

Abstract
Interleukin‐1 (IL‐1) is implicated in numerous pathologies, including multiple sclerosis and its animal model experimental autoimmune encephalomyelitis (EAE). However, the exact mechanism by which IL‐1 is involved in the generation of pathogenic T cells and in disease development remains largely unknown. We found that following EAE induction, pertussis toxin administration leads to IL‐1 receptor type 1 (IL‐1R1)‐dependent IL‐1β expression by myeloid cells in the draining lymph nodes. This myeloid‐derived IL‐1β did not vitally contribute to the generation and plasticity of Th17 cells, but rather promoted the expansion of a GM‐CSF + Th17 cell subset, thereby enhancing its encephalitogenic potential. Lack of expansion of GM‐CSF‐producing Th17 cells led to ameliorated disease in mice deficient for IL‐1R1 specifically in T cells. Importantly, pathogenicity of IL‐1R1‐deficient T cells was fully restored by IL‐23 polarization and expansion in vitro. Therefore, our data demonstrate that IL‐1 functions as a mitogenic mediator of encephalitogenic Th17 cells rather than qualitative inducer of their generation.
Keywords: autoimmunity, EAE, GM‐CSF, IL‐1, Th17
Subject Categories: Immunology
Introduction
Differentiated CD4 T cells play an essential role in defense against pathogens and are culprits in autoimmune disorders. Their implication among other diseases is well documented in multiple sclerosis (MS), an autoimmune dysfunction affecting the human central nervous system (CNS), and in its animal model experimental autoimmune encephalomyelitis (EAE) (Croxford et al, 2011). CD4 T‐cell involvement in CNS autoimmunity was initially associated with T helper cells producing IFNγ (Th1 cells) (Ando et al, 1989). However, with the discovery of cells expressing IL‐17 (Th17 cells) (Harrington et al, 2005; Park et al, 2005), this paradigm was challenged. Th17 cells represent a heterogeneous population of cells expressing not only IL‐17 but also additional cytokines, depending on the conditions available during their differentiation. Thus, Th17 cells were found to co‐express IFNγ (Ivanov et al, 2006), GM‐CSF (Codarri et al, 2011; El‐Behi et al, 2011), IL‐21 (Korn et al, 2007), IL‐22 (Kreymborg et al, 2007), and even the suppressive cytokine IL‐10 (McGeachy et al, 2007; Ghoreschi et al, 2010). Moreover, while the impact of IL‐17 itself for EAE pathogenesis is still controversial (Haak et al, 2009; Ishigame et al, 2009), GM‐CSF is indispensable for disease development (Codarri et al, 2011; El‐Behi et al, 2011). In line with that, several recent reports indicated GM‐CSF as an essential effector molecule also in MS (Hartmann et al, 2014; Noster et al, 2014; Li et al, 2015).
Th17 cell differentiation from naïve T cells can be initiated by TGFβ, the cytokine that is shared with the T regulatory (Treg) cell developmental program. However, Th17 cell commitment occurs only after subsequent inhibition of the Treg‐specific transcription factor FoxP3 by IL‐6 (Bettelli et al, 2006; Veldhoen et al, 2006) or IL‐21 (Korn et al, 2007). Th17 cells generated only in the presence of TGFβ1 and IL‐6 cannot transmit EAE (McGeachy et al, 2007) and require additional exposure to IL‐23 to gain pathogenic characteristics (Lee et al, 2012). Another pathway of Th17 cell development is independent of TGFβ and driven by a combination of IL‐6, IL‐23, and IL‐1β (Chung et al, 2009; Ghoreschi et al, 2010). Cells generated under these conditions express high levels of GM‐CSF (El‐Behi et al, 2011) and are highly pathogenic in EAE. Of note, the individual ablation of each aforementioned cytokine‐mediated signaling results in complete resistance of mice to EAE induction (Eugster et al, 1998; Cua et al, 2003; Matsuki et al, 2006; Sutton et al, 2006). Whereas deletion of IL‐6 prevented Th17 cell differentiation (Korn et al, 2007), IL‐23 showed a greater impact on expansion and stabilization of the Th17 cell phenotype (Langrish et al, 2005; Veldhoen et al, 2006). The contribution of IL‐1 signaling to the pathogenicity of T cells is still controversial with evidences supporting a role in both de novo generation and expansion of Th17 cells (Sutton et al, 2006; Chung et al, 2009; El‐Behi et al, 2011). The mechanism by which IL‐1 mediates pathogenicity in vivo could not yet be fully addressed, mainly due to the lack of suitable genetic tools, namely the conditional knockout of the IL‐1 receptor. There is only one known signaling receptor—IL‐1 receptor type 1 (IL‐1R1)—that is, however, broadly expressed by many cell types of immune and non‐immune origin (Boraschi & Tagliabue, 2013).
The induction of active EAE is achieved by the immunization with myelin oligodendrocyte glycoprotein (MOG), emulsified in complete Freund's adjuvant (CFA) and injections of pertussis toxin (PTx) (Mendel et al, 1995). PTx is commonly used in many autoimmune models to enhance disease susceptibility; however, its mode of action is not fully understood. In EAE, PTx application is associated with transient Treg cell suppression (Cassan et al, 2006; Chen et al, 2006), promotion of Th17 cell development (Chen et al, 2007), and contribution to blood–brain barrier disruption (Millward et al, 2007). On the other hand, PTx is also able to block leukocyte migration (Su et al, 2001), demonstrating its complex and diverse involvement in disease progression. Despite these proposed effects of PTx, many of them most likely are secondary events with largely unknown key intermediate factor(s). Importantly, PTx also was shown to stimulate expression and inflammasome‐mediated maturation of IL‐1β in myeloid cells (Dumas et al, 2014), suggesting that IL‐1β could be a potential candidate molecule through which PTx would, at least partially, promote EAE development.
In the present study, we aimed to investigate the requirement of IL‐1 signaling for T‐cell‐mediated autoimmunity. By using novel models of conditional IL‐1R1 deletion, we demonstrate that PTx treatment stimulates recruitment and expansion of myeloid cells expressing IL‐1β in the draining lymph nodes (dLN), in an IL‐1R1‐dependent manner. This myeloid cell‐derived IL‐1 stimulated expansion of GM‐CSF+ Th17 cells. The latter cells also developed in the absence of IL‐1 signaling but did not proliferate properly and therefore could not cause severe EAE. However, isolation and expansion of T cells devoid of IL‐1 signaling in the presence of IL‐23 resulted in recovery of pathogenicity. Thus, we could show that IL‐1 signaling in CD4 T cells is critical for their expansion, but not for differentiation to encephalitogenic Th17 cells.
Results
PTx administration promotes IL‐1β expression by myeloid cells during the induction phase of EAE in an IL‐1R1‐dependent manner
Recently, we generated a new mouse line that allows for the conditional deletion of the IL‐1 receptor type 1 (Abdulaal et al, 2016), the obligatory receptor for IL‐1α‐ and IL‐1β‐mediated signaling. By deleting the receptor in the germ line, using CMV‐Cre, we obtained mice that globally lack IL‐1R1 (hereafter termed IL‐1R1−/−). These mice differ from previously reported full knockout mice by the deletion of exon 5, which results in inactivation of all known isoforms for IL‐1R1 (Glaccum et al, 1997; Labow et al, 1997; Qian et al, 2012). As a result of complete inactivation of IL‐1 signaling, IL‐1R1−/− mice displayed impaired development of CD11b+ cells in secondary lymphoid organs under steady state conditions, including myeloid cells producing IL‐1β, as compared to wild‐type (WT) controls (Fig 1A–C).
Figure 1. Pertussis toxin induces IL‐1β‐expressing myeloid cells after MOG/CFA immunization dependent on IL‐1R1 signaling.
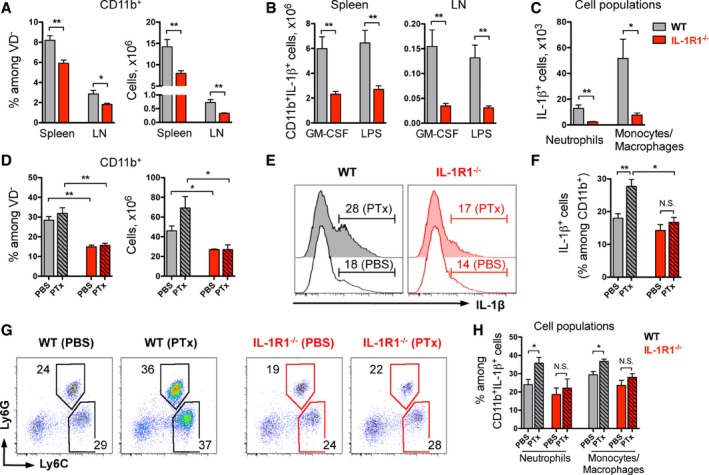
- Frequencies and total numbers (mean + SEM) of CD11b+ myeloid cells isolated from the spleen and peripheral LN of naïve non‐immunized mice.
- Total numbers (mean + SEM) of CD11b+ cells expressing IL‐1β shown in (A) and restimulated with indicated stimuli for 4 h.
- Quantification (mean + SEM) of myeloid cell populations expressing IL‐1β isolated from LN and restimulated with GM‐CSF shown in (B).
- Frequencies and total numbers (mean + SEM) of CD11b+ myeloid cells isolated from the spleen of immunized mice.
- Analysis of IL‐1β expression by CD11b+ cells isolated from dLN of mice treated as in (D). Data are representative FACS histogram overlays, gated on VD−CD11b+ cells with mean frequencies per group.
- Quantification (mean + SEM) of IL‐1β expression by CD11b+ cells shown in (E).
- Analysis of IL‐1β‐expressing CD11b+ cells shown in (E). Data are representative FACS plots, gated on VD−CD11b+IL‐1β+ cells with mean frequencies per group.
- Quantification (mean + SEM) of myeloid cell populations expressing IL‐1β shown in (G).
Experimental autoimmune encephalomyelitis can be induced in mice by immunization with the MOG peptide p35‐55 emulsified in CFA, along with PTx injections. To study the role of IL‐1 signaling in EAE induction, we immunized mice with MOG/CFA with or without addition of PTx. We found dramatically reduced frequencies and numbers of CD11b+ myeloid cells in the spleen of IL‐1R1‐deficient animals compared to wild‐type controls (Fig 1D). These experiments suggest that IL‐1 signaling might be involved in the recruitment of myeloid cells to the secondary lymphoid organs and their subsequent expansion after immunization.
To investigate whether mobilized myeloid cells not only respond to IL‐1 but also enhance the immune response by producing this cytokine, we measured the expression of IL‐1β upon immunization. Following stimulation with GM‐CSF, LPS, or PMA/ionomycin to trigger ex vivo isolated cells, we found that the vast majority of IL‐1β originated from CD11b+ cells (Fig EV1). Moreover, we noted a robust enhancement of IL‐1β expression by myeloid cells when WT animals were additionally treated with PTx, an effect that was completely absent in IL‐1R1‐deficient animals (Figs 1E and F, and EV1). Further analysis of the myeloid cell populations revealed that treatment of the mice with PTx resulted in increased frequencies of neutrophils and monocytes/macrophages among the cells expressing IL‐1β in the WT group, whereas it had a very limited effect on the same cell populations in IL‐1R1−/− mice (Fig 1G and H). In contrast to IL‐1β, the expression of IL‐1α in myeloid cells was not affected by PTx treatment (Fig EV2). However, in line with the IL‐1β data, IL‐1α‐expressing CD11b+ cells were dramatically reduced in mice deficient for IL‐1R1 (Fig EV2).
Figure EV1. Myeloid cells are the main source of IL‐1β upon MOG/CFA/PTx immunization.
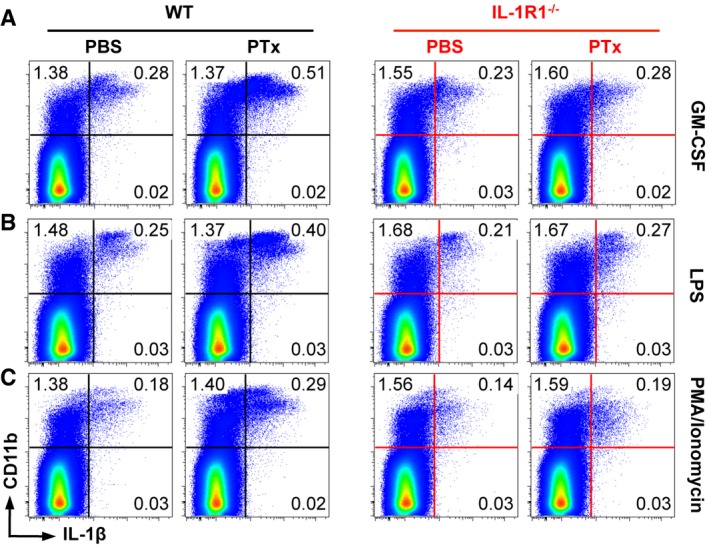
-
A–CAnalysis of IL‐1β expression by cells isolated from the dLN and stimulated with GM‐CSF (A), LPS (B), and PMA/ionomycin (C). Data are representative FACS plots gated on VD− cells with mean frequencies per group.
Figure EV2. Myeloid cells are the main source of IL‐1α upon MOG/CFA/PTx immunization.
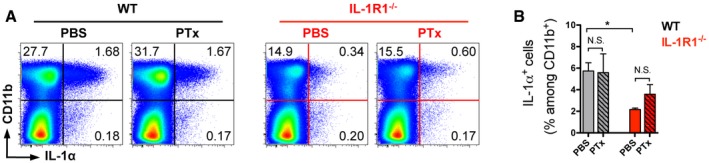
- Analysis of IL‐1α expression by cells isolated from the spleen and stimulated with GM‐CSF. Data are representative FACS plots, gated on VD− cells with mean frequencies per group.
- Frequencies (mean + SEM) of IL‐1α expression by CD11b+ cells shown in (A).
Collectively, these data show that PTx activates myeloid cells to produce IL‐1β in an IL‐1R1‐dependent manner after MOG/CFA immunization. In addition, our data suggest that IL‐1β production by myeloid cells is dependent on an autocrine stimulatory feedback loop.
Mouse and human Th17 cells express high levels of IL‐1R1
Development of EAE requires the generation of pathogenic CD4 T cells that initiate a coordinated immune attack of the CNS. To study the role of IL‐1 signaling in the development of a T‐cell‐mediated immune response, we investigated the expression of IL‐1R1 by different populations of CD4 T cells following immunization with MOG/CFA/PTx (Appendix Fig S1). We found that Th17 cells expressed the highest levels of IL‐1R1 compared to conventional Th1 or GM‐CSF‐producing T cells in both the dLN and the inflamed CNS of EAE‐diseased mice (Fig 2A and C). We also noted that FoxP3+ Treg cells hardly expressed any IL‐1R1 (Fig 2A and C). Importantly, IL‐17A‐expressing cells regardless of their IFNγ and/or GM‐CSF co‐expression equally displayed high levels of IL‐1R1 (Fig 2B and D).
Figure 2. IL‐1R1 expression by murine and human Th17 cells.
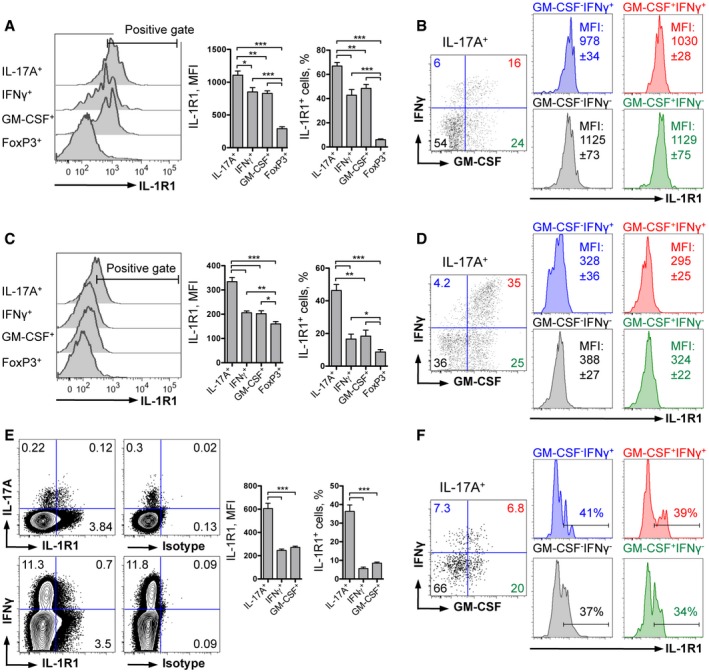
-
A–DAnalysis of IL‐1R1 expression by different populations of murine (A) CD4 T cells and (B) Th17 cells isolated from the dLN 9 days after immunization, and (C) CD4 T cells and (D) Th17 cells isolated from the CNS at the peak of EAE.
-
E, FAnalysis of IL‐1R1 expression by different populations of human (E) CD4 T cells and (F) Th17 cells isolated from the peripheral blood.
Also in humans, CD4 T‐cell development was shown to be dependent on IL‐1 signaling (Acosta‐Rodriguez et al, 2007; Zielinski et al, 2012). Although mouse models represent useful tools in investigating human pathologies, possible differences in cell biology can result in misleading translation of the findings to the clinic. In line with our mouse data, we found that a large proportion of human Th17 cells also expressed IL‐1R1, but very few Th1‐ and GM‐CSF‐positive cells do so (Fig 2E). When we further analyzed the different subpopulations of IL‐17A‐producing human CD4 T cells, we found that all of them, regardless of co‐expression of IFNγ and/or GM‐CSF, expressed similar levels of IL‐1R1 (Fig 2E and F). We conclude that both mouse and human subpopulations of Th17 cells, including the most autoaggressive GM‐CSF+ Th17 cells, can potentially respond to IL‐1 and therefore could be dependent on IL‐1 signaling.
PTx enhances the development of MOG‐specific GM‐CSF+ Th17 cells in an IL‐1R1‐dependent manner
To investigate the role of PTx‐induced IL‐1β expression on the development of autoreactive T cells, we compared cytokine production by MOG‐specific CD4 T cells generated after MOG/CFA immunization of mice with or without PTx injections. To visualize antigen‐specific T cells, we re‐activated spleen‐ and dLN‐derived lymphocytes with MOG peptide for 6 h and addressed the expression of CD40 ligand (CD40L) together with the cytokine staining. CD40L is transiently expressed by T cells after their activation and can be used to identify antigen‐specific T cells (Chattopadhyay et al, 2005; Frentsch et al, 2005). In WT mice, addition of PTx to the immunization protocol doubled numbers of MOG‐reactive Th17 cells (Fig 3A and B). This effect of PTx was strongly dependent on IL‐1R1, since we did not observe it in mice lacking the IL‐1 receptor. Instead, IL‐1R1−/− mice showed a dramatic reduction of antigen‐specific Th17 cells compared to WT animals, regardless of the addition of PTx (Fig 3A and B). As a negative control, we restimulated cells with the non‐relevant OVA peptide and could not detect OVA‐specific cells neither in WT nor in IL‐1R1‐deficient animals (Fig EV3).
Figure 3. GM‐CSF‐expressing Th17 cells are expanded by PTx in response to IL‐1 signaling.
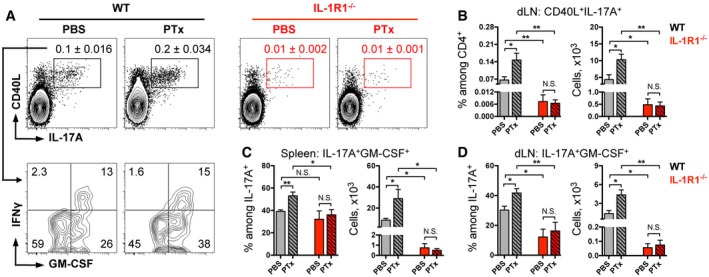
-
AAnalysis of cytokine expression by CD4 T cells isolated from the spleen. Data are representative FACS plots, gated on VD−TCRβ+CD4+CD44+ cells (upper row) with mean frequencies among CD4 T cells per group ± SEM and (lower row) with mean frequencies per group.
-
BFrequencies and total numbers (mean + SEM) of MOG‐specific Th17 cells isolated from the dLN.
-
C, DFrequencies and total numbers (mean + SEM) of MOG‐specific GM‐CSF+ Th17 cells isolated from the spleen (C) and dLN (D).
Figure EV3. IL‐1R1 deletion impairs MOG‐specific Th17 cell expansion.
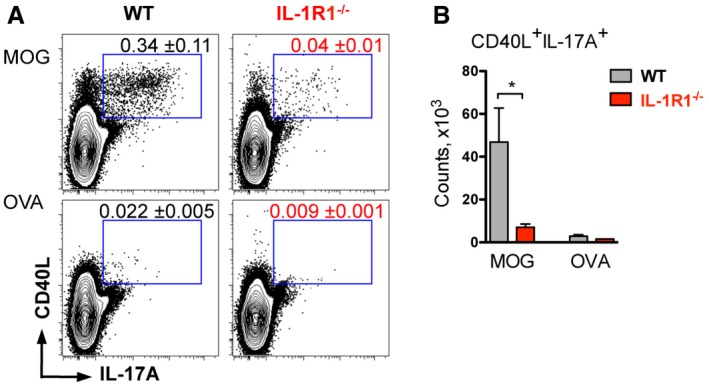
- Analysis of IL‐17A expression by CD4 T cells isolated from the spleen and restimulated with MOG or OVA for 6 h.
- Total cell numbers of antigen‐specific Th17 cells isolated from the spleen shown in (A).
Importantly, the stimulatory effect of PTx observed in WT mice was not restricted to Th17 cells but was also seen in CD4 T cells expressing IFNγ, GM‐CSF, or both (Fig EV4). These data are in agreement with our findings that IL‐1R1−/− mice displayed very limited numbers of such cells regardless of PTx administration (Fig EV4). Previously, our group and others have shown that in the context of EAE, IFNγ‐, and GM‐CSF‐positive cells are mainly (or even exclusively) originating from Th17 cells (Kurschus et al, 2010; Hirota et al, 2011; Brucklacher‐Waldert et al, 2016). Taken together, we conclude that deficiency in IL‐1 signaling (either by IL‐1R1 deletion or absence of PTx during immunization) results in reduced Th17 cells and, as expected, their IFNγ‐ and GM‐CSF‐expressing progenies, during priming of EAE.
Figure EV4. IL‐17A‐related cytokine expression by CD4 T cells is dependent on IL‐1R1 signaling.
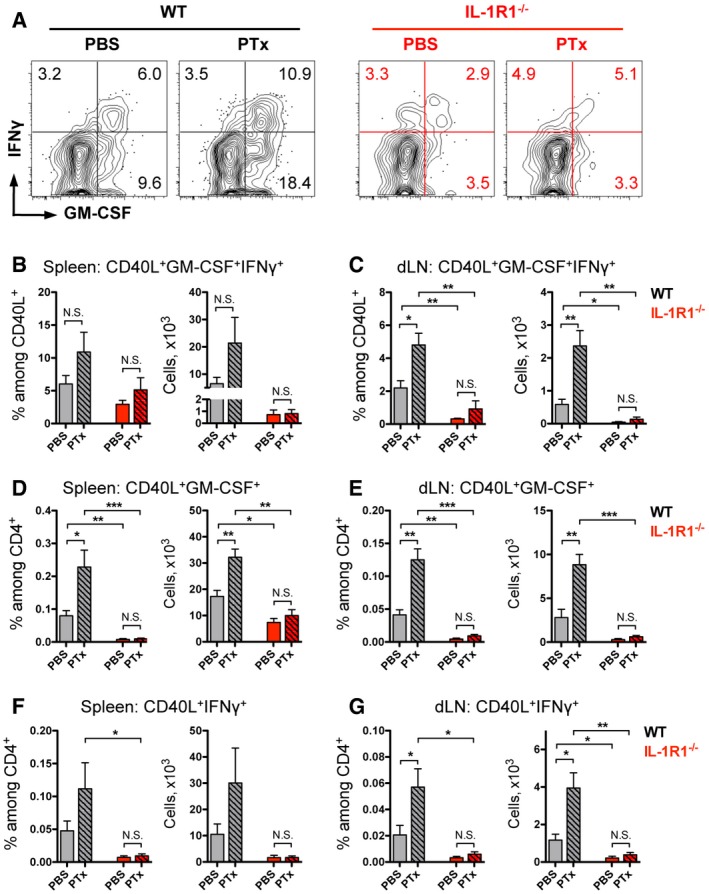
-
A–GAnalysis of cytokine expression by CD4 T cells isolated from the (A, B, D, F) spleen and (C, E, G) dLN (depicted in Fig 3). Frequencies and total numbers of MOG‐specific (B, C) GM‐CSF+IFNγ+ cells, (D, E) GM‐CSF+ cells, and (F, G) IFNγ+ cells. Data (A) are representative FACS plots, gated on VD−TCRβ+CD4+CD44+CD40L+ cells with mean frequencies among CD44+CD40L+ cells per group, and (B–G) bar diagram (mean + SEM).
As mentioned above, following EAE induction, Th17 cells go through a process of differentiation that results in the co‐expression of IFNγ and GM‐CSF, which are critical for the pathogenicity of CD4 T cells (Kurschus et al, 2010; Codarri et al, 2011; El‐Behi et al, 2011; Hirota et al, 2011). Therefore, we further analyzed antigen‐specific IL‐17A‐producing cells for the production of IFNγ and GM‐CSF. In WT mice, we found that frequencies and numbers of MOG‐specific Th17 cells co‐expressing GM‐CSF were significantly higher when mice were injected with PTx compared to PBS‐treated animals (Fig 3A, C and D). The vast majority of IFNγ‐positive Th17 cells also expressed GM‐CSF, while IFNγ single positive Th17 cells represented only a minor subpopulation regardless of the PTx administration (Fig 3A). Importantly, the stimulatory effect of PTx was not observed in mice deficient for IL‐1R1, in which autoreactive IL‐17A+ cells displayed impaired co‐expression of GM‐CSF in both the spleen and dLN (Fig 3C and D). Collectively, these findings suggest that PTx enhances the generation of pathogenic Th17 cells and particularly its GM‐CSF‐positive subset, by promoting their expansion via IL‐1 signaling.
Regulatory T cells are generated early after EAE induction (O'Connor & Anderton, 2008; Yogev et al, 2012), and their development was shown to be transiently suppressed by PTx (Cassan et al, 2006; Chen et al, 2006). However, we did not observe any effects of PTx administration or IL‐1R1 deletion on the numbers of FoxP3+ CD4 T cells in dLN and spleen when mice were analyzed 9 days after immunization (Appendix Fig S2A–C). Moreover, expression levels of Treg co‐stimulatory molecules GITR, ICOS, and CTLA‐4 were also unchanged in all groups of animals (Appendix Fig S2D and E). We conclude that neither IL‐1 signaling, nor PTx administration is critically influencing Treg cell development following EAE induction.
IL‐1 signaling in T cells is essential for expansion of antigen‐specific GM‐CSF+ Th17 cells
To investigate the exact role of IL‐1 in the development of autoreactive T cells and their pathogenicity, we specifically deleted the IL‐1 receptor type 1 in αβ‐TCR+ T cells by crossing conditional IL‐1R1fl/fl mice to CD4‐Cre mice, resulting in IL‐1R1∆T mice. These mice developed normally and did not display abnormalities in the immune system when kept under SPF conditions. However, CD4 T cells isolated from mutant mice failed to proliferate in response to IL‐1β administration in vitro (Mufazalov et al, 2016). Previously, it was reported that IL‐1R1 fully deficient mice show an impaired CD4 T‐cell‐mediated antigen response and that these mice are resistant to induction of EAE (Matsuki et al, 2006; Sutton et al, 2006; Chung et al, 2009). To test whether EAE resistance specifically stems from the lack of T‐cell responses to IL‐1, we immunized WT, IL‐1R1−/−, and IL‐1R1∆T mice with MOG/CFA along with two injections of PTx and investigated priming of CD4 T cells 9 days postimmunization. As expected, we could not detect IL‐1R1 on the mutant T cells isolated from either IL‐1R1∆T or IL‐1R1−/− mice (Fig 4A). We observed that not only global deletion of IL‐1R1, but also T‐cell‐specific deletion of the receptor resulted in a reduction of IL‐17A‐expressing T cells (Fig 4A).
Figure 4. T‐cell‐specific deletion of IL‐1R1 impairs Th17 cell expansion.
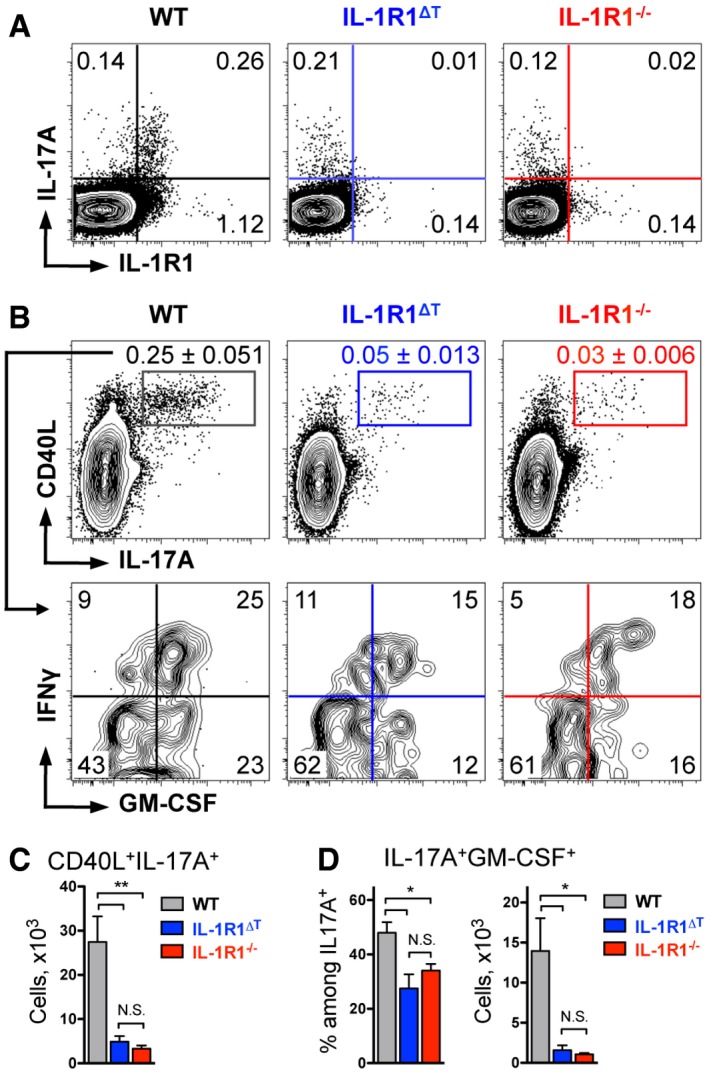
- Analysis of CD4 T cells isolated from the dLN and restimulated with PMA/ionomycin for 4 h. Data are representative FACS plots, gated on VD−CD4+ cells with mean frequencies per group.
- Analysis of cytokine expression by CD4 T cells isolated from the spleen and restimulated with MOG for 6 h. Data are representative FACS plots, gated on VD−TCRβ+CD4+CD44+ cells (upper row) with mean frequencies among CD4 T cells per group ± SEM and (lower row) with mean frequencies per group.
- Total cell numbers (mean + SEM) of MOG‐specific Th17 cells isolated from the spleen shown in (B, upper row).
- Frequencies and total numbers (mean + SEM) of MOG‐specific GM‐CSF+ Th17 cells isolated from the spleen shown in (B, lower row).
When we further analyzed antigen‐specific CD4 T cells, we found that germ line or T‐cell‐specific deletion of IL‐1R1 resulted in a similar decrease of autoreactive CD4 T cells compared to WT controls (Appendix Fig S3). Moreover, frequencies and numbers of MOG‐specific Th17 cells and Th17 cells co‐expressing GM‐CSF were equally reduced in both groups of IL‐1R1‐deficient animals (Fig 4B–D), suggesting that this is a T‐cell‐intrinsic phenomenon, since antigen‐presenting cells should not be targeted by CD4‐Cre‐mediated recombination. Importantly, although numbers of GM‐CSF+ Th17 cells were significantly reduced in both groups of mutant mice compared to WT controls, they were still detectable (Fig 4B and D). This observation is in favor of impaired proliferation or survival, rather than differentiation failure, of MOG‐specific Th17 cells devoid of IL‐1 signaling.
Development of neuroinflammation is dependent on a T‐cell‐intrinsic response to IL‐1
To investigate the consequences of impaired Th17 cell development for autoimmunity, we immunized IL‐1R1‐deficient mice with MOG/CFA along with PTx injections and followed the progression of EAE. We found that T‐cell‐specific deletion of IL‐1R1 resulted in a mild disease, which was nevertheless aggravated compared to mice fully deficient for IL‐1 signaling (Fig 5A–C). Of note, mice with global IL‐1R1 deletion were resistant to EAE over 28 days of examination, recapitulating the reported phenotype of previously published IL‐1R1‐deficient mice (Matsuki et al, 2006; Sutton et al, 2006).
Figure 5. Mice with T‐cell‐specific deletion of IL‐1R1 are only partially resistant to EAE induction.
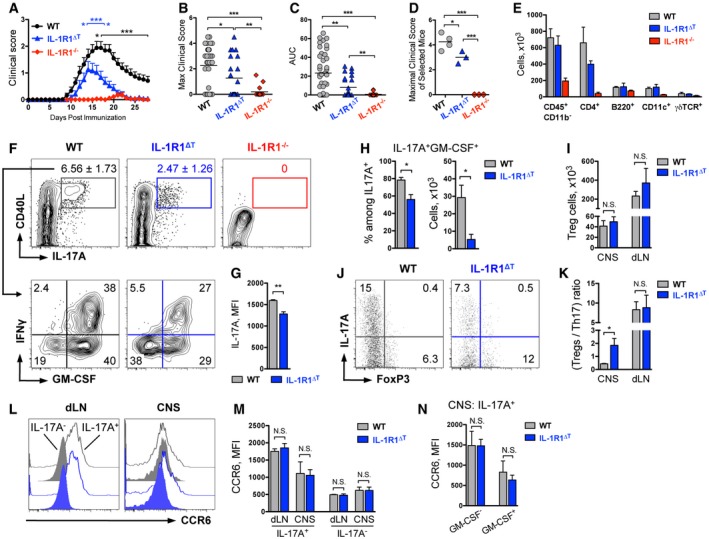
-
A–C(A) Mean, (B) maximal EAE clinical scores, and (C) area under the curve (AUC) of mice immunized with MOG/CFA/PTx. Data shown as (A) mean (+ SEM) and (B, C) individual plots with mean and are combined from at least four independent experiments and consist of n = 41 WT, n = 24 IL‐1R1ΔT, n = 18 IL‐1R1−/− mice. *P < 0.05, ***P < 0.001; two‐tailed unpaired t‐test. Black asterisks in (A) represent statistical analysis of IL‐1R1ΔT vs WT and blue asterisks of IL‐1R1ΔT vs IL‐1R1−/− groups.
-
D, E(D) Maximal EAE clinical scores (individual plots with mean) of selected mice and (E) total cell numbers (mean + SEM) of infiltrates isolated from CNS and used for further analysis.
-
F–HAnalysis of cytokine expression by CD4 T cells isolated from the CNS of mice shown in (D). Data (F) are representative FACS plots, gated on VD−CD90+CD4+CD44+ cells (upper row) with mean frequencies among CD4 T cells per group ± SEM and (lower row) with mean frequencies per group. (G) Mean fluorescent intensity (mean + SEM) of IL‐17A staining by MOG‐specific Th17 cells shown in (F, upper row). (H) Quantification of MOG‐specific GM‐CSF+ Th17 cells (mean + SEM) shown in (F, lower row).
-
I–KAnalysis of Treg cells isolated from mice shown in (D). (I) Total Treg cell numbers (mean + SEM) isolated from CNS and dLN. (J) Ratio of Treg and Th17 cells isolated from the CNS. Data are representative FACS plots, gated on VD−CD90+CD4+ cells with mean frequencies per group. (K) Quantification of Treg/Th17 cell ratio (mean + SEM) within CNS and dLN.
-
L–NAnalysis of CCR6 expression by CD4 T isolated from the dLN and CNS of EAE‐diseased mice. Data (L) are representative FACS histogram overlays of (black) control, (blue) IL‐1R1ΔT cells, gated on CD3+CD4+IL‐17A+ cells; and of (black filled) control, (blue filled) IL‐1R1ΔT cells, gated on CD3+CD4+IL‐17A− cells. (M) Mean fluorescent intensity (mean + SEM) of CCR6 staining by CD4 T cells shown in (L). (N) Mean fluorescent intensity (mean + SEM) of CCR6 staining by GM‐CSF+ and GM‐CSF− Th17 cells isolated from CNS shown in (L).
To investigate which cell types are responsible for the (mild) EAE symptoms seen in the IL‐1R1∆T mice, we analyzed immune cell populations in the secondary lymphoid organs and the CNS infiltrates of diseased animals depicted in Fig 5D. Sixteen days after immunization, when WT mice were at the peak of disease, we found no differences in T‐cell numbers in the spleen of immunized animals regardless of the genotype (Appendix Fig S4). However, when we analyzed the CNS samples, we found that IL‐1R1∆T mice had similar numbers of infiltrating leukocytes but a reduction of CD4 T cells compared to WT controls, while the mice with complete IL‐1R1 deficiency had almost no cell infiltrates in the CNS (Fig 5E). Furthermore, when compared to WT mice, we found a strong reduction in antigen‐specific Th17 cells in the CNS of IL‐1R1∆T animals and a complete absence of these cells in IL‐1R1−/− mice (Fig 5F). The presence of autoreactive Th17 cells in diseased IL‐1R1∆T mice, although in reduced numbers, suggests that also without IL‐1 signaling, encephalitogenic T cells can develop, as long as other cells in their environment are able to respond to IL‐1. In addition to reduced Th17 cell numbers, also the mean fluorescence intensity for IL‐17A staining was significantly reduced in cells isolated from IL‐1R1∆T animals, suggesting impaired IL‐17A expression on the single cell level (Fig 5G). When antigen‐specific Th17 cells were further analyzed, we found reduced frequencies and numbers of GM‐CSF+ IL‐17A+ cells in the IL‐1R1∆T mice (Fig 5F and H), correlating with the reduced disease severity of these mice.
In contrast to the effect on the development of pathogenic Th17 cells, T‐cell‐specific deletion of IL‐1R1 had no impact on the development of regulatory T cells (Fig 5I). Therefore, the ratio of FoxP3+ cells to Th17 cells shifted toward Treg cells in the CNS of IL‐1R1∆T mice (Fig 5J and K), reflecting impaired cytokine production by mutant cells. We conclude that IL‐1 signaling is not critical for Treg cell homeostasis.
Migration of inflammatory Th17 cells from the secondary lymphoid organs to the CNS was shown to be dependent on the expression of the chemokine receptor CCR6 (Reboldi et al, 2009; Comerford et al, 2010). To test whether the reduction in EAE severity observed in IL‐1R1∆T mice stems from altered trafficking of IL‐1R1‐deficient Th17 cells, we analyzed CCR6 expression in the dLN and CNS of EAE‐diseased mice. We found no differences in the expression of this chemokine receptor by IL‐1R1‐sufficient and IL‐1R1‐deficient Th17 cells (Fig 5L–N), suggesting normal CCR6‐dependant migratory capacities of IL‐1R1‐deficient T cells. Taken together, our data demonstrate that encephalitogenic T cells can develop in the absence of IL‐1 signaling, although in much lower numbers, which ultimately leads to the reduced EAE severity in IL‐1R1∆T mice.
Ex vivo expansion of Th17 cells in the presence of IL‐23 restores the pathogenic potential of IL‐1R1‐deficient T cells
To study the role of IL‐1 signaling in expansion of MOG‐specific Th17 cells, we isolated cells from MOG/CFA‐immunized WT mice and cultured them in the presence of MOG peptide and anti‐IFNγ. We detected a dramatic increase in the frequencies and numbers of Th17 cells in cultures supplemented with IL‐1β compared to cytokine‐free conditions (Fig 6A). Apart from IL‐1β, also IL‐23 was shown to play a critical role in the establishment of T‐cell‐mediated pathogenicity (Cua et al, 2003). When we added IL‐23 to the cultures, we observed an even stronger stimulatory effect for the expansion of Th17 cells, with the greatest outcome when both IL‐1β and IL‐23 were combined together (Fig 6A). Accordingly, numbers of GM‐CSF+, as well as GM‐CSF+IFNγ+ Th17 cells, were also increased in cultures supplemented with either IL‐1β or IL‐23 or both (Fig 6B and C). Therefore, we hypothesized that stimulation with IL‐23 may substitute the deficiency in IL‐1 signaling. Indeed, cells isolated from both IL‐1R1‐deficient strains, namely IL‐1R1∆T and IL‐1R1−/− mice, responded equally to IL‐23 and showed robust expression of IL‐17A after culturing for four days (Fig 6D and E). Exposure to IL‐23 also had a stimulatory effect on numbers of GM‐CSF‐producing cells among the MOG‐specific IL‐1R1‐deficient Th17 cells (Fig 6F). Moreover, IFNγ co‐expression by GM‐CSF+ Th17 cells was comparable between different genotypes when IL‐23 was added (Fig 6G).
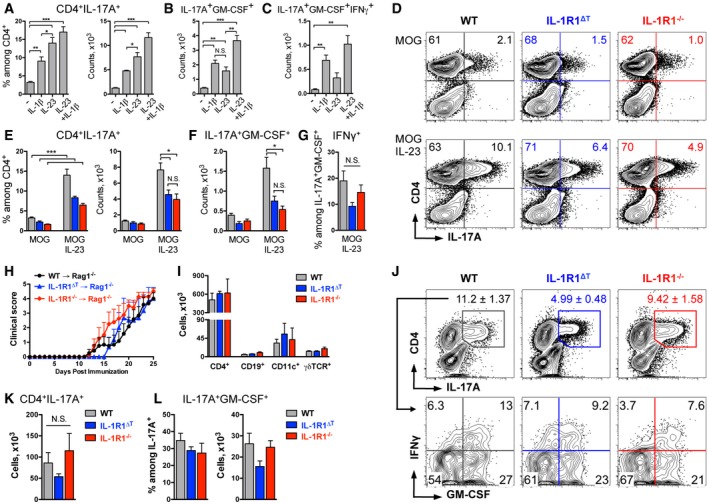
Figure 6. IL‐23 increases numbers of encephalitogenic IL‐1R1‐deficient Th17 cells.
-
A–CAnalysis of (A) IL‐17A expression by wild‐type CD4 T cells, followed with the analysis of (B) GM‐CSF and (C) GM‐CSF and IFNγ co‐expression by Th17 cells, shown as mean + SEM.
-
D–GAnalysis of cytokine expression by cells isolated from WT (black), IL‐1R1ΔT (blue), and IL‐1R1−/− (red) mice and cultured with or without IL‐23. Data (D) are representative FACS plots, gated on VD−TCRβ+ cells with mean frequencies per group. Quantification (mean + SEM) of (E) MOG‐specific Th17 cells, (F) GM‐CSF+ Th17 cells, and (G) GM‐CSF+IFNγ+ Th17 cells.
-
H, I(H) Mean EAE clinical scores (+ SEM) of Rag1−/− mice which received 1 × 105 IL‐17A+ cells of indicated genotypes and (I) total cell numbers (mean + SEM) of infiltrates isolated from CNS and used for further analysis.
-
J–LAnalysis of cytokine expression by CD4 T cells isolated from CNS of mice shown in (H) and restimulated with MOG for 6 h. (J) Data are representative FACS plots, gated on VD−TCRβ+ cells (upper row) with mean frequencies ± SEM, and (lower row) with mean frequencies per group. (K) Total cell numbers (mean + SEM) of Th17 cells shown in (J, upper row). (L) Frequencies and total numbers (mean + SEM) of GM‐CSF+ Th17 cells shown in (J, lower row).
As addition of IL‐23 led to higher numbers of IL‐1R1‐deficient GM‐CSF‐producing Th17 cells, we tested whether these cells are pathogenic by using a model of passive EAE by transfer of ex vivo reactivated T cells. For that we isolated cells from the spleen and dLN of WT, IL‐1R1∆T, and IL‐1R1−/− MOG/CFA‐immunized mice and polarized them in vitro in the presence of MOG peptide, anti‐IFNγ, and IL‐23, as described above. After four days of culture, the numbers of harvested cells were adjusted to 1 × 105 IL‐17A+ cells of each genotype and total cell preparations were transferred into Rag1−/− mice. These cells, regardless of the genotype, transmitted disease and caused strong EAE symptoms in recipient mice (Fig 6H), confirming the pathogenicity of IL‐1R1‐deficient T cells observed upon active immunization. At the peak of disease, we isolated cellular infiltrates from the CNS and found that CD4 T cells represented the major population of immune cells and were equally present in mice that received WT or IL‐1R1‐deficient cells (Fig 6I). Furthermore, we observed high numbers of IL‐17A+ CD4 T cells within the inflamed CNS in all groups of diseased animals (Fig 6J and K). In line with this, we did not find differences in the percentages and total numbers of GM‐CSF‐co‐expressing cells among the mice of the different groups (Fig 6J and L). CD4 T‐cell‐derived GM‐CSF was shown to activate CNS resident microglia cells (Ponomarev et al, 2007) as well as monocytes (Croxford et al, 2015) and to sustain neuroinflammation by promoting IL‐1β expression. Therefore, we investigated IL‐1β production by myeloid cells isolated from inflamed CNS and indeed detected large populations of CD11b+ cells expressing IL‐1β in a similar manner in all analyzed animals (Appendix Fig S5).
To directly study the role of IL‐23 in vivo, we induced EAE in WT and IL‐1R1∆T mice and administered the cytokine via i.p. injections. Constitutive treatment with IL‐23 had a mild, but stimulatory effect on EAE severity of both WT and IL‐1R1∆T mice (Fig EV5). Collectively, we conclude that the necessity of IL‐1 signaling for the expansion of encephalitogenic T cells can be substituted by IL‐23 in an adoptive transfer system and partially in vivo.
Figure EV5. IL‐23 promotes EAE severity in IL‐1R1‐deficient mice.
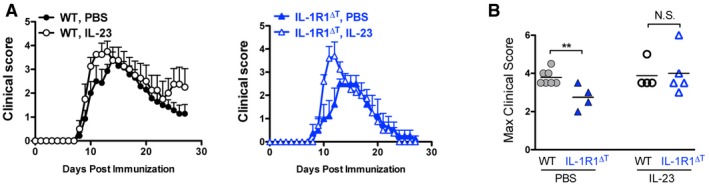
- EAE clinical scores (mean + SEM) of mice treated with IL‐23 or PBS.
- Maximal EAE clinical scores (individual plots with mean) of mice shown in (A).
Treg cells suppress disease in IL‐1R1‐deficient mice
Treg cells are the potent suppressors of autoimmunity and can be transiently depleted by injections of PC61 antibodies directed against CD25 (McGeachy et al, 2005). To study whether the small numbers of Th17 cells found after immunization of IL‐1R1−/− mice can be pathogenic also in active EAE, we depleted Tregs 5 days prior to immunization. This led to the development of strong EAE symptoms in IL‐1R1−/− mice treated with PC61, although with later disease onset compared to controls (Fig 7A). Nineteen days after immunization, we selected mice with similar clinical scores (Fig 7B) and analyzed them for cytokine‐producing T cells in the CNS. As seen in Fig 7C, we found no differences in numbers of Th17 cells deficient or sufficient for IL‐1 signaling. These data suggest that IL‐1 signaling is not critical for the suppressive capacity of Treg cells, which is in agreement with our findings that these cells hardly express the IL‐1R1 (Figs 2A and C, and 4A) and that IL‐1R1‐deficient Treg cells develop normally after EAE induction (Appendix Fig S2 and Fig 5I). Our data suggest that under IL‐1R1‐deficient conditions, Th17 cells cannot properly expand due to their impaired IL‐1 response and consequent suppression by Tregs.
Figure 7. Treg depletion results in EAE susceptibility of IL‐1R1‐deficient mice.
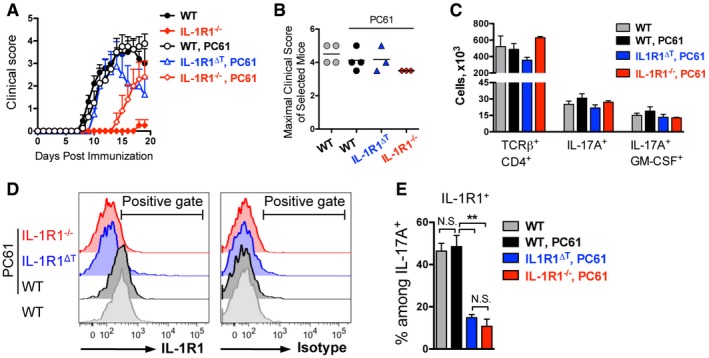
- Mean EAE clinical scores (+ SEM).
- Maximal EAE clinical scores (individual plots with mean) of selected mice.
- Total cell numbers (mean + SEM) of infiltrates isolated from the CNS of mice shown in (B).
- Analysis of IL‐1R1 expression by Th17 cells isolated from CNS of EAE‐diseased mice shown in (B). Data are representative FACS histogram overlays of cells isolated from (gray) WT mice treated with PBS, (black) WT, (blue) IL‐1R1ΔT, and (red) IL‐1R1−/− mice treated with PC61, gated on VD−TCRβ+CD4+IL‐17A+ cells.
- Quantification (mean + SEM) of IL‐1R1 expression by Th17 cells, shown in (D).
Importantly, IL‐1R1 expression by encephalitogenic Th17 cells was not altered in wild‐type mice treated with PC61 in comparison with the non‐treated group (Fig 7D and E). Furthermore, we could show that T cells isolated from the CNS of PC61‐treated IL‐1R1∆T mice expressed no or very little IL‐1R1 (Fig 7D and E), indicating that disease in these mice was not driven by cells that could potentially escape CD4‐Cre‐mediated IL‐1R1 deletion.
Discussion
The development of EAE requires CD4 T‐cell maturation in secondary lymphoid organs upon immunization. Here, we demonstrate that T‐cell‐intrinsic IL‐1 signaling is necessary to promote the expansion of encephalitogenic CD4 T cells. We show that PTx, an essential component of the immunization protocol, stimulates IL‐1β production by myeloid cells, which leads to proliferation of encephalitogenic GM‐CSF‐producing Th17 cells. Importantly, our data emphasize that IL‐1β signaling is not necessary for primary differentiation of pathogenic Th17 cells in vivo.
A recent study identified an important role of IL‐1 produced by neutrophils and monocyte‐derived myeloid cells during their transmigration into the CNS for EAE development (Levesque et al, 2016). We speculate that IL‐1 does not only activate the endothelial cells of the blood–spinal cord barrier, but promotes the secondary activation and expansion of CD4 T cells during the disease onset within the CNS. Here, we investigated IL‐1 production in secondary lymphoid organs during CD4 T‐cell priming and found that also at this stage, the myeloid cells represent the main source of IL‐1β in draining lymph nodes and spleen. Addition of PTx to the immunization protocol greatly increased the numbers of autoreactive CD4 T cells, although the primary effects of PTx on T cells were described to be rather negative due to suppressed proliferation and migration (Robbinson et al, 1996; Su et al, 2001). On the other hand, EAE cannot be induced using most protocols of active immunization without the addition of PTx (Stromnes & Goverman, 2006). Together, these observations suggested that PTx might promote EAE development through intermediate factor(s). In line with that, PTx‐induced IL‐6 was suggested as one of the factors that stimulates Th17 cell development (Chen et al, 2007). Later, it was found that IL‐6 in fact stimulates IL‐1R1 expression on Th17 cells rendering them responsive to IL‐1 (Chung et al, 2009). In agreement with our data, neutrophils, macrophages, and monocyte‐derived dendritic cells were shown to produce IL‐1 in response to PTx administration, thereby promoting CD4 T‐cell priming (Dumas et al, 2014; Lin et al, 2016; Ronchi et al, 2016). It was also reported that deletion of IL‐1 receptor antagonist, a key negative regulator of IL‐1 signaling, allows EAE development even without PTx (Matsuki et al, 2006). Together, these findings suggest that PTx promotes EAE development via IL‐1 signaling.
In line with previous reports (Chung et al, 2009; Guo et al, 2009; Martin et al, 2016), we found that Th17 cells, including its IFNγ‐ and GM‐CSF‐positive subpopulations, express the highest levels of IL‐1R1 among other CD4 T cells. Therefore, the stimulatory effect of myeloid cell‐derived IL‐1β was not only seen in the general expansion of MOG‐specific Th17 cells but was also observed in IL‐17A‐positive cells that co‐express GM‐CSF. The importance of IL‐1 signaling for GM‐CSF production by CD4 T cells in vivo has been reported previously (Lukens et al, 2012), but the question of its T‐cell‐intrinsic function remained open. Using a novel mouse strain with IL‐1R1 conditionally deleted in αβ‐TCR+ T cells, we could show that T cells, which have developed in the absence of IL‐1 signaling but in an otherwise IL‐1R1‐sufficient environment, also displayed reduced GM‐CSF expression in the Th17 subset. This notion is important in light of broad IL‐1R1 expression by different cell types and restricts its necessity directly to T cells. Of note, previous studies reported that T‐cell‐derived GM‐CSF is a potent inducer of IL‐1β expression by myeloid cells (Croxford et al, 2015), which then stimulates further expansion of GM‐CSF+ Th17 cells in a positive feedback loop, as we show here. Our data contribute in elucidating such GM‐CSF/IL‐1 proinflammatory axis (reviewed in Paré et al (2016)).
Interestingly, some IL‐1R1∆T mice developed a mild paralysis after EAE induction, showing for the first time that CD4 T cells indeed gain pathogenic potential also in the absence of IL‐1 signaling. This in vivo data suggested that IL‐1 is not an indispensible factor for pathogenicity and our following transfer experiments confirmed this notion. Cells, which differentiated independently of IL‐1 signaling, successfully transmitted EAE after polarization in the presence of IL‐23. It is broadly accepted that IL‐23 does not stimulate de novo Th17 cell generation, but rather stabilizes this phenotype and promotes Th17 cell expansion (Bettelli et al, 2006; Veldhoen et al, 2006; McGeachy et al, 2007). This led us to speculate that in our cultures, supplemented with a very limited cytokine cocktail, the main effect was the expansion of already existing pathogenic cells rather than their de novo generation. The observed pathogenicity of the transferred cells is in contrast to the reported inability of IL‐1R1‐deficient cells to transmit EAE into Rag1−/− mice (Chung et al, 2009). However, we believe that differences in the experimental setting may explain this discrepancy. Chung and co‐authors did not expand cells prior to transfer but rather immunized recipient animals, while we enriched transferred cell populations for IL‐17A‐positive cells after in vitro expansion. Nevertheless, their independent experiments with chimeric mice, and experiments reported by Croxford et al (2015), which both consisted of WT and IL‐1R1‐deficient bone marrow‐derived progenitors, showed that neuroinflammation driven by WT cells resulted also in recruitment of IL‐1R1‐deficient CD4 T cells into the CNS. Although IL‐1R1‐deficient CD4 T cells were reduced in numbers and showed impaired cytokine production, this finding is in full agreement with our data showing the presence of small but sufficient quantities of IL‐1R1‐deficient cells in the CNS of diseased IL‐1R1∆T mice.
The balance between Tregs and Th17 cells is crucial for development of autoimmunity. Analyzing both the preclinical and the effector phase of EAE, we did not find effects of disrupted IL‐1 signaling neither on peripheral nor on CNS infiltrated Tregs. However, we did find increased frequencies of Tregs among CD4 T‐cell CNS infiltrates in IL‐1R1∆T mice, reflecting the impaired effector T‐cell development in these animals. One might speculate that the shift of the Treg‐Th17 ratio in IL‐1R1∆T mice may even disguise the real encephalitogenic potential of effector T cells devoid of IL‐1 signaling. Our data are in line with an impaired Th17 response combined with increased frequencies of Treg cells seen in mice with T cells lacking MyD88, the adaptor molecule for Toll‐like receptor and IL‐1 receptor family signaling (Schenten et al, 2014). This study and our data together support the notion that IL‐1 signaling acts on precursor cells of Tregs, rather than on already developed regulatory T cells.
Importantly, mice completely lacking IL‐1 signaling did not show EAE symptoms or CNS infiltrates, unless regulatory T cells were ablated. This indicates that IL‐1 has multiple roles in the induction of EAE beyond expansion of encephalitogenic T cells. One of probably many roles might be the activation and recruitment of myeloid cell populations as shown here. Only under very restrictive conditions, when the suppressive arm was removed by Treg cell depletion, IL‐1R1−/− mice developed EAE. Together, our data clearly show that the main role of IL‐1 in terms of T helper cell‐mediated pathogenicity in mice is not primarily the differentiation of Th17 cells but rather the expansion or boosting of GM‐CSF‐producing encephalitogenic Th17 cells.
Using mouse models greatly contributed to the development of therapeutic interventions for human diseases. Suppression of IL‐1 signaling has beneficial effects in several pathologies, including those with autoimmune and inflammatory components (reviewed in Schett et al (2016)). Human Th17 cells were found to express high levels of IL‐1R1 (Lee et al, 2010) and IL‐1 was proposed as a critical factor in pathogenic Th17 cell development (Zielinski et al, 2012). Our data demonstrate that like the mouse counterpart, also human GM‐CSF+ Th17 cells express IL‐1R1, making them responsive to IL‐1. Importantly, IL‐1 might be essential not only for the proliferation of human Th17 cells but also for their differentiation (Acosta‐Rodriguez et al, 2007). Our data provide the mechanistic basis for better characterization of T cells recovered from MS patients, studies that are needed in order to explore the involvement of IL‐1 signaling in this human disease.
Materials and Methods
Mice
The Il1r1 fl/fl mouse strain with exon 5 of Il1r1 gene flanked by loxP sites was crossed to Cd4‐Cre and CMV‐Cre mice resulting in IL‐1R1ΔT and IL‐1R1−/− mouse strains, respectively (Mufazalov et al, 2016). Rag1−/− mice were obtained from in‐house breeding.
All mice were bred in‐house under SPF conditions. Age‐ and gender‐matched genetically modified animals carrying loxP sites without Cre transgene were used as controls. Experiments were performed with 6‐ to 14‐week‐old mice on C57BL/6 background in accordance with the guidelines of the central animal facility institution (TARC, University of Mainz).
Immunization, active EAE induction, and clinical assessment
Mice were immunized subcutaneously at the tail basis with 50 μg MOG35‐55 peptide (Gene Script) emulsified in complete Freund's adjuvant (Difco) supplemented with 1.1 mg of heat‐inactivated Mycobacterium tuberculosis (Difco). Along with immunization and on day 2 postimmunization, 200 ng of pertussis toxin (List Biological Laboratories) or Dulbecco's phosphate‐buffered saline (PBS, Sigma) was administered by intraperitoneal injection, when indicated. Mice were scored daily for clinical signs of EAE as described (Huppert et al, 2010) with modifications. The clinical assessment scale was graded from 0 to 6 as follows: 0, no disease; 0.5, partial loss of tail tonicity; 1, complete loss of tail tonicity; 1.5, partially impaired righting reflex on attempt to roll over (within 3 sec); 2, impaired righting reflex; 2.5, partial hindlimb paresis resulted in staggering gait; 3, hindlimb paresis; 3.5, hindlimb paralysis; 4, unilateral forelimb paresis; 4.5, complete forelimb paresis; 5, forelimb paresis resulted in inability to move the body; 5.5, moribund; and 6, dead animal. Calculation of the area under the curve (AUC) was performed as described elsewhere (Fleming et al, 2005).
Cytokines and cell culture
For in vitro culture, cells were isolated from emulsion‐draining lymph nodes (dLN) and spleen at day 10 after MOG/CFA immunization and mixed at 1–3 ratios. Cells were cultured at a concentration of 0.6 × 106 cells/well in 200 μl complete medium (RPMI medium containing 10% FCS, 2 mM L‐glutamine, 100 units/ml penicillin, 100 mg/ml streptomycin, 1 mM sodium pyruvate, 50 mM 2‐mercaptoethanol, 10 mM HEPES, and 1% non‐essential amino acids) supplemented with 10 μg/ml MOG35‐55 and 10 μg/ml α‐IFNγ antibodies (BioXCell) in 96‐well plates as triplicates. When indicated, cultures were additionally supplemented with 15 ng/ml IL‐23 (Miltenyi), 25 ng/ml IL‐1β (R&D Systems), or both. After culturing for 4 days, triplicates were pooled and restimulated in fresh medium containing MOG/brefeldin A for 6 h prior to cytokine analysis.
Transfer EAE
Donor mice were immunized with MOG/CFA as described above. On day 10 postimmunization, cells from dLN and spleen were isolated, mixed at 1 to 4 ratio, and cultured at a concentration of 5 × 106 cells/ml in complete medium supplemented with 10 μg/ml MOG35‐55, 10 μg/ml α‐IFNγ antibodies, and 15 ng/ml IL‐23 for 4 days. Afterward, total cell preparations containing 1 × 105 blasting MOG‐specific Th17 cells were washed twice in PBS and injected intraperitoneally into Rag1−/− mice. Along with cells transfer and on day 2 posttransfer, 200 ng of PTx was administered into recipient mice. Mice were scored for EAE signs as described above.
In vivo Treg depletion and IL‐23 treatment
For Treg cell depletion, mice were treated intraperitoneally with 0.5 mg PC61 (anti‐CD25 antibody, BioXCell) or with PBS five and three days prior to active EAE immunization. The treatment with 1 μg IL‐23 (Miltenyi) or with PBS was performed intraperitoneally at day 3, 5, 7, 9, 11, and 13 after active EAE immunization.
Organ preparation
Single cell suspensions of spleen and dLN were prepared by mechanical dissociation in PBS supplemented with 2% FCS. For the analysis of myeloid cells, spleens and dLN were digested with 2 mg/ml collagenase II (Sigma) and 40 μg/ml DNase I (Roche) for 20 min at 37°C and homogenized with syringe and needle (18G). To obtain CNS infiltrating lymphocytes, brain and spinal cord were isolated from transcardially perfused mice, pooled, enzymatically digested, and centrifuged in 30/37/70 Percoll gradient as previously described (Mufazalov & Waisman, 2016).
Cells restimulation prior to cytokine staining
For cytokine detection, cells were incubated at (4–20) × 106 cells/ml in complete culture medium. For IL‐1 detection, cells were incubated for 4 h in the presence of 2 μM monensin (BioLegend) and 20 ng/ml GM‐CSF (Peprotech), 500 ng/ml LPS (Sigma), or 50 ng/ml phorbol‐12‐myristate‐13‐acetate (PMA, Sigma) together with 500 ng/ml ionomycin (Invitrogen). For polyclonal stimulation of CD4 T cells, samples were incubated for 4 h in the presence of PMA, ionomycin together with monensin, or 1 μg/ml brefeldin A (AppliChem). For antigen‐specific CD4 T‐cell restimulation, cells were incubated for 6 h using a combination of 20 μg/ml MOG35‐55 or 20 μg/ml OVA (Calbiochem) in the presence of monensin or brefeldin A.
Antibodies and flow cytometry
Single cell suspensions were stained with antibodies (Appendix Table S1) specific to epitopes present at the cell surface together with fixable viability dyes (VD, eBioscience). Afterward, cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) or FoxP3 staining kit (eBiosciences) and stained intracellularly with antibodies (Appendix Table S2) according to the manufacturer's recommendations. Stained cells were acquired on FACS‐Canto II (BD Biosciences) and obtained data were further analyzed with FlowJo software. Gating strategy always considered cell size, excluded duplets, and defined living cells as viability dye negative (VD−).
Human PBMCs
Buffy coats of healthy human donors who gave their informed consent were obtained from the Center for Blood Transfusion University Medical Center Mainz, Germany. PBMCs were isolated by Ficoll‐Hypaque gradient centrifugation. Afterward, 2 × 106 cells were reactivated in complete medium with PMA, ionomycin, and monensin for 4 h prior to staining. For the surface staining, CD14, CD3, and CD4 (BioLegend) FACS antibodies were used. Anti‐IL‐17A, IFNγ, GM‐CSF (BioLegend), IL‐1R1, and goat IgG isotype control (R&D Systems) were applied after fixation with the cytokine staining kit (BD Biosciences).
Statistical analysis
Graphical and statistical analysis was done using Prism 5 software (GraphPad) and Excel (MS Office). Statistical significance was calculated using the two‐tailed unpaired t‐test. Values of P < 0.05, P < 0.01, and P < 0.001 were marked as *, **, and ***, respectively. N.S., not significant.
Author contributions
IAM designed experiments; IAM, CS, TR, JK, FW, LAG, and JH performed experiments; IAM analyzed data; IAM, FCK, and AW interpreted data; WM and EP supplied essential reagents; TR, WM, and EP corrected the manuscript; IAM, FCK, and AW wrote the manuscript; AW supervised study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank C. Hackenbruch, M. Wolk, K.‐S. Atretkhany, and A. Muratova for technical help. We are also thankful to N. Yogev for productive discussions. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grant WA 1600/8‐1 and SFB/TRR128 TP A07 to A.W. and TP A03 to F.C.K., and by the Gemeinnützige Hertie‐Stiftung to A.W.
The EMBO Journal (2017) 36: 102–115
Contributor Information
Ilgiz A Mufazalov, Email: ilgiz.mufazalov@uni-mainz.de.
Ari Waisman, Email: waisman@uni-mainz.de.
References
- Abdulaal WH, Walker CR, Costello R, Redondo‐Castro E, Mufazalov IA, Papaemmanouil A, Rothwell NJ, Allan SM, Waisman A, Pinteaux E, Muller W (2016) Characterization of a conditional interleukin‐1 receptor 1 mouse mutant using the Cre/LoxP system. Eur J Immunol 46: 912–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta‐Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1 beta and 6 but not transforming growth factor‐beta are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat Immunol 8: 942–949 [DOI] [PubMed] [Google Scholar]
- Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE (1989) Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th‐1 lymphokine subtype. Cell Immunol 124: 132–143 [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238 [DOI] [PubMed] [Google Scholar]
- Boraschi D, Tagliabue A (2013) The interleukin‐1 receptor family. Semin Immunol 25: 394–407 [DOI] [PubMed] [Google Scholar]
- Brucklacher‐Waldert V, Ferreira C, Innocentin S, Kamdar S, Withers DR, Kullberg MC, Veldhoen M (2016) Tbet or continued RORgammat expression is not required for Th17‐associated immunopathology. J Immunol 196: 4893–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassan C, Piaggio E, Zappulla JP, Mars LT, Couturier N, Bucciarelli F, Desbois S, Bauer J, Gonzalez‐Dunia D, Liblau RS (2006) Pertussis toxin reduces the number of splenic Foxp3+ regulatory T cells. J Immunol 177: 1552–1560 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay PK, Yu J, Roederer M (2005) A live‐cell assay to detect antigen‐specific CD4+ T cells with diverse cytokine profiles. Nat Med 11: 1113–1117 [DOI] [PubMed] [Google Scholar]
- Chen X, Howard OMZ, Oppenheim JJ (2007) Pertussis toxin by inducing IL‐6 promotes the generation of IL‐17‐producing CD4 cells. J Immunol 178: 6123–6129 [DOI] [PubMed] [Google Scholar]
- Chen X, Winkler‐Pickett RT, Carbonetti NH, Ortaldo JR, Oppenheim JJ, Howard OMZ (2006) Pertussis toxin as an adjuvant suppresses the number and function of CD4(+)CD25(+) T regulatory cells. Eur J Immunol 36: 671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity 30: 576–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) ROR gamma t drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560–567 [DOI] [PubMed] [Google Scholar]
- Comerford I, Bunting M, Fenix K, Haylock‐Jacobs S, Litchfield W, Harata‐Lee Y, Turvey M, Brazzatti J, Gregor C, Nguyen P, Kara E, McColl SR (2010) An immune paradox: how can the same chemokine axis regulate both immune tolerance and activation?: CCR6/CCL20: a chemokine axis balancing immunological tolerance and inflammation in autoimmune disease. BioEssays 32: 1067–1076 [DOI] [PubMed] [Google Scholar]
- Croxford AL, Kurschus FC, Waisman A (2011) Mouse models for multiple sclerosis: historical facts and future implications. Biochim Biophys Acta 1812: 177–183 [DOI] [PubMed] [Google Scholar]
- Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B (2015) The cytokine GM‐CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43: 502–514 [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin‐23 rather than interleukin‐12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421: 744–748 [DOI] [PubMed] [Google Scholar]
- Dumas A, Amiable N, de Rivero Vaccari JP, Chae JJ, Keane RW, Lacroix S, Vallieres L (2014) The inflammasome pyrin contributes to pertussis toxin‐induced IL‐1beta synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog 10: e1004150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol 12: 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A (1998) IL‐6‐deficient mice resist myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis. Eur J Immunol 28: 2178–2187 [DOI] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG (2005) Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol 170: 71–84 [DOI] [PubMed] [Google Scholar]
- Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, Scheffold A, Thiel A (2005) Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med 11: 1118–1124 [DOI] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W et al (2010) Generation of pathogenic T(H)17 cells in the absence of TGF‐beta signalling. Nature 467: 967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ (1997) Phenotypic and functional characterization of mice that lack the type I receptor for IL‐1. J Immunol 159: 3364–3371 [PubMed] [Google Scholar]
- Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, Paul W (2009) IL‐1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USA 106: 13463–13468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A (2009) IL‐17A and IL‐17F do not contribute vitally to autoimmune neuro‐inflammation in mice. J Clin Invest 119: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6: 1123–1132 [DOI] [PubMed] [Google Scholar]
- Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, Gran B, Piehl F, Olsson T, Codarri L, Becher B (2014) Multiple sclerosis‐associated IL2RA polymorphism controls GM‐CSF production in human TH cells. Nat Commun 5: 5056 [DOI] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (2011) Fate mapping of IL‐17‐producing T cells in inflammatory responses. Nat Immunol 12: 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR (2010) Cellular mechanisms of IL‐17‐induced blood‐brain barrier disruption. Faseb J 24: 1023–1034 [DOI] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y (2009) Differential roles of interleukin‐17A and ‐17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30: 108–119 [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell 126: 1121–1133 [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL‐21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448: 484–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B (2007) IL‐22 is expressed by TH17 cells in an IL‐23‐dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol 179: 8098–8104 [DOI] [PubMed] [Google Scholar]
- Kurschus FC, Croxford AL, Heinen AP, Wortge S, Ielo D, Waisman A (2010) Genetic proof for the transient nature of the Th17 phenotype. Eur J Immunol 40: 3336–3346 [DOI] [PubMed] [Google Scholar]
- Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, McIntyre KW (1997) Absence of IL‐1 signaling and reduced inflammatory response in IL‐1 type I receptor‐deficient mice. J Immunol 159: 2452–2461 [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201: 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WW, Kang SW, Choi J, Lee SH, Shah K, Eynon EE, Flavell RA, Kang I (2010) Regulating human Th17 cells via differential expression of IL‐1 receptor. Blood 115: 530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK (2012) Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13: 991–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque SA, Pare A, Mailhot B, Bellver‐Landete V, Kebir H, Lecuyer MA, Alvarez JI, Prat A, de Rivero Vaccari JP, Keane RW, Lacroix S (2016) Myeloid cell transmigration across the CNS vasculature triggers IL‐1beta‐driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med 213: 929–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, Moore CS, Michel L, Althekair F, Rajasekharan S, Gommerman JL, Prat A, Fillatreau S, Bar‐Or A (2015) Proinflammatory GM‐CSF‐producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 7: 310ra166 [DOI] [PubMed] [Google Scholar]
- Lin CC, Bradstreet TR, Schwarzkopf EA, Jarjour NN, Chou C, Archambault AS, Sim J, Zinselmeyer BH, Carrero JA, Wu GF, Taneja R, Artyomov MN, Russell JH, Edelson BT (2016) IL‐1‐induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J Exp Med 213: 251–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD (2012) Inflammasome‐derived IL‐1beta regulates the production of GM‐CSF by CD4(+) T cells and gammadelta T cells. J Immunol 188: 3107–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BN, Wang C, Zhang CJ, Kang Z, Gulen MF, Zepp JA, Zhao J, Bian G, Do JS, Min B, Pavicic PG Jr, El‐Sanadi C, Fox PL, Akitsu A, Iwakura Y, Sarkar A, Wewers MD, Kaiser WJ, Mocarski ES, Rothenberg ME et al (2016) T cell‐intrinsic ASC critically promotes T(H)17‐mediated experimental autoimmune encephalomyelitis. Nat Immunol 17: 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y (2006) Abnormal T cell activation caused by the imbalance of the IL‐1/IL‐1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol 18: 399–407 [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak‐Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF‐beta and IL‐6 drive the production of IL‐17 and IL‐10 by T cells and restrain T(H)‐17 cell‐mediated pathology. Nat Immunol 8: 1390–1397 [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Stephens LA, Anderton SM (2005) Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol 175: 3025–3032 [DOI] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben‐Nun A (1995) A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H‐2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol 25: 1951–1959 [DOI] [PubMed] [Google Scholar]
- Millward JM, Caruso M, Campbell IL, Gauldie J, Owens T (2007) IFN‐gamma‐induced chemokines synergize with pertussis toxin to promote T cell entry to the central nervous system. J Immunol 178: 8175–8182 [DOI] [PubMed] [Google Scholar]
- Mufazalov IA, Regen T, Schelmbauer C, Kuschmann J, Muratova AM, Nikolaev A, Muller W, Pinteaux E, Waisman A (2016) Generation of a novel T cell specific Interleukin‐1 receptor Type 1 conditional knock out mouse reveals intrinsic defects in survival, expansion and cytokine production of CD4 T cells. PLoS One 11: e0161505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufazalov IA, Waisman A (2016) Isolation of central nervous system (CNS) infiltrating cells. Methods Mol Biol 1304: 73–79 [DOI] [PubMed] [Google Scholar]
- Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, Chang HD, Radbruch A, Zielinski CE (2014) IL‐17 and GM‐CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med 6: 241ra280 [DOI] [PubMed] [Google Scholar]
- O'Connor RA, Anderton SM (2008) Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol 193: 1–11 [DOI] [PubMed] [Google Scholar]
- Paré A, Mailhot B, Lévesque SA, Lacroix S (2016) Involvement of the IL‐1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: breaking the vicious cycle between IL‐1b and GM‐CSF. Brain Behav Immun doi: 10.1016/j.bbi.2016.07.146 [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras‐Vasconcelos J, Verthelyi D, Dittel BN (2007) GM‐CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 178: 39–48 [DOI] [PubMed] [Google Scholar]
- Qian J, Zhu L, Li Q, Belevych N, Chen Q, Zhao F, Herness S, Quan N (2012) Interleukin‐1R3 mediates interleukin‐1‐induced potassium current increase through fast activation of Akt kinase. Proc Natl Acad Sci USA 109: 12189–12194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F (2009) C‐C chemokine receptor 6‐regulated entry of TH‐17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10: 514–523 [DOI] [PubMed] [Google Scholar]
- Robbinson D, Cockle S, Singh B, Strejan GH (1996) Native, but not genetically inactivated, pertussis toxin protects mice against experimental allergic encephalomyelitis. Cell Immunol 168: 165–173 [DOI] [PubMed] [Google Scholar]
- Ronchi F, Basso C, Preite S, Reboldi A, Baumjohann D, Perlini L, Lanzavecchia A, Sallusto F (2016) Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin‐driven IL‐1beta production by myeloid cells. Nat Commun 7: 11541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten D, Nish SA, Yu S, Yan XT, Lee HK, Brodsky I, Pasman L, Yordy B, Wunderlich FT, Bruning JC, Zhao HY, Medzhitov R (2014) Signaling through the adaptor molecule MyD88 in CD4(+) T cells is required to overcome suppression by regulatory T cells (vol 40, pg 78, 2014). Immunity 40: 814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett G, Dayer JM, Manger B (2016) Interleukin‐1 function and role in rheumatic disease. Nat Rev Rheumatol 12: 14–24 [DOI] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM (2006) Active induction of experimental allergic encephalomyelitis. Nat Protoc 1: 1810–1819 [DOI] [PubMed] [Google Scholar]
- Su SB, Silver PB, Zhang M, Chan CC, Caspi RR (2001) Pertussis toxin inhibits induction of tissue‐specific autoimmune disease by disrupting G protein‐coupled signals. J Immunol 167: 250–256 [DOI] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC (2006) A crucial role for interleukin (IL)‐1 in the induction of IL‐17‐producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203: 1685–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity 24: 179–189 [DOI] [PubMed] [Google Scholar]
- Yogev N, Frommer F, Lukas D, Kautz‐Neu K, Karram K, Ielo D, von Stebut E, Probst HC, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A (2012) Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD‐1 receptor(+) regulatory T cells. Immunity 37: 264–275 [DOI] [PubMed] [Google Scholar]
- Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F (2012) Pathogen‐induced human TH17 cells produce IFN‐gamma or IL‐10 and are regulated by IL‐1beta. Nature 484: 514–518 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File