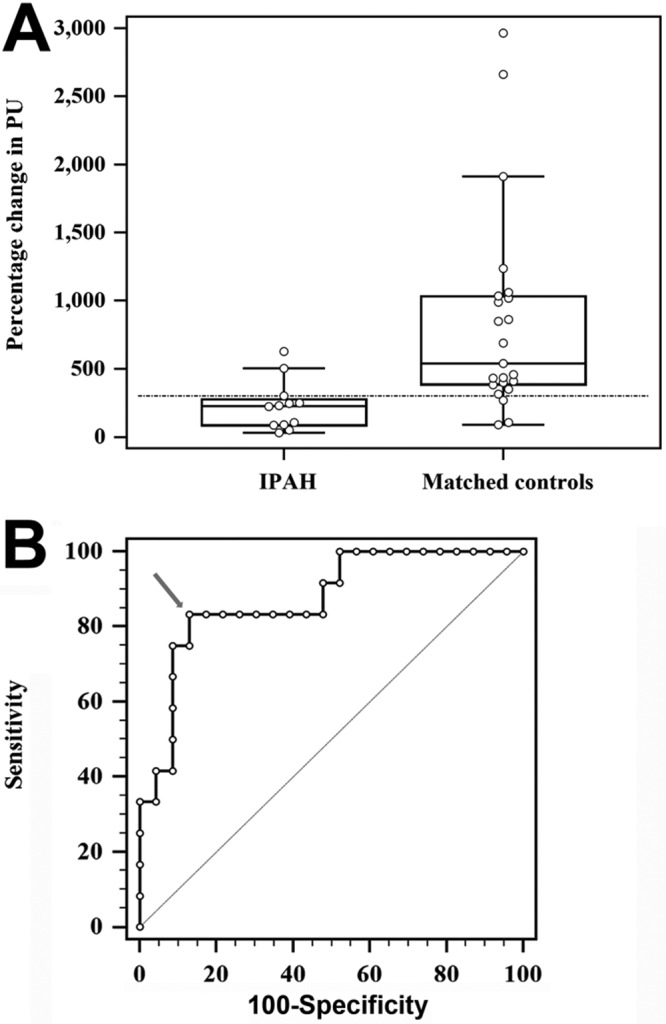
Peripheral vascular response to the iontophoresis of treprostinil in IPAH patients and controls. A, Box-and-whisker plot contrasting IPAH patients not receiving PGI2 analogs against controls. The horizontal line marks the 300% change in PUs obtained with the Youden index. B, The arrow marks the 300% change in PUs as the best cutoff to differentiate IPAH patients from controls. The area under the receiver operating characteristic curve is 0.87 (95% confidence interval: 0.72–0.96, P < 0.0001). IPAH: idiopathic pulmonary arterial hypertension; PGI2: prostacyclin; PUs: perfusion units.
Figure 3.
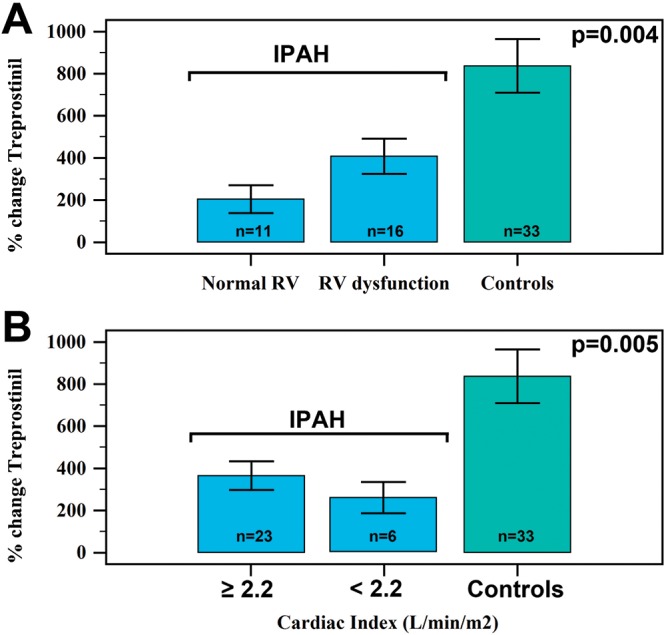
Treprostinil response (measured as percent change in perfusion units) based on right ventricular (RV) function (A) and cardiac index (B). IPAH: idiopathic pulmonary arterial hypertension patients.
Figure 4.
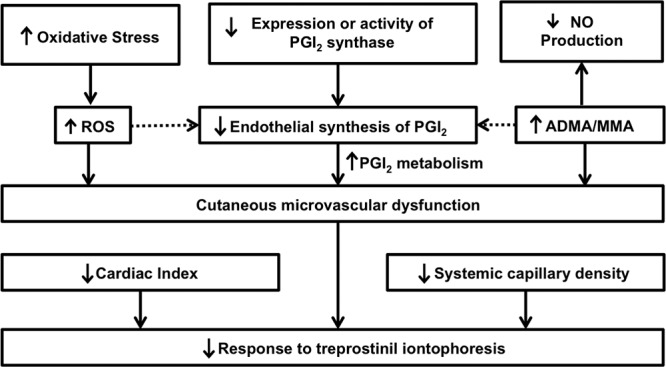
Mechanisms potentially involved in the lower response to treprostinil iontophoresis in idiopathic pulmonary arterial hypertension patients. ADMA: asymmetric dimethylarginine; MMA: N-monomethylarginine; NO: nitric oxide; PGI2: prostacyclin; ROS: reactive oxygen species.
Exhaled-breath analysis
Exhaled-breath analysis is a noninvasive process with significant clinical potential due to its ease of monitoring concentrations of substances previously difficult to analyze in adults and children with disease. Within the exhaled breath, volatile organic compounds (VOCs) are present.81,82 On the basis of the VOCs present, exhaled breath can be useful in generating a “breathprint” capable of distinguishing patients with diseases such as asthma, lung cancer, liver disease, and diabetes as well as other metabolic disorders. Interestingly, its use for clinical purposes has been practiced since ancient Greece, and more than 200 different compounds have been detected in human breath.83,84 Some have been documented for their potential relevance in human disease.85-88 This process has been made possible by nanosensor and nanodetector technology (i.e., the electronic nose) and mass spectrometry (e.g., gas chromatography–mass spectrometry, selected ion flow tube–mass spectrometry) that has fine-tuned the analysis of exhaled-breath VOCs at very low concentrations (typically around parts per billion). Using these tools, analysis of exhaled-breath VOCs has the potential to uncover alterations in multiple metabolic processes in an inexpensive, rapid, and noninvasive manner.
Exhaled-breath analysis in patients with PAH started by determining exhaled NO levels.89 NO is an important pathobiologic mediator of PH. In addition to being a vasodilator, NO is involved in endothelial cell proliferation and angiogenesis. Patients with PAH have low fractional exhaled NO (FeNO) values13 in exhaled breath and low amounts of NO reaction products in their lungs. Therapies that modulate the NO pathway are mainstays in PH treatment, including phosphodiesterase type 5 inhibitors and sGC stimulators. This suggests that FeNO levels may have utility in identifying patients who may benefit from the different medications that modulate different parts of the NO pathway.
Furthermore, the NO deficiency state in patients with PAH also improves with other therapies that do not necessarily directly target the NO pathway, such as prostacyclins and endothelin receptor antagonists. Interestingly, FeNO-level changes in response to therapy appear to have a prognostic significance in PAH. Higher FeNO levels after treatment are associated with improved survival in PAH.90 This suggests that serial monitoring of FeNO levels may be useful as a prognostic indicator in managing the disease.
More recently, we have been able to study the full spectrum of VOCs in the breath and not just single molecules such as NO. Like the headspace of the blood, our exhaled breath contains a vast array of substances and molecules that hold great promise for monitoring our health and for the diagnosis and management of various lung and systemic diseases.85,91,92 With continued advances in technology, essentially anything in the blood that is potentially volatile or has a volatile metabolite can be measured in exhaled breath. Sensor array (electronic nose) devices can be trained to recognize patterns, or “smellprints,” in exhaled breath that allow identification of certain diseases or disorders. However, this technology is not well suited to identify the specific compounds that contribute to a recognized pattern. The mass spectrometry approach to breath analysis, on the other hand, allows the identification of specific individual compounds in the breath, but it is not well suited to recognize patterns commonly seen in disease.
In our group, we have used both methods to analyze breath and have come to recognize the strengths and weaknesses of both approaches. More recently, we started to use an approach that combines the strengths of both methods. By approaching each compound (or peak) on the mass spectrometry output as its own sensor, we are able to recognize patterns, or “breathprints,” in mass spectrometry data in a way similar to how the sensor arrays (or electronic noses) recognize “smellprints.” Unlike pattern recognition by the sensor array–based systems, the major strength of our approach is that we are able to identify the individual components that contribute to each pattern we recognize. With this best-of-both-worlds approach, we are able to identify unique “breathprints” in patients with liver disease (fetor hepaticus) as well as those for heart and lung disease.85,88,93,94 We are further able to analyze these patterns to identify single molecules in the breath of these patients and link them to the underlying pathobiology of the disease.85,86,88,95,96
In a single-center cohort study of PAH patients, we correlated exhaled-breath VOCs with clinical parameters.85 In comparison to those in healthy controls, the concentrations of the VOCs (2-propanol, acetaldehyde, ammonia, ethanol, pentane, 1-decene, 1-octene, and 2-nonene) were altered in patients with PAH (Fig. 5). We found that exhaled-breath ammonia was higher in patients with PAH (median [interquartile range]: 94.7 [70–129] vs. 60.9 [46–77] ppb, P < 0.001) and was associated with elevated right atrial pressure (ρ = 0.57, P < 0.001), mean pulmonary artery pressure (ρ = 0.43, P = 0.015), cardiac index (ρ = −0.39, P = 0.03), pulmonary vascular resistance (ρ = 0.40, P = 0.04), mixed venous oxygen (ρ = −0.59, P < 0.001), and RV dilation (ρ = 0.42, P = 0.03). In addition, the PAH VOC “breathprint” classified 82.8% of the individuals in an independent validation cohort (n = 14), as determined by discriminant analysis. Consistent with this finding in PAH, we have shown that patients with acute decompensated heart failure have a distinct “breathprint,” as compared to healthy control subjects.94 These data, combined, indicate that breath analytes generate a disease “breathprint” distinctly different from that of healthy subjects. Future work will be focused on devising methods to phenotype the different types of PAH patients.
Figure 5.

Representative areas of the average selected ion flow tube–mass spectrometry for the pulmonary arterial hypertension (PAH) and control groups. All peaks are from the NO+ precursor ion spectrum. The concentrations of specific compounds (PAH in red and controls in blue) are calculated on the basis of counts relative to the precursor counts (details in Cikach et al.85).
One of the major challenges of using exhaled-breath analysis in medicine is that exhaled breath includes not only substances that we produce endogenously as part of our normal (or disease-related) metabolism but also substances from many other sources. Since we are constantly inhaling air from our environment, as we breathe in the ambient air, exhaled breath can also reflect our environmental exposure(s). Furthermore, our breath contains volatile compounds produced by our “internal environment”: the bacteria in our gut and mouth. In addition to the volatile by-products produced (from our diet, medications, drugs, or toxins), the analytical matrix generated from the VOCs can be very complex and difficult to sift through. Despite these challenges, the breath analysis has great potential to revolutionize and personalize our approach to medical testing.
Conclusion
PAH is the one of the best studied of PVDs and can have multiple etiologies and complex pathobiologic heterogeneity. Consequently, developing treatment options for PAH have been a daunting and slow process. Traditional approaches to evaluation and treatment offer little or no personalization. Novel methods are needed to identify new PH phenotypes that transcend the current WHO classification. This may allow us to develop newer precision medicine–based approaches to evaluation and therapy. We have provided some details about our current approach, which includes metabolic studies, examination of lung/systemic circulation, and exhaled-breath analysis, and how these have given us a better understanding of disease mechanisms. These approaches are paving the way to personalized medicine in treating PAH and other PVDs in the future.
Source of Support: This project was supported by the following grants from the National Institutes of Health: K99/R00 Pathway to Independence Award 1K99HL131866 to JWB, Mentored Patient-Oriented Research grant K23HL125697 to GAH, R01 grant 1R01HL130307 to ART, and R01 and Programs of Excellence in Glycosciences grants (1R01HL130209 and 1P01HL10714, respectively), both from the National Heart, Lung, and Blood Institute, to RAD. Other support was provided by a Center of Excellence in Pulmonary Vascular Disease Lerner Research Institutional grant to all the authors.
Conflict of Interest: None declared.
References
- 1.Butrous G, Ghofrani HA, Grimminger F. Pulmonary vascular disease in the developing world. Circulation 2008;118(17):1758–1766. [DOI] [PubMed]
- 2.Dweik RA, Rounds S, Erzurum SC, Archer S, Fagan K, Hassoun PM, Hill NS, et al. An official American Thoracic Society Statement: pulmonary hypertension phenotypes. Am J Respir Crit Care Med 2014;189(3):345–355. [DOI] [PMC free article] [PubMed]
- 3.Koress C, Swan K, Kadowitz P. Soluble guanylate cyclase stimulators and activators: novel therapies for pulmonary vascular disease or a different method of increasing cGMP? Curr Hypertens Rep 2016;18(5):42. doi:10.1007/s11906-016-0645-6. [DOI] [PubMed]
- 4.Elhwuegi AS. The wonders of phosphodiesterase-5 inhibitors: a majestic history. Ann Med Health Sci Res 2016;6(3):139–145. [DOI] [PMC free article] [PubMed]
- 5.Barnett CF, Alvarez P, Park MH. Pulmonary arterial hypertension: diagnosis and treatment. Cardiol Clin 2016;34(3):375–389. [DOI] [PubMed]
- 6.Hoeper MM, McLaughlin VV, Dalaan AM, Satoh T, Galiè N. Treatment of pulmonary hypertension. Lancet Respir Med 2016;4(4):323–336. [DOI] [PubMed]
- 7.Ghamra ZW, Dweik RA. Primary pulmonary hypertension: an overview of epidemiology and pathogenesis. Cleve Clin J Med 2003;70(suppl 1):S2–S8. [DOI] [PubMed]
- 8.Abenheim L, Moride Y, Rich S, Chaslerie A, Brenot F, Higenbottam T, Oakley C, et al. The International Primary Pulmonary Hypertension Study (IPPHS). Chest 1994;105(2 suppl):37S–41S. [DOI] [PubMed]
- 9.Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997;336(2):111–117. [DOI] [PubMed]
- 10.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Investig 2012;122(12):4306–4313. [DOI] [PMC free article] [PubMed]
- 11.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004;351(16):1655–1665. [DOI] [PubMed]
- 12.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43(12 suppl):S13–S24. [DOI] [PubMed]
- 13.Kaneko FT, Arroliga AC, Dweik RA, Comhair SA, Laskowski D, Oppedisano R, Thomassen MJ, Erzurum SC. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med 1998;158(3):917–923. [DOI] [PubMed]
- 14.Austin ED, Lahm T, West J, Tofovic SP, Johansen AK, Maclean MR, Alzoubi A, Oka M. Gender, sex hormones and pulmonary hypertension. Pulm Circ 2013;3(2):294–314. [DOI] [PMC free article] [PubMed]
- 15.Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D4–D12. [DOI] [PMC free article] [PubMed]
- 16.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 2007;104(4):1342–1347. [DOI] [PMC free article] [PubMed]
- 17.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res 2014;115(1):148–164. [DOI] [PubMed]
- 18.Dweik RA, Laskowski D, Özkan M, Farver C, Erzurum SC. High levels of exhaled nitric oxide (NO) and NO synthase III expression in lesional smooth muscle in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol 2001;24(4):414–418. [DOI] [PubMed]
- 19.Barnes JW, Kucera ET, Tian L, Mellor NE, Dvorina N, Baldwin WW III, Aldred MA, et al. Bone morphogenic protein type 2 mutation-independent mechanisms of disrupted bone morphogenetic protein signaling in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2016;55(4):564–575. [DOI] [PMC free article] [PubMed]
- 20.Barnes J, Dweik RA. Is pulmonary hypertension a metabolic disease? Am J Respir Crit Care Med 2014;190(9):973–975. [DOI] [PMC free article] [PubMed]
- 21.Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2008;178(6):558–564. [DOI] [PMC free article] [PubMed]
- 22.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl) 2013;91(10):1185–1197. [DOI] [PMC free article] [PubMed]
- 23.Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, Huertas A, Humbert M, Montani D. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev 2013;22(130):543–551. [DOI] [PMC free article] [PubMed]
- 24.Cottrill KA, Chan SY. Metabolic dysfunction in pulmonary hypertension: the expanding relevance of the Warburg effect. Eur J Clin Investig 2013;43(8):855–865. [DOI] [PMC free article] [PubMed]
- 25.Connelly MA, Gruppen EG, Otvos JD, Dullaart RP. Inflammatory glycoproteins in cardiometabolic disorders, autoimmune diseases and cancer. Clin Chim Acta 2016;459:177–186. [DOI] [PubMed]
- 26.Almeida A, Kolarich D. The promise of protein glycosylation for personalised medicine. Biochim Biophys Acta 2016;1860(8):1583–1595. [DOI] [PubMed]
- 27.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 1993;3(2):97–130. [DOI] [PMC free article] [PubMed]
- 28.Häuselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol 2014;4:28. doi:10.3389/fonc.2014.00028. [DOI] [PMC free article] [PubMed]
- 29.da Fonseca LM, da Silva VA, Freire-de-Lima L, Previato JO, Mendonça-Previato L, Capella MA. Glycosylation in cancer: interplay between multidrug resistance and epithelial-to-mesenchymal transition? Front Oncol 2016;6:158. doi:10.3389/fonc.2016.00158. [DOI] [PMC free article] [PubMed]
- 30.Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in cancer biology: linking metabolism and signaling. J Mol Biol 2016;428(16):3282–3294. [DOI] [PMC free article] [PubMed]
- 31.Kizuka Y, Taniguchi N. Enzymes for N-glycan branching and their genetic and nongenetic regulation in cancer. Biomolecules 2016;6(2):25. doi:10.3390/biom6020025. [DOI] [PMC free article] [PubMed]
- 32.Taparra K, Tran PT, Zachara NE. Hijacking the hexosamine biosynthetic pathway to promote EMT-mediated neoplastic phenotypes. Front Oncol 6:85. doi:10.3389/fonc.2016.00085. [DOI] [PMC free article] [PubMed]
- 33.Boon L, Ugarte-Berzal E, Vandooren J, Opdenakker G. Glycosylation of matrix metalloproteases and tissue inhibitors: present state, challenges and opportunities. Biochem J 2016;473(11):1471–1482. [DOI] [PMC free article] [PubMed]
- 34.Tolg C, McCarthy JB, Yazdani A, Turley EA. Hyaluronan and RHAMM in wound repair and the “cancerization” of stromal tissues. BioMed Res Int 2014;2014:103923. doi:10.1155/2014/103923. [DOI] [PMC free article] [PubMed]
- 35.Aytekin M, Comhair SA, de la Motte C, Bandyopadhyay SK, Farver CF, Hascall VC, Erzurum SC, Dweik RA. High levels of hyaluronan in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Biol 2008;295(5):L789–L799. [DOI] [PMC free article] [PubMed]
- 36.Lauer ME, Aytekin M, Comhair SA, Loftis J, Tian L, Farver CF, Hascall VC, Dweik RA. Modification of hyaluronan by heavy chains of inter-α-inhibitor in idiopathic pulmonary arterial hypertension. J Biol Chem 2014;289(10):6791–6798. [DOI] [PMC free article] [PubMed]
- 37.Barnes JW, Tian L, Heresi GA, Farver CF, Asosingh K, Comhair SA, Aulak KS, Dweik RA. O-linked β-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015;131(14):1260–1268. [DOI] [PMC free article] [PubMed]
- 38.Cai A, Li X, Zhong Q, Li M, Wang R, Liang Y, Chen W, et al. Associations of high HDL cholesterol level with all-cause mortality in patients with heart failure complicating coronary heart disease. Medicine (Baltimore) 2016;95(28):e3974. doi:10.1097/MD.0000000000003974. [DOI] [PMC free article] [PubMed]
- 39.Annema W, von Eckardstein A. Dysfunctional high-density lipoproteins in coronary heart disease: implications for diagnostics and therapy. Transl Res 2016;173:30–57. [DOI] [PubMed]
- 40.Holvoet P, De Keyzer D, Jacobs DR Jr. Oxidized LDL and the metabolic syndrome. Future Lipidol 2008;3(6):637–649. [DOI] [PMC free article] [PubMed]
- 41.Lam DW, LeRoith D. Metabolic syndrome. In: De Groot LJ, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, Korbonits M, et al., eds. Endotext. South Dartmouth, MA: MDText.com. 2000–. https://www.ncbi.nlm.nih.gov/books/NBK278936.
- 42.Hoenig MR, Sellke FW. Insulin resistance is associated with increased cholesterol synthesis, decreased cholesterol absorption and enhanced lipid response to statin therapy. Atherosclerosis 2010;211(1):260–265. [DOI] [PubMed]
- 43.Zhao QH, Peng FH, Wei H, He J, Chen FD, Di RM, Jiang X, et al. Serum high-density lipoprotein cholesterol levels as a prognostic indicator in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol 2012;110(3):433–439. [DOI] [PubMed]
- 44.Naderi N, Boobejame P, Bakhshandeh H, Amin A, Taghavi S, Maleki M. Insulin resistance in pulmonary arterial hypertension, is it a novel disease modifier? Res Cardiovasc Med 2014;3(3):e19710. doi:10.5812/cardiovascmed.19710. [DOI] [PMC free article] [PubMed]
- 45.Heresi GA, Aytekin M, Newman J, DiDonato J, Dweik RA. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med 2010;182(5):661–668. [DOI] [PMC free article] [PubMed]
- 46.Larsen CM, McCully RB, Murphy JG, Kushwaha SS, Frantz RP, Kane GC. Usefulness of high-density lipoprotein cholesterol to predict survival in pulmonary arterial hypertension. Am J Cardiol 2016;118(2):292–297. [DOI] [PubMed]
- 47.Cracowski JL, Labarère J, Renversez JC, Degano B, Chabot F, Humbert M. Plasma levels of high-density lipoprotein cholesterol are not associated with survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(1):107; author reply 107–108. [DOI] [PubMed]
- 48.Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 2015;5(2):269–278. [DOI] [PMC free article] [PubMed]
- 49.Brittain EL, Talati M, Fessel JP, Zhu H, Penner N, Calcutt MW, West JD, et al. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation 2016;133(20):1936–1944. [DOI] [PMC free article] [PubMed]
- 50.Ross DJ, Hough G, Hama S, Aboulhosn J, Belperio JA, Saggar R, Van Lenten BJ, et al. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm Circ 2015;5(4):640–648. [DOI] [PMC free article] [PubMed]
- 51.Sharma S, Umar S, Potus F, Iorga A, Wong G, Meriwether D, Breuils-Bonnet S, et al. Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 2014;130(9):776–785. [DOI] [PMC free article] [PubMed]
- 52.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator–activated receptor-γ activation. Circulation 2007;115(10):1275–1284. [DOI] [PubMed]
- 53.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Investig 2008;118(5):1846–1857. [DOI] [PMC free article] [PubMed]
- 54.Xie D, Bollag WB. Obesity, hypertension and aldosterone: is leptin the link? J Endocrinol 2016;230(1):F7–F11. [DOI] [PMC free article] [PubMed]
- 55.Ku IA, Farzaneh-Far R, Vittinghoff E, Zhang MH, Na B, Whooley MA. Association of low leptin with cardiovascular events and mortality in patients with stable coronary artery disease: the Heart and Soul Study. Atherosclerosis 2011;217(2):503–508. [DOI] [PMC free article] [PubMed]
- 56.Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med 2014;20(7):759–763. [DOI] [PMC free article] [PubMed]
- 57.Hall ME, Harmancey R, Stec DE. Lean heart: role of leptin in cardiac hypertrophy and metabolism. World J Cardiol 2015;7(9):511–524. [DOI] [PMC free article] [PubMed]
- 58.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev 2008;88(2):389–419. [DOI] [PMC free article] [PubMed]
- 59.Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, Fabre D, et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J 2012;40(4):895–904. [DOI] [PubMed]
- 60.Tonelli AR, Aytekin M, Feldstein AE, Dweik RA. Leptin levels predict survival in pulmonary arterial hypertension. Pulm Circ 2012;2(2):214–219. [DOI] [PMC free article] [PubMed]
- 61.Dweik RA, Erzurum SC. Effects of nitric oxide and cyclic GMP on smooth muscle proliferation. In: Moss J, ed. LAM and other diseases characterized by smooth muscle proliferation. New York: Dekker, 1999:333–349.
- 62.Smiljić S, Nestorović V, Savić S. Modulatory role of nitric oxide in cardiac performance. Med Pregl 2014;67(9–10):345–352. [PubMed]
- 63.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J 2012;33(7):829–837. [DOI] [PMC free article] [PubMed]
- 64.Cooper CJ, Landzberg MJ, Anderson TJ, Charbonneau F, Creager MA, Ganz P, Selwyn AP. Role of nitric oxide in the local regulation of pulmonary vascular resistance in humans. Circulation 1996;93(2):266–271. [DOI] [PubMed]
- 65.Gaston B, Drazen JM, Loscalzo J, Stamler JS. The biology of nitrogen oxides in the airways. Am J Respir Crit Care Med 1994;149(2):538–551. [DOI] [PubMed]
- 66.Lei J, Vodovotz Y, Tzeng E, Billiar TR. Nitric oxide, a protective molecule in the cardiovascular system. Nitric Oxide 2013;35:175–185. [DOI] [PubMed]
- 67.Fagan KA, Fouty BW, Tyler RC, Morris KG Jr., Hepler LK, Sato K, LeCras TD, et al. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Investig 1999;103(2):291–299. [DOI] [PMC free article] [PubMed]
- 68.Dababneh L, Cikach F, Alkukhun L, Dweik RA, Tonelli AR. Sublingual microcirculation in pulmonary arterial hypertension. Ann Am Thorac Soc 2014;11(4):504–512. [DOI] [PMC free article] [PubMed]
- 69.Tonelli AR, Alkukhun L, Cikach F, Ahmed M, Dweik RA. Are transcutaneous oxygen and carbon dioxide determinations of value in pulmonary arterial hypertension? Microcirculation 2015;22(4):249–256. [DOI] [PMC free article] [PubMed]
- 70.Tonelli AR, Ahmed MK, Alkukhun L, Cikach F, Aulak K, Dweik RA. Treprostinil iontophoresis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192(8):1014–1016. [DOI] [PMC free article] [PubMed]
- 71.Potus F, Malenfant S, Graydon C, Mainguy V, Tremblay È, Breuils-Bonnet S, Ribeiro F, et al. Impaired angiogenesis and peripheral muscle microcirculation loss contribute to exercise intolerance in pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;190(3):318–328. [DOI] [PubMed]
- 72.Peled N, Bendayan D, Shitrit D, Fox B, Yehoshua L, Kramer MR. Peripheral endothelial dysfunction in patients with pulmonary arterial hypertension. Respir Med 2008;102(12):1791–1796. [DOI] [PubMed]
- 73.Friedman D, Szmuszkovicz J, Rabai M, Detterich JA, Menteer J, Wood JC. Systemic endothelial dysfunction in children with idiopathic pulmonary arterial hypertension correlates with disease severity. J Heart Lung Transplant 2012;31(6):642–647. [DOI] [PMC free article] [PubMed]
- 74.Hofstee HM, Vonk Noordegraaf A, Voskuyl AE, Dijkmans BA, Postmus PE, Smulders YM, Serné EH. Nailfold capillary density is associated with the presence and severity of pulmonary arterial hypertension in systemic sclerosis. Ann Rheum Dis 2009;68(2):191–195. [DOI] [PubMed]
- 75.Restrepo RD, Hirst KR, Wittnebel L, Wettstein R. AARC clinical practice guideline: transcutaneous monitoring of carbon dioxide and oxygen: 2012. Respir Care 2012;57(11):1955–1962. [DOI] [PubMed]
- 76.Franzeck UK, Bollinger A, Huch R, Huch, A. Transcutaneous oxygen tension and capillary morphologic characteristics and density in patients with chronic venous incompetence. Circulation 1984;70(5):806–811. [DOI] [PubMed]
- 77.Stern MD. In vivo evaluation of microcirculation by coherent light scattering. Nature 1975;254(5495):56–58. [DOI] [PubMed]
- 78.Kruger A, Stewart J, Sahityani R, O’Riordan E, Thompson C, Adler S, Garrick R, Vallance P, Goligorsky MS. Laser Doppler flowmetry detection of endothelial dysfunction in end-stage renal disease patients: correlation with cardiovascular risk. Kidney Int 2006;70(1):157–164. [DOI] [PubMed]
- 79.Holowatz LA, Thompson-Torgerson CS, Kenney WL. The human cutaneous circulation as a model of generalized microvascular function. J Appl Physiol 2008;105(1):370–372. [DOI] [PubMed]
- 80.Khan F, Patterson D, Belch JJ, Hirata KK, Lang CC. Relationship between peripheral and coronary function using laser Doppler imaging and transthoracic echocardiography. Clin Sci 2008;115(9):295–300. [DOI] [PubMed]
- 81.Dweik RA, Amann A. Exhaled breath analysis: the new frontier in medical testing. J Breath Res 2008;2(3):030301. doi:10.1088/1752-7163/2/3/030301. [DOI] [PMC free article] [PubMed]
- 82.Mashir A, Dweik RA. Exhaled breath analysis: the new interface between medicine and engineering. Adv Powder Technol 2009;20(5):420–425. [DOI] [PMC free article] [PubMed]
- 83.Mathew TL, Pownraj P, Abdulla S, Pullithadathil B. Technologies for clinical diagnosis using expired human breath analysis. Diagnostics (Basel) 2015;5(1):27–60. [DOI] [PMC free article] [PubMed]
- 84.Cao W, Duan Y. Breath analysis: potential for clinical diagnosis and exposure assessment. Clin Chem 2006;52(2):800–811. [DOI] [PubMed]
- 85.Cikach FS Jr., Tonelli AR, Barnes J, Paschke K, Newman J, Grove D, Dababneh L, Wang S, Dweik RA. Breath analysis in pulmonary arterial hypertension. Chest 2014;145(3):551–558. [DOI] [PMC free article] [PubMed]
- 86.Alkhouri N, Cikach F, Eng K, Moses J, Patel N, Yan C, Hanouneh I, Grove D, Lopez R, Dweik R. Analysis of breath volatile organic compounds as a noninvasive tool to diagnose nonalcoholic fatty liver disease in children. Eur J Gastroenterol Hepatol 2014;26(1):82–87. [DOI] [PubMed]
- 87.Kurada S, Alkhouri N, Fiocchi C, Dweik R, Rieder F. Review article: breath analysis in inflammatory bowel diseases. Aliment Pharmacol Ther 2015;41(4):329–341. [DOI] [PubMed]
- 88.Patel N, Alkhouri N, Eng K, Cikach F, Mahajan L, Yan C, Grove D, Rome ES, Lopez R, Dweik RA. Metabolomic analysis of breath volatile organic compounds reveals unique breathprints in children with inflammatory bowel disease: a pilot study. Aliment Pharmacol Ther 2014;40(5):498–507. [DOI] [PMC free article] [PubMed]
- 89.Barnes PJ, Dweik RA, Gelb AF, Gibson PG, George SC, Grasemann H, Pavord ID, et al. Exhaled nitric oxide in pulmonary diseases: a comprehensive review. Chest 2010;138(3):682–692. [DOI] [PubMed]
- 90.Özkan M, Dweik RA, Laskowski D, Arroliga AC, Erzurum SC. High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy. Lung 2001;179(4):233–243. [DOI] [PubMed]
- 91.Dweik RA. The great challenge for exhaled breath analysis: embracing complexity, delivering simplicity. J Breath Res 2011;5(3):030201. doi:10.1088/1752-7155/5/3/030201. [DOI] [PMC free article] [PubMed]
- 92.Cikach FS Jr., Dweik RA. Cardiovascular biomarkers in exhaled breath. Prog Cardiovasc Dis 2012;55(1):34–43. [DOI] [PMC free article] [PubMed]
- 93.Alkhouri N, Eng K, Cikach F, Patel N, Yan C, Brindle A, Rome E, et al. Breathprints of childhood obesity: changes in volatile organic compounds in obese children compared with lean controls. Pediatr Obes 2015;10(1):23–29. [DOI] [PMC free article] [PubMed]
- 94.Samara MA, Tang WH, Cikach F Jr., Gul Z, Tranchito L, Paschke KM, Viterna J, Wu Y, Laskowski D, Dweik RA. Single exhaled breath metabolomic analysis identifies unique breathprint in patients with acute decompensated heart failure. J Am Coll Cardiol 2013;61(13):1463–1464. [DOI] [PMC free article] [PubMed]
- 95.Alkhouri N, Singh T, Alsabbagh E, Guirguis J, Chami T, Hanouneh I, Grove D, Lopez R, Dweik R. Isoprene in the exhaled breath is a novel biomarker for advanced fibrosis in patients with chronic liver disease: a pilot study. Clin Transl Gastroenterol 2015;6:e112. doi:10.1038/ctg.2015.40. [DOI] [PMC free article] [PubMed]
- 96.Eng K, Alkhouri N, Cikach F, Patel N, Yan C, Grove D, Lopez R, Rome E, Dweik RA. Analysis of breath volatile organic compounds in children with chronic liver disease compared to healthy controls. J Breath Res 2015;9(2):026002. doi:10.1088/1752-7155/9/2/026002. [DOI] [PubMed]