

Abstract
Mechanism of photo-induced electron transfer and the subsequent hole transfer in DNA has been studied extensively, but so far we are not aware of any reliable report of the observation of the long-distance hole-transfer process. In this article, we demonstrate the results of direct observation for the long-distance hole transfer in double-helical DNA over 100 Å with time-resolved transient absorption measurements. DNA conjugated with naphthalimide (NI) and phenothiazine (PTZ) (which worked as electron-acceptor and donor molecules, respectively) at both 5′ ends was synthesized to observe the hole-transfer process. Site-selective charge injection into G by means of the adenine-hopping process was accomplished by excitation of NI with a 355-nm laser flash. Transient absorption around 400 nm, which was assigned to the NI radical anion, was observed immediately after the irradiation of a laser flash, indicating that the charge separation between NI and the nearest G occurred. Then, the transient absorption of the PTZ radical cation (PTZ•+) at 520 nm was emerged, which was attributed to the hole transfer through DNA to the PTZ site. By monitoring the time profiles of the transient absorption of PTZ•+ for NI-A6-(GA)n-PTZ and NI-A6-(GT)n-PTZ (n = 2, 3, 4, 6, 8, 12) (base sequences correspond to those for DNA modified with NI), the long-distance hole-transfer process from G to PTZ, which occurred in the time scale of microsecond to millisecond, was observed directly. By assuming an average distance of 3.4 Å between base-pairs, total distance reaches 100 Å for n = 12 sequences. Our results clearly show the direct observation of the long-distance hole transfer over 100 Å.
Double-helical DNA, which has unique structural properties, is a promising candidate for molecular electronic devices and a scaffold for two- and three-dimensional nanostructures (1, 2). Mechanistic studies of charge transfer in DNA have attracted considerable attention because of their relevance to the development of DNA molecular wires (3) and the involvement in DNA oxidative lesion and strand cleavage (4, 5). There are many mechanistic studies for the “one-dimensional” conductivity in the double helix (6–8). Factors controlling charge-transfer properties in DNA (such as electronic coupling between donor and acceptor, structural dynamics, reorganization energy, etc.) have been discussed (9–14), and the mechanism for charge transfer, especially hole transfer over long distances in DNA, is the subject of experimental and theoretical studies.
Generally, experimental studies of charge transfer in DNA have been performed by time-resolved measurements and electrophoresis analysis (15–19). A model of a multistep hole-hopping mechanism, which was proposed by Giese and coworkers (20), is the most widely adopted. An alternative mechanism in which delocalized hole migrates by means of a polaron-like mechanism was proposed by Schuster and coworkers (18). Strand-cleavage studies using PAGE analysis have provided valuable information about the sequence- and distance-dependence of hole migration for various DNA structures, such as not only double-helical DNA but also triplex and quadraplex DNA (21–25).
Time-resolved measurements during laser flash photolysis are useful for investigating the kinetics of the charge-transfer process in DNA. Lewis et al. (15) and Wasielewski and coworkers (26) reported the kinetic study of single-step hole transfer in DNA by means of time-resolved transient absorption measurements. They determined the hole-transfer rate from G to GG by monitoring the decay of stilbene radical anion. Shafirovich et al. (27, 28) have investigated the distance-dependence of hole transfer from the radical cation of 2-aminopurine to guanine across variable numbers of AT base pairs by means of two-photon ionization with time-resolved transient absorption measurements.
However, experimental evidence for the long-distance hole transfer in DNA was based primarily on the strand-cleavage studies. Gel electrophoretic analysis clearly demonstrated that the hole can migrate over 100 Å, whereas time-resolved transient absorption measurement was limited to the kinetics of the single-step (26) or short-distance (29, 30) hole transfer. To our knowledge, there is no reliable report for the kinetics of the multistep long-distance hole transfer because of the difficulty of efficiently generating a hole in DNA, and therefore, the kinetics of the long-distance hole transfer still remains unclear.
Previously, we demonstrated that the adenine hopping (A-hopping) occurred very rapidly (>108 s–1) and can be applied to generate a long-lived charge-separated state in DNA (31, 32). Accordingly, it is expected that a long-lived hole escaped from charge recombination can be generated effectively in DNA by using the A-hopping mechanism, which enables us to observe the hole-transfer process occurring in the much slower time scale. In this article, we clearly show the direct observation of the long-distance hole transfer in DNA over 100 Å as well as the sequence- and distance-dependence of the hole transfer.
Materials and Methods
DNA Synthesis. DNA used in this study was synthesized on a DNA synthesizer (Applied Biosystems) with standard solid-phase techniques and purified on an HPLC system (Jasco, Tokyo) with a reverse-phase C-18 column with an acetonitrile/50 mM ammonium formate gradient. Duplex solutions (20 mM sodium phosphate buffer, pH 7.0/100 mM sodium chloride) were prepared by mixing equimolar amounts of the desired oligode-oxynucleotide complements and annealing gradually with cooling from 80°C to room temperature. DNA conjugated with phenothiazine (PTZ) and naphthalimide (NI) at the 5′ end were synthesized according to the procedure described in refs. 33 and 34.
Laser Flash Photolysis Experiments. The experiments were carried out at 23°C with a duplex concentration of 100 μM in 20 mM sodium phosphate, pH 7.0/100 mM sodium chloride. Nanosecond transient absorption measurements were performed by using the LFP technique (31, 35). The third-harmonic oscillation (355 nm, full width at half maximum of 4 ns, 20 mJ per pulse) from a Q-switched neodymium-doped yttrium aluminum garnet (Nd:YAG) laser (Surelight, Continuum, Santa Clara, CA) was used to excite the NI site selectively because NI showed strong absorption around 355 nm (ε355 ≈ 8 × 103 M–1·cm–1) and PTZ showed no absorption at wavelengths longer than 320 nm (32). A xenon flash lamp (XBO-450, Osram, Berlin) was focused into the sample solution as the probe light for the transient absorption measurement. Time profiles of the transient absorption in the UV-visible region were measured with a monochromator (G250, Nikon) equipped with a photomultiplier (R928, Hamamatsu Photonics, Hamamatsu City, Japan) and digital oscilloscope (TDS-580D, Tektronix). Obtained kinetic signals were simply analyzed by single-exponential fits to determine the apparent hole-transfer mechanism (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site).
Results and Discussion
Strategy for the Direct Observation of Long-Distance Hole Transfer Through Double-Helical DNA. We have demonstrated (32) that the long-lived charge-separated state can be generated efficiently in DNA conjugated with naphthaldiimide (NDI) and PTZ by using A-hopping processes after the excitation of the NDI site with a 355-nm laser pulse. Charge-separation process between donor and acceptor based on A-hopping is advantageous for generating a long-lived hole compared with a superexchange-type mechanism because the multistep process weakly depends on the distance between donor and acceptor (36). Our approach for the time-resolved transient absorption measurements of the hole transfer is shown schematically in Fig. 1. The chemical structures of NI and PTZ linked to 5′ terminus are shown in Fig. 2a. Planar molecules, NI and PTZ, are expected to associate with DNA terminus by end capping. Increase of the stability of double-helical DNA attached with NI or PTZ has been reported (34, 37). Schuster and coworkers (38) also reported a similar association mode with respect to anthraquinone derivatives attached to the 5′ end of duplex DNA. The DNA sequences used in this study are shown in Fig. 2b. DNA conjugated with NI and PTZ as an electron acceptor and donor, respectively, was synthesized according to the procedure described in refs. 33 and 34.
Fig. 1.

Strategy for the kinetic study of long-distance hole-transfer process. A molecular representation for 30-mer DNA conjugated with NI (blue) and PTZ (red), which work as electron acceptor and donor, respectively, is shown. A schematic representation for hole injection by means of A-hopping and long-distance hole-transfer process in DNA is also shown.
Fig. 2.

Chemical structures of photosensitizer (NI) and hole probe molecule (PTZ), and DNA sequences. (a) Chemical structures of NI and PTZ, which are attached to 5′-terminus of each DNA strand through alkyl linker, respectively. (b) DNA sequences used in this study.
Radical anion of NI (NI•–) and radical cation of PTZ (PTZ•+) have a unique characteristic absorption in the visible region, show little spectral overlap, and therefore, can be used to monitor the charge-separated state and the hole-migration process after the laser excitation with time-resolved transient absorption measurements. Photophysical and redox properties for NI, PTZ, and nucleobases have been established (39–42). From the reduction potential of –1.0 V (vs. normal hydrogen electrode) for NI and the excitation energy of 3.4 eV for NI in the singlet excited state (1NI*), reduction potential of 1NI* is calculated to be 2.4 eV. Because a high reduction potential of 1NI* is enough to oxidize all nucleobases, the charge separation between 1NI* and A is expected to occur initially after the excitation of the NI site when NI is stacked with the AT base pair.
Irradiation of the NI site with a 355-nm laser flash causes charge separation between NI and adjacent A to give NI•– and A radical cation (A•+), respectively. A part of a hole escapes from the initial charge-recombination process by means of A-hopping and is trapped at G. When G radical cation (G•+) is generated far from NI•–, charge recombination proceeds by either strongly distance-dependent superexchange or A-hopping after slow A oxidation by G•+ (43). Thus, the charge-separated state persists more than several hundred microseconds, allowing the direct observation of the long-distance hole-transfer process.
Direct Observation of Hole-Transfer Process Through Double-Helical DNA. Immediately after the excitation of the NI site for (GT)4 with a 355-nm laser flash, characteristic transient absorption spectrum with a peak at 400 nm assigned to NI•– and characterless and weak transient absorption around 500 nm were observed. Considering that NI•– shows very weak absorption at wavelengths longer than 450 nm, the absorption around 500 nm can be assigned to G•+ or deprotonated G• (–H+) (44). These results showed that the charge separation (hole injection) occurred within a laser flash duration (<5 ns) (Fig. 3). Then, a transient absorption spectrum with a peak around 500 nm, assigned to PTZ•+, emerged in the time scale of several microseconds, which corresponded to the hole transfer through DNA to the PTZ site (Fig. 3 Inset). It was difficult to observe the kinetics of decay of G•+ or deprotonated G• (–H+), corresponding to the hole transfer from G•+ to PTZ, because the extinction coefficients of G•+ and G• (–H+) are much smaller than those of NI•– and PTZ•+ (44).
Fig. 3.

Transient absorption spectra observed at 200 ns (black), 2 μs (red), 5 μs (green), and 10 μs (blue) after a 355-nm laser flash (4 ns, 20 mJ·pulse–1), with excitation of the NI site during the laser flash photolysis of NI- and PTZ-conjugated DNA. (Inset) Time profiles observed at 400 (black) and 520 (blue) nm assigned to the transient absorption peak for NI•– and PTZ•+, respectively, after irradiation of NI with a 355-nm laser flash. Sample solution contained 100 μM duplex DNA, 20 mM sodium phosphate buffer (pH 7.0), and 100 mM sodium chloride.
The transient absorption of NI•– monitored at 400 nm persisted over several hundred microseconds when NI was separated from the nearest G by six AT base pairs. Slow decay of NI•– is probably attributed to the charge recombination by an intermolecular collisional process. Time profiles monitored at 520 nm, which corresponded to the hole-transfer process, were independent of the concentration of DNA in the range of 10–100 μM, suggesting that the formation of PTZ•+ corresponds to the intramolecular process (that is, hole-transfer process through DNA). In addition, transient absorption monitored at 200 ns for DNA conjugated with only NI was similar with that for duplex DNA modified with NI and PTZ, showing that the initial charge-separation process between NI and the nearest G is not affected by the presence of PTZ located at opposite 5′ end (Fig. 7, which is published as supporting information on the PNAS web site). These results indicate that the charge separation between NI and the nearest G occurs initially and that the hole migrates along the DNA to reach PTZ.
Time profiles of the transient absorption of PTZ•+ monitored at 520 nm for (GT)n and (GA)n sequences are shown in Fig. 4. Formation of the absorption at 520 nm within a laser pulse (5 ns) was observed because of the G• (–H+) generated within the laser flash duration by charge-separation process through A-hopping, as shown in Fig. 1. By increasing of the number of GA repeats, the formation rate of PTZ•+ for these sequences was slightly decreased. Apparent hole-transfer rate constants (kht) for (GT)n and (GA)n sequences obtained with single-exponential fits are shown in Table 1. In a precise sense, it may be inappropriate to analyze the data with single-exponential fitting because long-range hole transfer occurs through a multistep process. However, simple analysis based on single-exponential fitting can be applied approximately to evaluate the observed long-range hole-transfer process. Fitting results based on kinetic modeling of multistep hole-hopping process are given in Supporting Materials and Methods, Scheme 1, and Fig. 8, which are published as supporting information on the PNAS web site. The obtained rate constants clearly show that the hole-transfer process depends slightly on the distance, suggesting the multistep hopping process, but are highly sequence-dependent. The kht value for the (GA)n sequence was ≈50-fold larger than that for the (GT)n sequence. It is known that the energy gap between the initial and bridge states affects the electronic coupling (i.e., the electron-transfer rate in D-B-A system; ref. 14). Considering that the ionization potential of A is lower than that of T, the hole transfer for G-A-G sequence is expected to be faster than that for G-T-G sequence. Our results are in good agreement with the prediction, and similar results were reported in other photoinduced electron-transfer systems (26). In contrast, delocalization of a hole over several nucleobases may affect the hole-transfer process (45). Conwell and Basko (12) showed on the basis of a simple tight-binding model that wave function of a hole on G surrounded by A extended to neighboring A. This delocalization of a hole over neighboring A, which has lower oxidation potential than T, might be attributed to the difference in single-step hole-transfer rates. In addition, it is suggested that thermal fluctuations of DNA and its environment can affect the charge-transfer process through double helix. Molecular dynamics simulation shows that the electronic coupling between neighboring base pairs is extremely sensitive to the conformational dynamics of DNA. Therefore, the local dynamics of DNA, which depends on the sequence, should be considered to explain the sequence effect of hole transfer in DNA (11, 46, 47).
Fig. 4.

Time profiles of the transient absorption monitored at 520 nm assigned to PTZ•+ after the 355-nm laser flash excitation for (GA)n (a) and (GT)n (b) sequences. n corresponds to the n of (GA)n or (GT)n. Sample solution contained 100 μM duplex DNA, 20 mM sodium phosphate buffer (pH 7.0), and 100 mM sodium chloride.
Table 1. Rate constants of hole transfer for (GT)n and (GA)n sequences.
| (GT)n | kht*, 105 s-1 | (GA)n | kht*, 105 s-1 |
|---|---|---|---|
| 2 | 11 | 2 | 570 |
| 3 | 5.9 | 3 | 230 |
| 4 | 2.2 | 4 | 130 |
| 6 | 0.67 | 6 | 31 |
| 8 | 0.31 | 8 | 13 |
| 12 | 0.12 | 12 | 6.2 |
Hole-transfer rate constants were determined by fitting the formation of PTZ·+ to a single-exponential function. Experimental error was estimated to be within 10%.
Assuming an average distance of 3.4 Å between base-pairs, total distance reaches 100 Å for n = 12 for both (GA)n and (GT)n sequences. Specifically, our results indicate that the long-distance hole transfer over 100 Å was observed directly with time-resolved transient absorption measurements. To our knowledge, this is the first example for the direct observation of the long-distance hole-transfer process in DNA over 100 Å. Spectroscopic studies for charge-transfer process over several ten angstroms by the flash-quench technique have been reported by Barton and coworkers (48, 49). By monitoring the formation of methylindole radical upon the excitation of ruthenium complex modified to the DNA end, they found that charge transfer occurred faster than 107 s–1 and was independent of the distance between methylindole and ruthenium complex, which was varied by increasing the number of AT base pairs. In contrast, our results show that long-range hole transfer occurs in a much slower time scale and that the single-step hole-transfer process depends on the intervening nucleobases between guanine bases. Therefore, difference in the hole-transfer rates can be attributed to the intervening sequence. Also, it is suggested that the dynamic fluctuation and domain formation defined by DNA sequence and local dynamics affect the charge-transfer process (50).
Mechanistic Analysis of Multistep Hole-Transfer Process. If the hole transfer from G•+ nearest NI to PTZ occurs by random walk process through hopping between Gs, the observed rate constant kht follows:
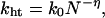 |
[1] |
where k0 is the rate constant of the single-step hole transfer between Gs, N is a hopping number, and η is a proportional factor that takes the value of 2 in the ideal case of random walk process (36, 51–54). Plots of ln kht vs. ln N are shown in Fig. 5. The η value of 2.0 ± 0.1 for both the (GT)n and (GA)n sequences obtained from the slopes was nearly the same as the ideal value, providing the kinetic evidence that the long-distance hole transfer occurs by means of the multistep hopping between Gs. From the value of kht at n = 1 for G-An-G, single-step hole transfer from G to G across a single AT base pair is determined to be 7.6 × 107 s–1, which is similar to the reported value of 5 × 107 s–1 for the hole transfer from G to GG (15).
Fig. 5.

Plots of ln kht vs. ln N for (GA)n (•) and (GT)n (○) sequences. Apparent rate constants of hole transfer (kht) were obtained with a single exponential fit, and N corresponds to the hopping number.
Influence of G·T and A·C Mismatch on Hole-Transfer Process. Single-base mismatches, G·T and A·C, were introduced in the middle of (GA)6 sequence to investigate the effect of base paring on the hole-transfer process. The time profiles of the transient absorption at 520 nm assigned to PTZ•+ for fully matched and mismatch sequences are shown in Fig. 6. Introduction of mismatch site in DNA caused the considerable decrease of the hole-transfer rate. The kht values for the A·C and G·T mismatches were obtained from the analysis of the formation of PTZ•+ with single-exponential fit to be 0.82 × 105 s–1 and 3.6 × 105 s–1, respectively. These processes involving the mismatch sequences were 100-fold slower than those for fully matched sequences. The highly sequence-dependent and mismatch-sensitive hole-transfer kinetics demonstrates that the hole transfer in DNA is governed by the base stacking, which alters the electronic coupling between Gs (55, 56).
Fig. 6.

Time profiles of the transient absorption at 520 nm assigned to PTZ•+ for DNA conjugates containing A·C (red) and G·T (blue) mismatch. For comparison, the time profile for the (GA)6 sequence, which is same as these conjugates except for the mismatch site, is shown (black). Sample solution contained 100 μM duplex DNA, 20 mM sodium phosphate buffer (pH 7.0), and 100 mM sodium chloride.
Concluding Remarks. In this article, we showed the direct observation for the hole-transfer process over 100 Å through double-helical DNA, which occurred at the apparent rate constants of 1.2 × 104 – 5.7 × 107 s–1 in the time scale of microseconds to milliseconds with time-resolved transient absorption measurement. The hole-transfer kinetics depended highly on the DNA sequence, and the hole-transfer rate of GAG sequence was ≈50-fold larger than that of the GTG sequence, demonstrating that the conductivity of the DNA double strand could be easily controlled by designing the DNA sequence. In addition, it was found that the hole transfer for (GA)n and (GT)n sequences showed weak distance-dependence, and the kinetic evidence for the occurrence of the multistep hole transfer was provided from the analysis of the kht values based on the multistep mechanism.
Our approach, which allows probing the kinetics in the nanosecond-to-millisecond time window, may be applied to investigate not only the kinetics in the various kinds of DNA structures (such as secondary structure, triplex, and quadruplex) but also the role of hole-transfer process in biological system (57–59). Furthermore, given a certain sequence and length of DNA, an unique hole-transfer kinetic can be obtained. The direct measurements of the long-distance hole transfer through DNA may allow high-throughput readout of the stored information.
Supplementary Material
Acknowledgments
This work was supported partly by a Grant-in-Aid for Scientific Research on Priority Area (417); the 21st Century Center of Excellence (COE) Research Program; and grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of the Japanese government.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NI, naphthalimide; PTZ, phenothiazine; A-hopping, adenine-hopping.
References
- 1.Sxzztorhoff, J. J. & Mirkin, C. A. (1999) Chem. Rev. 99, 1849–1862. [DOI] [PubMed] [Google Scholar]
- 2.Seeman, N. C. (1998) Angew. Chem. Int. Ed. 37, 3220–3238. [DOI] [PubMed] [Google Scholar]
- 3.Porath, D., Bezryadin, A., de Vries, S. & Dekker, C. (2000) Nature 403, 635–638. [DOI] [PubMed] [Google Scholar]
- 4.Armitage, B. (1998) Chem. Rev. 98, 1171–1200. [DOI] [PubMed] [Google Scholar]
- 5.Burrows, C. J. & Muller, J. G. (1998) Chem. Rev. 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 6.Schuster, G. B. (2000) Acc. Chem. Res. 33, 253–260. [DOI] [PubMed] [Google Scholar]
- 7.Giese, B. (2000) Acc. Chem. Res. 33, 631–636. [DOI] [PubMed] [Google Scholar]
- 8.Jortner, J., Bixon, M., Voityuk, A. A. & Roesch, N. (2002) J. Phys. Chem. A 106, 7599–7606. [Google Scholar]
- 9.Davis, W. B., Hess, S., Naydenova, I., Haselsberger, R., Ogrodnik, A., Newton, M. D. & Michel-Beyerle, M. E. (2002) J. Am. Chem. Soc. 124, 2422–2423. [DOI] [PubMed] [Google Scholar]
- 10.Beljonne, D., Pourtois, G., Ratner, M. A. & Bredas, J. L. (2003) J. Am. Chem. Soc. 125, 14510–14517. [DOI] [PubMed] [Google Scholar]
- 11.Troisi, A. & Orlandi, G. (2002) J. Phys. Chem. B 106, 2093–2101. [Google Scholar]
- 12.Conwell, E. M. & Basko, D. M. (2001) J. Am. Chem. Soc. 123, 11441–11445. [DOI] [PubMed] [Google Scholar]
- 13.LeBard, D. N., Lilichenko, M., Matyushov, D. V., Berlin, Y. A. & Ratner, M. A. (2003) J. Phys. Chem. B 107, 14509–14520. [Google Scholar]
- 14.Lewis, F. D., Liu, J. Q., Weigel, W., Rettig, W., Kurnikov, I. V. & Beratan, D. N. (2002) Proc. Natl. Acad. Sci. USA 99, 12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis, F. D., Liu, X. Y., Liu, J. Q., Miller, S. E., Hayes, R. T. & Wasielewski, M. R. (2000) Nature 406, 51–53. [DOI] [PubMed] [Google Scholar]
- 16.Kelley, S. O. & Barton, J. K. (1999) Science 283, 375–381. [DOI] [PubMed] [Google Scholar]
- 17.Kelley, S. O. & Barton, J. K. (1998) Chem. Biol. 5, 413–425. [DOI] [PubMed] [Google Scholar]
- 18.Henderson, P. T., Jones, D., Hampikian, G., Kan, Y. Z. & Schuster, G. B. (1999) Proc. Natl. Acad. Sci. USA 96, 8353–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakatani, K., Dohno, C. & Saito, I. (1999) J. Am. Chem. Soc. 121, 10854–10855. [Google Scholar]
- 20.Meggers, E., Michel-Beyerle, M. E. & Giese, B. (1998) J. Am. Chem. Soc. 120, 12950–12955. [Google Scholar]
- 21.Delaney, S. & Barton, J. K. (2003) Biochemistry 42, 14159–14165. [DOI] [PubMed] [Google Scholar]
- 22.Nunez, M. E., Noyes, K. T., Gianolio, D. A., McLaughlin, L. W. & Barton, J. K. (2000) Biochemistry 39, 6190–6199. [DOI] [PubMed] [Google Scholar]
- 23.Dohno, C., Nakatani, K. & Saito, I. (2002) J. Am. Chem. Soc. 124, 14580–14585. [DOI] [PubMed] [Google Scholar]
- 24.Kan, Y. Z. & Schuster, G. B. (1999) J. Am. Chem. Soc. 121, 11607–11614. [Google Scholar]
- 25.Lewis, F. D., Wu, Y., Hayes, R. T. & Wasielewski, M. R. (2002) Angew. Chem. Int. Ed. 41, 3485–3487. [DOI] [PubMed] [Google Scholar]
- 26.Lewis, F. D., Liu, J., Zuo, X., Hayes, R. T. & Wasielewski, M. R. (2003) J. Am. Chem. Soc. 125, 4850–4861. [DOI] [PubMed] [Google Scholar]
- 27.Shafirovich, V., Cadet, J., Gasparutto, D., Dourandin, A., Huang, W. D. & Geacintov, N. E. (2001) J. Phys. Chem. B 105, 586–592. [Google Scholar]
- 28.Shafirovich, V., Dourandin, A., Huang, W. D., Luneva, N. P. & Geacintov, N. E. (1999) J. Phys. Chem. B 103, 10924–10933. [Google Scholar]
- 29.Kawai, K., Takada, T., Tojo, S., Ichinose, N. & Majima, T. (2001) J. Am. Chem. Soc. 123, 12688–12689. [DOI] [PubMed] [Google Scholar]
- 30.Takada, T., Kawai, K., Tojo, S. & Majima, T. (2003) J. Phys. Chem. B 107, 14052–14057. [Google Scholar]
- 31.Kawai, K., Takada, T., Tojo, S. & Majima, T. (2003) J. Am. Chem. Soc. 125, 6842–6843. [DOI] [PubMed] [Google Scholar]
- 32.Takada, T., Kawai, K., Cai, X., Sugimoto, A., Fujitsuka, M. & Majima, T. (2004) J. Am. Chem. Soc. 126, 1125–1129. [DOI] [PubMed] [Google Scholar]
- 33.Tierney, M. T. & Grinstaff, M. W. (2000) J. Org. Chem. 65, 5355–5359. [DOI] [PubMed] [Google Scholar]
- 34.Kawai, K., Kimura, T., Kawabata, K., Tojo, S. & Majima, T. (2003) J. Phys. Chem. B. 107, 12838–12841. [Google Scholar]
- 35.Kawai, K., Wata, Y., Hara, M., Tojo, S. & Majima, T. (2002) J. Am. Chem. Soc. 124, 3586–3590. [DOI] [PubMed] [Google Scholar]
- 36.Bixon, M., Giese, B., Wessely, S., Langenbacher, T., Michel-Beyerle, M. E. & Jortner, J. (1999) Proc. Natl. Acad. Sci. USA 96, 11713–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawai, K., Takada, T., Tojo, S. & Majima, T. (2002) Tetrahedron Lett. 43, 89–91. [Google Scholar]
- 38.Sartor, V., Henderson, P. T. & Schuster, G. B. (1999) J. Am. Chem. Soc. 121, 11027–11033. [Google Scholar]
- 39.Rogers, J. E., Weiss, S. J. & Kelly, L. A. (2000) J. Am. Chem. Soc. 122, 427–436. [Google Scholar]
- 40.Seidel, C. A. M., Schulz, A. & Sauer, M. H. M. (1996) J. Phys. Chem. 100, 5541–5553. [Google Scholar]
- 41.Steenken, S. & Jovanovic, S. V. (1997) J. Am. Chem. Soc. 119, 617–618. [Google Scholar]
- 42.Rogers, J. E. & Kelly, L. A. (1999) J. Am. Chem. Soc. 121, 3854–3861. [Google Scholar]
- 43.Giese, B., Amaudrut, J., Kohler, A. K., Spormann, M. & Wessely, S. (2001) Nature 412, 318–320. [DOI] [PubMed] [Google Scholar]
- 44.Candeias, L. P. & Steenken, S. (1993) J. Am. Chem. Soc. 115, 2437–2440. [Google Scholar]
- 45.Conwell, E. M. & Rakhmanova, S. V. (2000) Proc. Natl. Acad. Sci. USA 97, 4556–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voityuk, A. A., Siriwong, K. & Roesch, N. (2001) Phys. Chem. Chem. Phys. 3, 5421–5425. [Google Scholar]
- 47.Voityuk, A. A., Siriwong, K. & Rosch, N. (2004) Angew. Chem. Int. Ed. 43, 624–627. [DOI] [PubMed] [Google Scholar]
- 48.Yoo, J., Delaney, S., Stemp, E. D. A. & Barton, J. K. (2003) J. Am. Chem. Soc. 125, 6640–6641. [DOI] [PubMed] [Google Scholar]
- 49.Pascaly, M., Yoo, J. & Barton, J. K. (2002) J. Am. Chem. Soc. 124, 9083–9092. [DOI] [PubMed] [Google Scholar]
- 50.O'Neill, M. A., Becker, H. C., Wan, C. Z., Barton, J. K. & Zewail, A. H. (2003) Angew. Chem. Int. Ed. 42, 5896–5900. [DOI] [PubMed] [Google Scholar]
- 51.Giese, B., Wessely, S., Spormann, M., Lindemann, U., Meggers, E. & Michel-Beyerle, M. E. (1999) Angew. Chem. Int. Ed. 38, 996–998. [DOI] [PubMed] [Google Scholar]
- 52.Jortner, J., Bixon, M., Langenbacher, T. & Michel-Beyerle, M. E. (1998) Proc. Natl. Acad. Sci. USA 95, 12759–12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kendrick, T. & Giese, B. (2002) Chem. Commun., 2016–2017. [DOI] [PubMed]
- 54.Behrens, C., Burgdorf, L. T., Schwogler, A. & Carell, T. (2002) Angew. Chem. Int. Ed. 41, 1763–1766. [DOI] [PubMed] [Google Scholar]
- 55.Bhattacharya, P. K. & Barton, J. K. (2001) J. Am. Chem. Soc. 123, 8649–8656. [DOI] [PubMed] [Google Scholar]
- 56.Ropp, P. A. & Thorp, H. H. (1999) Chem. Biol. 6, 599–605. [DOI] [PubMed] [Google Scholar]
- 57.Nunez, M. E., Noyes, K. T. & Barton, J. K. (2002) Chem. Biol. 9, 403–415. [DOI] [PubMed] [Google Scholar]
- 58.Nakatani, K., Dohno, C., Ogawa, A. & Saito, I. (2002) Chem. Biol. 9, 361–366. [DOI] [PubMed] [Google Scholar]
- 59.Kawai, K., Takada, T., Nagai, T., Cai, X., Sugimoto, A., Fujitsuka, M. & Majima, T. (2003) J. Am. Chem. Soc. 125, 16198–16199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}