

Abstract
Comparative genomics of closely related bacterial isolates is a powerful method for uncovering virulence and other important genome elements. We determined draft sequences (8-fold coverage) of the genomes of strains JD1 and N40 of Borrelia burgdorferi sensu stricto, the causative agent of Lyme disease, and we compared the predicted genes from the two genomes with those from the previously sequenced B31 genome. The three genomes are closely related and are evolutionarily approximately equidistant (≈0.5% pairwise nucleotide differences on the main chromosome). We used a Poisson model of nucleotide substitution to screen for genes with elevated levels of nucleotide polymorphisms. The three-way genome comparison allowed distinction between polymorphisms introduced by mutations and those introduced by recombination using the method of phylogenetic partitioning. Tests for recombination suggested that patches of high-density nucleotide polymorphisms on the chromosome and plasmids arise by DNA exchange. The role of recombination as the main mechanism driving B. burgdorferi diversification was confirmed by multilocus sequence typing of 18 clinical isolates at 18 polymorphic loci. A strong linkage between the multilocus sequence genotypes and the major alleles of outer-surface protein C (ospC) suggested that balancing selection at ospC is a dominant force maintaining B. burgdorferi diversity in local populations. We conclude that B. burgdorferi undergoes genome-wide genetic exchange, including plasmid transfers, and previous reports of its clonality are artifacts from the use of geographically and ecological isolated samples. Frequent recombination implies a potential for rapid adaptive evolution and a possible polygenic basis of B. burgdorferi pathogenicity.
Keywords: balancing selection, ospC, Lyme disease, Stevens' test
Comparative genomics of closely related species is a powerful method for tracking microbial epidemics (1, 2), uncovering microbial virulence factors (3–5), and annotating genes and other conserved elements in genomes (6, 7). A powerful method of bacterial genotyping is multilocus sequence typing (MLST), which is the comparative sequencing of selected genes (5, 8–10). (Note that in this article by MLST we refer to the comparative sequencing of multiple loci at large, not necessarily housekeeping genes.) We have taken a comparative genomics approach to identify polymorphic ORFs in the genomes of Borrelia burgdorferi sensu stricto, which is the predominant bacterial species causing Lyme disease in North America (11). Measuring rates of sequence evolution and selective constraints (12, 13) is a means of uncovering virulence factors and candidates for vaccine, diagnostics, and therapeutics.
Early population studies of B. burgdorferi using multilocus enzyme electrophoresis (MLEE) (14) and DNA sequences (15) found little evidence for genetic exchange among different isolates. In fact, one report (16) concluded that B. burgdorferi was among the most clonal of bacterial species. However, these studies were based on archival strains isolated from several worldwide locations, and the conclusions on clonality described above may have reflected a high degree of geographic structuring of this obligate parasitic species (11, 17) rather than a lack of genetic exchange within local populations (18). Indeed, analysis of outer-surface protein C (ospC) genes from local B. burgdorferi isolates in North America (19) and Europe (20) suggested extensive recombination at this locus. Crossspecies gene transfer has been suggested at other loci as well (21), and Stevenson and Miller (22) concluded that intracellular transfers of cp32 plasmids have occurred.
Key questions remain in regard to the rate, extent, and mechanism of genetic exchanges among B. burgdorferi clones. At issue are questions of whether there are loci other than ospC that show selectively maintained and recombination-mediated variation and whether large (>1-kb) pieces of DNA or whole plasmids can be transferred among clones (20). Such questions may best be answered by surveying closely related B. burgdorferi isolates on a genome-wide scale.
Here, we report the identification of highly polymorphic loci identified by means of a comparison of three closely related genomes and their use in the study of population structure of B. burgdorferi. We found that incorporation of divergent alleles through homologous recombination is the most likely cause of clusters of nucleotide polymorphisms in the B. burgdorferi genome. This article also provides evidence that genomic diversity in natural populations of B. burgdorferi is maintained by balancing selection at ospC predominantly and, to a lesser extent, at a limited number of other plasmid-borne loci. The discovery of genome-wide genetic exchange among local B. burgdorferi clones suggests that new adaptive features, such as its human virulence, could emerge quickly.
Materials and Methods
DNA Methods. We determined 8-fold coverage draft sequences of B. burgdorferi JD1 and N40 genomes as described (23, 24). Genome assembly of JD1 and N40 (in particular, the assembly of the highly paralogous cp32 plasmids) is not yet complete. Sequencing of selected genes (both strands) from additional clinical isolates was performed by the Stony Brook University Core DNA Sequencing Facility (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site).
Identification of Orthologous ORF Pairs. We used nucmer of the mummer software package (25) to align the JD1 and N40 assemblies with the finished B31 genome. JD1/N40 scaffolds that uniquely (1:1) match the B31 genome were identified as orthologous genome segments. JD1/N40 plasmids were named by using the B31 nomenclature when plasmid orthology is established. JD1/N40 scaffolds matching multiple B31 plasmids (such as scaffolds homologous to the cp32 plasmids of B31), and scaffolds with no matches were excluded in this study. For each pair of orthologous genome segments, we identified nonoverlapping ≥250-bp ORFs on both genomes by using glimmer (26). Syntenic ORFs in two orthologous genome segments are considered “orthologous ORFs” (e.g., see Fig. 4, which is published as supporting information on the PNAS web site).
Identification of High-Density Polymorphisms. Nucleotide sequences of orthologous ORF pairs were aligned according to the clustalw (27) alignment of translated amino acid sequences. Amino acid variability, nucleotide variability, and the rate of synonymous to nonsynonymous substitutions [Ka/Ks ratio; method of Nei and Gojobori (28) by using snap (29)] were calculated for each pair. For each pair of orthologous ORFs, we obtained the expected number of nucleotide differences (Dexp) as the product of the average ORF differences between two genomes and the length of aligned nucleotides. The Dexp value was compared with the observed number of nucleotide differences (Dobs) under the assumption of neutral nucleotide substitutions. The probability of Dobs differences (approximate to the number of substitutions for low-divergence alleles) or more was calculated from the upper tail of the Poisson distribution as follows:
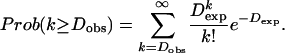 |
We defined high-density nucleotide polymorphisms (HDNPs) as ORFs with more nucleotide differences than the neutral expectation (P < 0.001).
MLST. We performed multilocus genomic typing (see Supporting Materials and Methods for procedures) on an additional 18 North American B. burgdorferi sensu stricto clinical isolates (Table 3, which is published as supporting information on the PNAS web site) by using a selected library of 18 polymorphic loci (loci and PCR primers are given in Table 4, which is published as supporting information on the PNAS web site) identified by means of genome comparisons.
Tests of Recombination. We applied a modification (30) of Stevens' test (31) and Sawyer's test (32) to distinguish polymorphisms introduced by recombination and those introduced by mutational substitutions. These tests identify recombination based on significant clustering of nucleotide substitutions in each of the three phylogenetic partitions (see Results). For MLST analysis, multiple alignments of nucleotide sequences were obtained for each locus according to the clustalw (27) alignments of translated amino acid sequences. Gene phylogeny at each locus was estimated by using mrbayes 2.1 (33), based on a nucleotide evolution model with site-specific rate of evolution at each codon position. A majority-rule consensus tree was obtained from the converged chains. Posterior probabilities were obtained as measures of branch support. We defined major-group alleles as major lineages on the gene tree. Recombination events were identified by using the following criteria. Recombination causes incongruent gene trees among loci (34). Multiple nucleotide substitutions at one locus between two strains that are identical in other loci are most likely results of lateral gene transfer (35). For single-nucleotide differences, alleles unique among strains are mutational substitutions, whereas alleles shared among divergent clonal groups are introduced by recombination (35).
Data Availability. The B31 genome has been deposited in the GenBank database [National Center for Biotechnology Information (NCBI) taxonomy ID 224326]. Nucleotide sequences of MLST loci have been deposited in GenBank under the accession nos. AY696304–AY696571. Alignments and Bayesian trees of MLST sequences are available from the authors.
Results
Strain Choice and Genome Coverage. B. burgdorferi N40 is a tick isolate obtained from Westchester County, NY (36), and JD1 is a nymphal tick isolate obtained from Ipswich, MA (37). Together with the completed genome of strain B31 (23, 24) [a tick isolate obtained from Shelter Island, NY (38)], these three strains cover a wide range of population-level genetic diversity. This diversity is based on ospC typing [B31, JD1, and N40 belong to ospC major groups A, D, and E, respectively (19)], on rDNA spacer typing [restriction fragment length polymorphism types I, II, and III (39)], and on chromosomal pulsed-field gel electrophoresis typing [MluI pulsed-field gel electrophoresis types B, C, and E (40, 41)].
The draft genome sequences for N40 and JD1 are incomplete, so the comparisons in this study cover ≈80% of chromosomal ORFs and approximately one half of the plasmids in B31 (Tables 1 and 2). Plasmid profiles differ among the genomes, and the assembly of JD1 and N40 plasmids (cp32s in particular) is not yet complete. ORFs that are not in our comparison are those that are missing from one of the sequences (because of either genetic differences among the three strains or incompleteness of the draft sequences) or have multiple nucmer hits (e.g., multiple JD1/N40 matches to cp32 in B31). Nonetheless, the three-way comparison of plasmidborne ORFs covered a high proportion of the two universally present plasmids, lp54 (46 of 57 ORFs of ≥250 bp) and cp26 (21 of 26 long ORFs).
Table 1. Summary of ORF variability.
| Genomes | Orthologous scaffolds* | No. of ORF alignments† | Nucleotide difference, % | Amino acid difference, % | Ka/Ks |
|---|---|---|---|---|---|
| Pairwise comparisons | |||||
| B31 and JD1 | Main chromosome | 705 (88.6%) | 0.4615 | 0.4618 | 0.1353 |
| lp54,cp26,cp9,lp17,lp25,lp28-2,lp28-4,lp38,lp36 | 100 (42.0%) | 0.8131 | 0.9553 | 0.2003 | |
| B31 and N40 | Main chromosome | 706 (88.7%) | 0.5706 | 0.5681 | 0.1344 |
| lp54,cp26,lp25,lp28-3,lp28-4,lp36 | 146 (84.4%) | 2.0342 | 3.0364 | 0.3999 | |
| JD1 and N40 | Main chromosome | 692 (86.9%) | 0.6083 | 0.5882 | 0.1279 |
| lp54,cp26,lp25,lp28-4,lp36 | 83 (55.3%) | 1.8611 | 2.6895 | 0.3577 | |
| HDNPs vs. non-HDNPs on lp54 and cp26 | |||||
| B31 and N40 | BBA24,68,69,70 | 4 (7.0%) | 24.7 | 42.6 | 0.7145 |
| Rest of lp54 | 45 (78.9%) | 0.3380 | 0.4334 | 0.1937 | |
| ospC | 1 (3.8%) | 17.1 | 25.8 | 0.5750 | |
| Rest of cp26 | 22 (84.6%) | 0.7832 | 0.7457 | 0.1236 | |
Table 2. Three-way comparisons for the B31, JD1, and N40 genomes.
| Orthologous scaffolds* | No. of ORF alignments† | Partition 1‡ | Partition 2 | Partition 3 |
|---|---|---|---|---|
| Main chromosome | 675 (84.8%) | 0.0036 | 0.0026 | 0.0021 |
| lp54,cp26,lp25,lp28-4,lp36 | 79 (52.7%) | 0.0126 | 0.0066 | 0.0031 |
No. of alignments of orthologous ORFs, given as percentage of total ≥250-bp ORFs predicted on B31 chromosome or plasmids.
Genome Divergence. Genomes of B31, N40, and JD1 are closely related (Tables 1 and 2), and nearly all differences are single-nucleotide polymorphisms (SNPs) with only two nucleotide states [there are a few three-state polymorphisms and insertion/deletions (indels)]. The three genomes are approximately equally distant from each other, with N40 slightly more diverged from B31 (0.77% overall nucleotide difference) and JD1 (0.80%) than B31 and JD1 are diverged from each other (0.51%). We categorized each SNP into one of the following three possible phylogenetic partitions (31). Partition 1 SNPs are those with one state shared by B31 and JD1, with N40 showing another state, designated as [(B31, JD1), N40]; partition 2 SNPs are designated as [(B31, N40), JD1]; and partition 3 SNPs are designated as [(JD1, N40), B31]. As a result of the low (≈0.5%) sequence divergence and the approximately equal distances among the three genomes (essentially a “star phylogeny”), partition 1–3 substitutions can be regarded conveniently as lineage-specific substitutions in N40, JD1, and B31, respectively. The implicit assumption is that at each SNP, the most common nucleotide state among the three genomes is the ancestral one.
Plasmid ORFs appear to evolve, on average, 2- to 4-fold faster than the average chromosomal ORFs, and the apparent accelerated evolution of the N40 lineage is mainly due to substitutions on the plasmids (Tables 1 and 2). Selective constraint for amino acid replacement, as measured by the Ka/Ks ratio, for the plasmid ORFs is 2-fold weaker than that for the chromosomal ORFs (Tables 1 and 2). However, plasmid sequences are not uniformly more variable or unconstrained than chromosomal ORFs. For instance, most ORFs on the two universally present plasmids, lp54 and cp26, are as conserved as chromosomal ORFs, except for BBA24 [encoding decorin-binding protein (42)], BBA68 [encoding BbCRASP-1, (43)], BBA69, BBA70, and BBB19 (ospC) (Tables 1 and 2).
Frequency distributions of sequence differences closely follow the Poisson expectations (Fig. 5, which is published as supporting information on the PNAS web site), suggesting that most nucleotide substitutions are selectively neutral. ORFs containing HDNPs were identified in a small proportion (n = 49, 6.9%) of the chromosomal ORFs and a much larger proportion (n = 42, 28%) of the plasmid ORFs (Table 5, which is published as supporting information on the PNAS web site). From this HDNP list, we chose 11 of the most polymorphic ORFs and six polymorphic plasmid ORFs found in an earlier study (W.-G.Q., J.F.B., and B.J.L., unpublished data) as markers for genomic typing of clinical isolates (described below). These most variable ORFs encompass one B31 chromosomal locus (BB0082) and 17 loci on nine linear and circular plasmids.
Evidence for Recombination from Three-Way Genome Comparison. Nucleotide differences of HDNPs among three pairs of genome comparisons are shown in Fig. 1. The overall pattern is that most HDNPs show high variability in only two of the three pairwise comparisons. This pattern indicates that most nucleotide substitutions occurred during the evolution of only one of the three possible lineages. For example, the most variable loci on the main chromosome (BB0082, BB0218, BB0348, BB0550, and BB0684) varied little between B31 and JD1. Likewise, BB0276 and BB0492 are unchanged between B31 and N40. The high number of clustered nucleotide substitutions at these HDNP loci, in conjunction with the mostly single-lineage origin of these polymorphisms, strongly suggests that most, if not all, of these HDNPs were introduced en masse by genetic exchange rather than by multiple point mutations. (As examples, the significant clustering of single-partition polymorphisms at BB0032, the BB0082–BB0084 region, and BB0833 are shown in Fig. 6, which is published as supporting information on the PNAS web site.) Indeed, tests of recombination (31, 32) using three-way alignments of the HDNP loci show significantly nonrandom runs of single-partition substitutions on 11 chromosomal and 5 plasmid ORFs, supporting the origin of these HDNPs by recombination. However, such tests of recombination are known to be conservative and to vastly underestimate the true magnitude of recombination (44). A few ORFs showing hypervariability in all three pairwise comparisons [prominent examples are BB0144, coding for a putative glycine/betaine/l-proline ATP-binding cassette transporter, and BBB19 (ospC)] are likely to be results of multiple recombination events at the same loci.
Fig. 1.

Variability of HDNP loci. Pairwise nucleotide differences of B. burgdorferi HDNPs are shown. Data for some pairs are missing (no vertical bar). (A) HDNPs on the main chromosome. (B) Plasmid HDNPs. Note that most ORFs are polymorphic in two of the three pairwise genome comparisons and varied little in the third pairwise comparison (low vertical bar). BBB19 (ospC), BBA24, and BB0144 stand out as the only ORFs that are significantly (P < 0.001) variable in all three pairwise comparisons. Note the scale difference in the vertical axis between A and B. *, ORFs used in MLST.
Fig. 1 shows that the largest fraction of the HDNPs was introduced during the evolution of the N40 lineage (high red and blue bars with a low green bar). For instance, the HDNPs at the right end of lp54, encompassing BBA68, BBA69, and BBA70, were apparently introduced by a single event of recombination into N40. Likewise, part or all of cp26 was introduced by one event into JD1, and another event converted the lp25 in JD1. The consecutive runs of HDNPs sharing a single phylogenetic partition on these plasmids (Fig. 1) suggest that whole-plasmid transfers between B. burgdorferi strains may well be responsible for these information-transfer events. Plasmids that are not in our study are largely those that are homologous to the cp32 plasmids in B31. Had these plasmids been included, our conclusion of large-scale genetic exchange mediated by plasmid transfer would likely have been strengthened, given the recent results of Stevenson and Miller (22).
MLST Analysis of Genomes and Clinical Isolates. The central role of recombination in driving B. burgdorferi adaptive diversification was confirmed by genomic typing at one chromosomal and seventeen plasmid-borne polymorphic loci for 18 additional B. burgdorferi sensu stricto isolates from human skin and blood (Table 3). These isolates were chosen to include multiple representatives of 11 genetically diverse ospC types [major groups A–U of ospC (19)]. Many ospC types were represented twice or more to maximize the chance of detecting recent mutation and recombination. The MLST system (8, 9) designed for the genomic typing of pathogenic bacteria typically uses selected housekeeping genes. However, this strategy is not suitable for genomic typing of B. burgdorferi because its genome contains a large number of plasmids that carry only very few housekeeping genes (23, 24, 45). Therefore, identification of events such as genetic exchanges by way of plasmid transfer requires the inclusion of nonhousekeeping markers to cover the wide range of replicons.
Comparison of these 18 genes across 21 strains led us to several insights. First, none of the 17 non-ospC ORFs that were analyzed shows hypervariability like ospC. Only a small number (two to five, mostly two) of alleles are present at each HDNP loci except ospC, which has 11 “major-group” alleles (Fig. 2). Even based on incomplete genome comparisons and a limited number of isolates, the absence of ospC-like hypervariability among these most variable markers is significant and suggests that ospC is under the strongest form of balancing selection across the genome. [Recombination at vlsE generates transient diversity during infection (46), unlike the evolutionarily stable balanced polymorphisms at ospC.] Second, gene trees (shown in Fig. 7, which is published as supporting information on the PNAS web site) among MLST loci, especially those on different plasmids, are often incongruent, strongly indicating gene exchange (34). Other evidence for recombination includes the presence of all four combinations of alleles (most frequent alleles, brown and green in Fig. 2) at pairs of loci, such as BBC02/BBD14, BBD14/BBG32, and BBE17/BBG32. Incongruent gene genealogies and allele reassortment often occur among loci located on different plasmids, further supporting the hypothesis that plasmid transfer is the main mechanism of recombination. Third, strains of the same ospC types generally share the same MLST genotype (Fig. 2). The fact that genetic homogeneity among the MLST genotypes sharing the same ospC is disrupted only rarely by gene exchange at non-ospC markers suggests that B. burgdorferi population structure is dominated by balanced polymorphisms at ospC. Therefore, the ospC groups could be viewed as evolutionarily stable “clonal complexes” (9) within B. burgdorferi populations. Last, we observed breakdown of the homogeneity of ospC clonal complexes by recent events of recombination (boxed region in Fig. 2). One such event occurred at BBQ49 among 132a, 132b, and B31 within the ospC group A complex. Other events transferred fragments spanning lp54 BBA68, BBA69, and BBA70; a divergent lp28–4 BBI38 allele into the N40 genome; and an lp36 BBJ19 allele into the 136b genome. The putative direction of these lateral gene transfers was determined by assuming that the most common allelic type among the B31, JD1, and N40 genomes is the ancestral type.
Fig. 2.

Multilocus sequence types of the three genomes and 18 clinical isolates. For each gene (column), colors represent various major-group alleles. Blank spaces indicate that data were not obtained because of either a lack of amplification or multiple amplicons in PCR. Isolates of the same ospC types (labeled on the right) share alleles across all surveyed loci except at loci enclosed in the red boxes, to which divergent alleles were introduced by recent genetic exchange.
Relative Rates of Recombination and Mutation. Rates of recombination relative to mutation were estimated by counting the number of recombination and mutation events among strains belonging to the same ospC clonal complexes (35, 47). All SNPs at BB0082 are shown in Fig. 8, which is published as supporting information on the PNAS web site. Six ospC clonal complexes (A, B, E, G, I, and K) are represented with two or more isolates. Isolates within the A, E, and K complexes show no polymorphism. Isolates within the complexes B, G, and I are segregated with one identical SNP at site 17, which is likely introduced by recombination. Only one mutational event was found within the G complex (at site 823) because this SNP is not found in any other clonal complexes. Therefore, there were three events of recombination and one event of mutation during the recent divergence of isolates within the B, G, and I clonal complexes at this chromosomal locus, resulting in a recombination to mutation (r/m) ratio of 3:1. By using SNPs from one chromosomal locus and 16 plasmid loci (excluding ospC), we found a total of six mutations and 16 recombination events (r/m, 2.7:1). These values are smaller than the ratio found in more freely recombining bacterial species, such as Neisseria meningitidis and Streptococcus pneumoniae (r/m, 5:1 to 10:1; ref. 48), and larger than the ratio in more clonal species, such as Staphylococcus aureus (r/m, 1:15; ref. 49).
Discussion
Scale-Dependent Population Structure of B. burgdorferi. Bacterial species show different degrees of clonality (17, 50). Many bacterial species show a freely recombining population structure, in which genetic exchange among bacterial cells is frequent enough that there are no stable clones. Examples are N. meningitidis, S. pneumoniae, Staphylococcus pyogenes (9), Helicobacter pylori (51, 52), and the Bacillus cereus species group (53). Other species show a more clonal population structure, such as Haemophilus influenzae, Escherichia coli (9), Salmonella enterica (54), and Sta. aureus (49). An often neglected aspect in the discussion of bacterial clonality is the choice of isolates used in such an analysis. Frank (18) recognized the effect of the geographic and ecological sampling scale in characterizing the population structure of microbial species. For instance, a critical factor in understanding the role of recombination in the clonal divergence in E. coli was the sampling of isolates within the same subgroup (47). MacLeod et al. (55) showed that proper characterization of the genetic structure of Trypanosoma brucei depends critically on recognizing population subdivisions due to geographical and ecological isolation.
Cross-population sampling is likely the main reason most previous studies of B. burgdorferi population structure (e.g., ref. 14) failed to show recombination beyond a few selected loci. These studies used laboratory archival strains that were obtained from diverse locations on two continents and belonged to different species in the B. burgdorferi species complex. We were able to observe past genetic exchange in B. burgdorferi sensu stricto by comparing isolates from endemic regions in the northeastern United States. B. burgdorferi from these regions forms a geographically and ecologically uniform metapopulation, which is founded on a single rather recent ancestral population (56) and maintained in a similar enzootic transmission cycle with the white-footed mouse (Peromyscus leucopus) as the main reservoir and the northern lineage of black-legged ticks (Ixodes scapularis) as the main vector (57).
The scale-dependence of bacterial population structures is expected to be a more prominent characteristic in obligate parasitic and geographically structured species (such as B. burgdorferi) than in free-living and more cosmopolitan species (such as E. coli). B. burgdorferi is a vector-borne pathogen, further restricting the chance of direct encounter and gene transfers between local clones. Even among B. burgdorferi clones from the same geographic region, there could be niche differentiation and ecological isolation, resulting in a lack of gene flow between sympatric clones or species. Our study shows that whole-genome sequencing of a small number of local clones (preferably with different ospC types) will be an effective means for fine-scale characterization of local B. burgdorferi population structure in other endemic regions of Lyme disease. We conclude from this study and previous studies that, although B. burgdorferi shows a high degree of clonality between geographically or ecologically isolated populations (15, 20), it is quite capable of genome-wide genetic exchange within the same ecological populations.
Selective Maintenance of ospC Clonal Complexes. Recombination is a powerful facilitator of species adaptation. Rare beneficial alleles are more likely to be fixed in a population by avoiding “Muller's Ratchet” (58), the mutational meltdown and rapid fitness loss of an asexual species. More important, beneficial combinations of alleles among loci can arise quickly through random genome assortment. The most popular MLST system of detecting recombination in a bacterial species uses neutral variations of housekeeping genes (8, 59). Use of neutral markers allows estimation of the rate and fragment size of lateral gene transfers. Feil et al. (35) defined a bacterial clonal complex as a group of isolates identical in their MLST genotypes and the immediate descendant variants of these isolates. The adaptive relevance of clonal complexes defined by shared neutral variations is generally not known, and the target of selection, if present, could not be identified. In contrast, we used a set of mostly nonhousekeeping genes in consideration of the highly segmented nature of B. burgdorferi genomes and the potential clinical significance of nonneutral variations. Our study shows that ospC is likely to be a dominant locus in the selective maintenance of B. burgdorferi diversity in nature. Previous evidence for the extraordinarily strong balancing selection at ospC includes the within-population diversity and geographic uniformity of ospC allele frequencies (19, 56). Hypervariable sites of ospC have been mapped to its surface (60). The age of balanced polymorphisms may predate divergence among B. burgdorferi species (20). Comparative genomics shows that the balancing selection present at ospC may be the strongest across the genome. This conclusion is mainly based on the observations that (i) we failed to find ospC-like hypervariability at this genome-wide, albeit incomplete, survey of ORF variability, and (ii) MLST genotypes are largely definable by their ospC types.
We also conclude that, although the strongest form of balancing selection in B. burgdorferi may be operating at ospC, highly divergent HDNPs at other plasmid loci are likely maintained by balancing selection as well. Our reasoning is as follows. On the main chromosome, the HDNPs are predominantly synonymous polymorphisms, whereas on the plasmids the level of nonsynonymous substitutions is almost as high as that of synonymous substitutions (Fig. 3). The high Ka/Ks ratio of plasmid HDNPs may be due either to positive selection or relaxed selective constraint. However, one might expect to observe a high number of sequence variants at these loci if these sequences were to diverge through relaxation of selection. Instead, we observed only two to three major-group alleles of ORFs in multiple isolates of these HDNP markers. Sequences of plasmid non-ospC HDNPs are highly divergent, suggesting that these are ancient alleles that have been maintained in population for a long time. Maintenance of divergent alleles in high frequencies is a hallmark of balanced polymorphisms (e.g., ospC, or Adh in Drosophila melanogaster; ref. 61). However, balancing selection at these non-ospC plasmid HDNPs is not as dominant as selection at ospC in influencing the B. burgdorferi population structure.
Fig. 3.

Rates of synonymous and nonsynonymous substitutions in HDNP ORFs. Ka vs. Ks of HDNPs between B31 and N40 are shown. (A) Chromosomal HDNPs. ORFs are mostly far below the Ka = Ks neutrality line (solid line) and there is no Ka ≈ Ks correction (regression line in dashes; R2 not significant), indicating strong selective constraints on amino acid substitution. (B). Plasmid HDNPs. ORFs are closer to the Ka = Ks neutrality line and Ka is strongly correlated (R2 = 0.83, P « 0.001) with Ks, likely resulting from balancing selection. Note the difference in scale between A and B.
Plasmid HDNPs as Virulence and Phylogenetic Markers. Based on the putative selective prominence of ospC and other plasmid HDNPs, we suggest that these plasmid ORFs, mostly with unknown functions, may play a role in interaction with the host and, therefore, may be potential virulence factors. The HDNPs that we identified include (besides ospC) genes such as dbpA (BBA24), whose product is involved in host-cell binding (42), and BBA68, which encodes the complement resistance protein BbCRASP-1 (43), but not ospA or ospB (BBA15/16) outer-surface protein genes. Associative studies of the alleles of plasmid HDNPs with Lyme disease symptomology may clarify the role of potential virulence factors. The present study provides a library of potential markers for the study of B. burgdorferi virulence in humans (39, 62).
However, the HDNPs revealed from genome comparisons should not be used to infer the overall phylogenetic relationships among B. burgdorferi isolates. Frequent recombination precludes the reconstruction of intraspecific phylogenies for B. burgdorferi isolates based on such polymorphisms. Nonetheless, plasmid HNDPs may prove to be valuable markers for classifying B. burgdorferi isolates immunologically (18) and clinically.
In summary, comparative genomics and MLST analysis of closely related isolates revealed genome-wide genetic exchange and plasmid transfers in B. burgdorferi. The mechanisms performing such transfers remain unproven. However, we note that B. burgdorferi carries the requisite homologous-recombination machinery to incorporate genetic material that it might take up (63, 64), bacteriophage-mediated transduction has been demonstrated in the laboratory (65), and both ticks and reservoir hosts are capable of being simultaneously infected by multiple ospC clones (19, 56) (D. Brisson and D. E. Dykhuizen, personal communication). Balanced polymorphisms at ospC and other plasmid HDNP loci, which appear to be cornerstones of B. burgdorferi adaptation, would not be maintained independently if B. burgdorferi were a predominantly clonal species, in which genetic variability is easily lost by selective sweeps and genetic drift (66–68).
Supplementary Material
Acknowledgments
We thank Dan Dykhuizen (Stony Brook University) for comments and discussions, three anonymous reviewers for highly constructive critiques, and Martin Schriefer (Center for Disease Control and Prevention, Atlanta, GA) and Steven Barthold (University of California, Davis) for providing B. burgdorferi strains. This work was supported by a grant from the Lyme Disease Association, Inc. (Jackson, NJ) and National Institutes of Health Grants AI37256 (to J.J.D. and B.J.L.) and AI49003 (to S.R.C.). W.-G.Q. was supported by Howard Hughes Medical Institute Undergraduate Science Education Program in Biology Grant 52002679 and by National Institutes of Health Research Centers in Minority Institutions Award RR03037.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MLST, multilocus sequence typing; MLEE, multilocus enzyme electrophoresis; SNP, single-nucleotide polymorphism; HDNP, high-density nucleotide polymorphism.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY696304–AY696571).
References
- 1.Ruan, Y. J., Wei, C. L., Ee, A. L., Vega, V. B., Thoreau, H., Su, S. T., Chia, J. M., Ng, P., Chiu, K. P., Lim, L., et al. (2003) Lancet 361, 1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Read, T. D., Salzberg, S. L., Pop, M., Shumway, M., Umayam, L., Jiang, L., Holtzapple, E., Busch, J. D., Smith, K. L., Schupp, J. M., et al. (2002) Science 296, 2028–2033. [DOI] [PubMed] [Google Scholar]
- 3.Perna, N. T., Plunkett, G., III, Burland, V., Mau, B., Glasner, J. D., Rose, D. J., Mayhew, G. F., Evans, P. S., Gregor, J., Kirkpatrick, H. A., et al. (2001) Nature 409, 529–533. [DOI] [PubMed] [Google Scholar]
- 4.Read, T. D., Peterson, S. N., Tourasse, N., Baillie, L. W., Paulsen, I. T., Nelson, K. E., Tettelin, H., Fouts, D. E., Eisen, J. A., Gill, S. R., et al. (2003) Nature 423, 81–86. [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann, R. D., Alland, D., Eisen, J. A., Carpenter, L., White, O., Peterson, J., DeBoy, R., Dodson, R., Gwinn, M., Haft, D., et al. (2002) J. Bacteriol. 184, 5479–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423, 241–254. [DOI] [PubMed] [Google Scholar]
- 7.Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. (2002) Nature 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 8.Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., Zhang, Q., Zhou, J., Zurth, K., Caugant, D. A., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 3140–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feil, E. J. & Spratt, B. G. (2001) Annu. Rev. Microbiol. 55, 561–590. [DOI] [PubMed] [Google Scholar]
- 10.Morelli, G., Malorny, B., Muller, K., Seiler, A., Wang, J. F., del Valle, J. & Achtman, M. (1997) Mol. Microbiol. 25, 1047–1064. [DOI] [PubMed] [Google Scholar]
- 11.Baranton, G., Marti Ras, N. & Postic, D. (1998) Wien Klin. Wochenschr. 110, 850–855. [PubMed] [Google Scholar]
- 12.Hurst, L. D. (2002) Trends Genet. 18, 486–487. [DOI] [PubMed] [Google Scholar]
- 13.Jordan, I. K., Rogozin, I. B., Wolf, Y. I. & Koonin, E. V. (2002) Theor. Popul. Biol. 61, 435–447. [DOI] [PubMed] [Google Scholar]
- 14.Balmelli, T. & Piffaretti, J. C. (1996) Int. J. Syst. Bacteriol. 46, 167–172. [DOI] [PubMed] [Google Scholar]
- 15.Dykhuizen, D. E., Polin, D. S., Dunn, J. J., Wilske, B., Preac-Mursic, V., Dattwyler, R. J. & Luft, B. J. (1993) Proc. Natl. Acad. Sci. USA 90, 10163–10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maynard Smith, J. & Smith, N. H. (1998) Mol. Biol. Evol. 15, 590–599. [DOI] [PubMed] [Google Scholar]
- 17.Maynard Smith, J., Feil, E. J. & Smith, N. H. (2000) BioEssays 22, 1115–1122. [DOI] [PubMed] [Google Scholar]
- 18.Frank, S. A. (2002) Immunology and Evolution of Infectious Disease (Princeton Univ. Press, Princeton). [PubMed]
- 19.Wang, I. N., Dykhuizen, D. E., Qiu, W., Dunn, J. J., Bosler, E. M. & Luft, B. J. (1999) Genetics 151, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dykhuizen, D. E. & Baranton, G. (2001) Trends Microbiol. 9, 344–350. [DOI] [PubMed] [Google Scholar]
- 21.Marconi, R. T., Samuels, D. S., Landry, R. K. & Garon, C. F. (1994) J. Bacteriol. 176, 4572–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevenson, B. & Miller, J. C. (2003) J. Mol. Evol. 57, 309–324. [DOI] [PubMed] [Google Scholar]
- 23.Fraser, C. M., Casjens, S., Huang, W. M., Sutton, G. G., Clayton, R., Lathigra, R., White, O., Ketchum, K. A., Dodson, R., Hickey, E. K., et al. (1997) Nature 390, 580–586. [DOI] [PubMed] [Google Scholar]
- 24.Casjens, S., Palmer, N., van Vugt, R., Huang, W. M., Stevenson, B., Rosa, P., Lathigra, R., Sutton, G., Peterson, J., Dodson, R. J., et al. (2000) Mol. Microbiol. 35, 490–516. [DOI] [PubMed] [Google Scholar]
- 25.Delcher, A. L., Phillippy, A., Carlton, J. & Salzberg, S. L. (2002) Nucleic Acids Res. 30, 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. (1999) Nucleic Acids Res. 27, 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nei, M. & Gojobori, T. (1986) Mol. Biol. Evol. 3, 418–426. [DOI] [PubMed] [Google Scholar]
- 29.Korber, B. (2000) in Computational and Evolutionary Analysis of HIV Molecular Sequences, eds. Rodrigo, A. G. & Leam, G. H. (Kluwer, Dordrecht, The Netherlands), pp. 55–72.
- 30.Kuhner, M. K., Lawlor, D. A., Ennis, P. D. & Parham, P. (1991) Tissue Antigens 38, 152–164. [DOI] [PubMed] [Google Scholar]
- 31.Stephens, J. C. (1985) Mol. Biol. Evol. 2, 539–556. [DOI] [PubMed] [Google Scholar]
- 32.Sawyer, S. (1989) Mol. Biol. Evol. 6, 526–538. [DOI] [PubMed] [Google Scholar]
- 33.Huelsenbeck, J. P. & Ronquist, F. (2001) Bioinformatics 17, 754–755. [DOI] [PubMed] [Google Scholar]
- 34.Dykhuizen, D. E. & Green, L. (1991) J. Bacteriol. 173, 7257–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feil, E. J., Smith, J. M., Enright, M. C. & Spratt, B. G. (2000) Genetics 154, 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barthold, S. W., Moody, K. D., Terwilliger, G. A., Duray, P. H., Jacoby, R. O. & Steere, A. C. (1988) J. Infect. Dis. 157, 842–846. [DOI] [PubMed] [Google Scholar]
- 37.Piesman, J., Mather, T. N., Sinsky, R. J. & Spielman, A. (1987) J. Clin. Microbiol. 25, 557–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgdorfer, W., Barbour, A. G., Hayes, S. F., Benach, J. L., Grunwaldt, E. & Davis, J. P. (1982) Science 216, 1317–1319. [DOI] [PubMed] [Google Scholar]
- 39.Wormser, G. P., Liveris, D., Nowakowski, J., Nadelman, R. B., Cavaliere, L. F., McKenna, D., Holmgren, D. & Schwartz, I. (1999) J. Infect. Dis. 180, 720–725. [DOI] [PubMed] [Google Scholar]
- 40.Casjens, S., Delange, M., Ley, H. L., III, Rosa, P. & Huang, W. M. (1995) J. Bacteriol. 177, 2769–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathiesen, D. A., Oliver, J. H., Jr., Kolbert, C. P., Tullson, E. D., Johnson, B. J., Campbell, G. L., Mitchell, P. D., Reed, K. D., Telford, S. R., III, Anderson, J. F., et al. (1997) J. Infect. Dis. 175, 98–107. [DOI] [PubMed] [Google Scholar]
- 42.Guo, B. P., Norris, S. J., Rosenberg, L. C. & Hook, M. (1995) Infect. Immun. 63, 3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraiczy, P., Hellwage, J., Skerka, C., Becker, H., Kirschfink, M., Simon, M. M., Brade, V., Zipfel, P. F. & Wallich, R. (2004) J. Biol. Chem. 279, 2421–2429. [DOI] [PubMed] [Google Scholar]
- 44.Posada, D. & Crandall, K. A. (2001) Proc. Natl. Acad. Sci. USA 98, 13757–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casjens, S. (2000) J. Mol. Microbiol. Biotechnol. 2, 401–410. [PubMed] [Google Scholar]
- 46.Zhang, J. R. & Norris, S. J. (1998) Infect. Immun. 66, 3698–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guttman, D. S. & Dykhuizen, D. E. (1994) Science 266, 1380–1383. [DOI] [PubMed] [Google Scholar]
- 48.Spratt, B. G., Hanage, W. P. & Feil, E. J. (2001) Curr. Opin. Microbiol. 4, 602–606. [DOI] [PubMed] [Google Scholar]
- 49.Feil, E. J., Cooper, J. E., Grundmann, H., Robinson, D. A., Enright, M. C., Berendt, T., Peacock, S. J., Smith, J. M., Murphy, M., Spratt, B. G., et al. (2003) J. Bacteriol. 185, 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spratt, B. G. & Maiden, M. C. (1999) Philos. Trans. R. Soc. London B 354, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smeets, L. C., Arents, N. L., van Zwet, A. A., Vandenbroucke-Grauls, C. M., Verboom, T., Bitter, W. & Kusters, J. G. (2003) Infect. Immun. 71, 2907–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suerbaum, S., Smith, J. M., Bapumia, K., Morelli, G., Smith, N. H., Kunstmann, E., Dyrek, I. & Achtman, M. (1998) Proc. Natl. Acad. Sci. USA 95, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helgason, E., Tourasse, N. J., Meisal, R., Caugant, D. A. & Kolsto, A. B. (2004) Appl. Environ. Microbiol. 70, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown, E. W., Kotewicz, M. L. & Cebula, T. A. (2002) Mol. Phylogenet. Evol. 24, 102–120. [DOI] [PubMed] [Google Scholar]
- 55.MacLeod, A., Tweedie, A., Welburn, S. C., Maudlin, I., Turner, C. M. & Tait, A. (2000) Proc. Natl. Acad. Sci. USA 97, 13442–13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu, W. G., Dykhuizen, D. E., Acosta, M. S. & Luft, B. J. (2002) Genetics 160, 833–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane, R. S., Piesman, J. & Burgdorfer, W. (1991) Annu Rev Entomol 36, 587–609. [DOI] [PubMed] [Google Scholar]
- 58.Muller, H. J. (1964) Mutat. Res. 1, 2–9. [DOI] [PubMed] [Google Scholar]
- 59.Robertson, G. A., Thiruvenkataswamy, V., Shilling, H., Price, E. P., Huygens, F., Henskens, F. A. & Giffard, P. M. (2004) J. Med. Microbiol. 53, 35–45. [DOI] [PubMed] [Google Scholar]
- 60.Kumaran, D., Eswaramoorthy, S., Luft, B. J., Koide, S., Dunn, J. J., Lawson, C. L. & Swaminathan, S. (2001) EMBO J. 20, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kreitman, M. (1983) Nature 304, 412–417. [DOI] [PubMed] [Google Scholar]
- 62.Seinost, G., Dykhuizen, D. E., Dattwyler, R. J., Golde, W. T., Dunn, J. J., Wang, I. N., Wormser, G. P., Schriefer, M. E. & Luft, B. J. (1999) Infect. Immun. 67, 3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuels, D. S., Mach, K. E. & Garon, C. F. (1994) J. Bacteriol. 176, 6045–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosa, P., Samuels, D. S., Hogan, D., Stevenson, B., Casjens, S. & Tilly, K. (1996) J. Bacteriol. 178, 5946–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eggers, C. H., Kimmel, B. J., Bono, J. L., Elias, A. F., Rosa, P. & Samuels, D. S. (2001) J. Bacteriol. 183, 4771–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dykhuizen, D. (1992) in Encyclopedia of Microbiology, ed. Lederberg, J. (Academic, London), Vol. 3, pp. 351–355. [Google Scholar]
- 67.Cohan, F. M. (1994) Trends Ecol. Evol. 9, 175–180. [DOI] [PubMed] [Google Scholar]
- 68.Guttman, D. S. (1997) Trends Ecol. Evol. 12, 16–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}