

Abstract
High-count monoclonal B-cell lymphocytosis (MBL) is an asymptomatic expansion of clonal B-cells in the peripheral blood without other manifestations of chronic lymphocytic leukemia (CLL). Yearly, 1% of MBLs evolve to CLL requiring therapy; thus being critical to understand the biologic events that determine which MBLs progress to intermediate/advanced CLL. In this study, we performed targeted deep-sequencing on 48 high-count MBLs, 47 of them with 2-4 sequential samples analyzed, exploring the mutation status of 21 driver genes and evaluating clonal evolution. We found somatic non-synonymous mutations in 25 MBLs(52%) at the initial time-point analyzed, including 13(27%) with >1 mutated gene. In cases that subsequently progressed to CLL, mutations were detected 41 months (median) prior to progression. Excepting NOTCH1, TP53 and XPO1, which showed a lower incidence in MBL, genes were mutated with a similar prevalence to CLL, indicating the early origin of most driver mutations in the MBL/CLL continuum. MBLs with mutations at the initial time-point analyzed were associated with shorter time-to-treatment (TTT). Furthermore, MBLs showing subclonal expansion of driver mutations on sequential evaluation had shorter progression time to CLL and shorter TTT. These findings support that clonal evolution have prognostic implications already at the pre-malignant MBL stage, anticipating which individuals will progress earlier to CLL.
Keywords: Chronic lymphocytic leukemia, monoclonal B-cell lymphocytosis, targeted sequencing, Clonal evolution
Introduction
Chronic Lymphocytic Leukemia (CLL) has a heterogeneous clinical course, ranging from an indolent to a very aggressive disease requiring therapy shortly after diagnosis.(1, 2) The discovery of the CLL genetic landscape has significantly improved our understanding of which patients are more likely to experience an aggressive disease. In addition to the well-established role of TP53 in clinical outcome, mutations in NOTCH1, SF3B1, and BIRC3 have been recently associated with differential prognosis and subsequently incorporated in the risk stratification.(3-7) Other studies have identified additional genes recurrently mutated in CLL, suggesting their association with disease pathogenesis.(8-13) Moreover, the clonal architecture of CLL before and after therapy have been analyzed by several groups, and these studies demonstrate that the presence of subclonal mutations in driver genes adversely impacts clinical outcome.(8, 14) In addition, the use of chemo-immunotherapy in progressive CLL can be associated with the expansion of previously minor, and potentially highly fit, subclones.(8, 15-17)
CLL is preceded by a pre-malignant state known as high-count monoclonal B-cell lymphocytosis (MBL).(18, 19) MBL is an asymptomatic expansion of clonal B-cells with less than 5×109/L B-cells in the peripheral blood and without other manifestations of CLL.(20) In contrast to CLL, our knowledge regarding the genetics landscape of MBL is still limited. Initial cross-sectional studies involving 63 individuals have evaluated NOTCH1, SF3B1 and TP53 mutation status in high-count MBL cohorts, detecting a mutation frequency between 1% (SF3B1 and TP53) and 3% (NOTCH1).(21, 22) We performed the first comprehensive, whole exome sequencing (WES) study sequentially evaluating eight high-count MBLs at two time points.(23) The study detected mutations in putative CLL driver genes in half of the cases and confirmed the existence of clonal heterogeneity at a median of 5 years before progression to CLL.(23) Another recent study employed whole genome sequencing identified a similar overall mutation burden between MBL and CLL, but with a significantly lower number of affected driver genes in MBL.(12) The first studies showing the impact of NOTCH1, TP53 and SF3B1 mutations in the MBL progression have been recently published.(24, 25)
Approximately 1-2% of high-count MBLs evolve to CLL requiring treatment per year.(20) (26) Thus, identifying the biologic and genetic events that determine which MBLs will have a more aggressive clinical phenotype (i.e. progression to CLL and need for therapy) is critical to understanding the mechanisms of disease progression. In this study we performed a longitudinal sequencing analysis on a cohort of 48 high-count MBL, evaluating the mutation status of 21 genes at 2-4 time points in 47 out of 48 cases, and assessed the clonal heterogeneity and clonal dynamics over time. Furthermore, we evaluated the role of driver mutations and clonal evolution with progression to intermediate/advanced stage CLL (Rai stage I-IV) and time to treatment (TTT). Overall, we found driver mutations in 52% of MBL cases, on average 41 months before progression to intermediate/advanced stage CLL. Furthermore, the identification of driver mutations and subclonal expansion at MBL stage was associated with shorter TTT.
Material and Methods
Patient samples
We analyzed 48 high-count MBL individuals (17 female, 31 male), including 47 with longitudinal blood samples available. The study included results from eight cases previously published by us(23). All individuals provided written informed consent for the collection and use of samples for research purposes according to the Declaration of Helsinki. Clinical and prognostic information regarding age at MBL diagnosis, gender, B-cell count, absolute lymphocyte count (ALC), Rai stage, CD38, CD49d, ZAP70, IGHV mutation status and FISH status, date of CLL diagnosis and date of first-line treatment were acquired from clinical records and/or research studies. All clinical characteristics were assessed at the time of MBL diagnosis. Clinical information and the time-points analyzed for this cohort are summarized in Table 1, Supplementary Table S1 and Supplementary Figure S1.
Table 1.
Descriptive demographics of the MBL cohort.
| Clinical Variables at diagnosis | Cases, n=48 (%) | |
|---|---|---|
| Gender | Male | 31 (65) |
| Female | 17 (35) | |
| Age | Median (range) | 66.5 (44-80) |
| ZAP70 | Positive | 12 (25) |
| Negative | 36 (75) | |
| CD49* | Positive | 13 (28) |
| Negative | 35 (72) | |
| CD38 | Positive | 15 (31) |
| Negative | 33 (69) | |
| IGHV* | Unmutated | 16 (35) |
| Mutated | 32 (65) | |
| FISH* 17p or 11q- | Present | 5 (11) |
| Absent | 42 (89) | |
It was not possible to obtain diagnosis information for CD49 status in 2 cases, IGHV status in 2 cases and FISH in one case, respectively.
Sample preparation
The CD5+/CD19+ B lymphocyte population was enriched from peripheral blood mononuclear cells (PBMC) using anti-human CD5 APC and anti-human CD19 PE (e-bioscience, San Diego, CA). Flow cytometry sorting was performed using FACS Aria II SORP (BD Biosciences) and data analyzed using Cell Quest software. After enrichment, an average of 95% of CD19+/CD5+ cells was obtained. We used the values of the CD19+/CD5+ fraction to calculate the leukemic B-cell fraction and reduce any significant contamination of non-clonal B-cells in each biopsy. The variant reads (VR) frequency was corrected using the following formula: corrected VR = initial VR × (100 ÷ percentage of the CD19+/CD5+ fraction). The negative fraction was also collected and used as germ line controls in the sequencing analysis. DNA was extracted using the Gentra Puregene Cell Kit (Qiagen).
Targeted deep sequencing
We analyzed 152 samples from 48 cases (104 tumor B-cell samples and 48 germ line controls) using semiconductor-sequencing technology (IonTorrent PGM) as per manufacturer's protocol.(27) Twenty-four genes, previously found to be significantly mutated in CLL and indicated as putative CLL driver genes,(8, 9, 11, 28) were selected for targeted deep sequencing (TDS; Supplementary Table S2). Three genes (NOTCH2, NRAS and RB1) showed significant dips in coverage, not reaching sufficient probe depth and precluding the analysis of the entire coding regions. For that reason, we eliminated them from the subsequent analyses. All coding regions were amplified in 200bp amplicons by multiplex PCR using customized oligos (Ion Ampliseq designer). Libraries were templated and enriched using IonOneTouch2 and IonOneTouch ES automated systems, respectively. Samples were sequenced using the 318TM chip (ThermoFisher). Raw data was aligned and indexed in BAM and BAI files using the IonTorrent suite. Variants were called using IonTorrent Somatic VariantCaller version 4.6.0.7 and low stringency settings (ThermoFisher). VCF files were annotated using a standalone application developed by the Mayo Clinic Bioinformatics Program called BioR (Biological Annotation Data Repository)(29). BioR includes gene annotation from NCBI/Ensembl and UCSC, Gene annotated pathways (KEGG), tissue specificity, GeneCards and Gene Ontology (GO), dbSNP, GWAS catalog, HapMap, and 1000 Genomes. Furthermore, it provides annotation from the Catalogue of Somatic Mutations in Cancer (COSMIC, Wellcome Trust Sanger Institute), SIFT, and PolyPhen-2. Pair-wise analyses were performed comparing each tumor sample with paired germ line sample, and all variants found in the germ line samples were filtered out. Additionally, somatic variants with a Mapping Quality <20 or read depth <10X were removed. Finally, variants of significant interest were visually inspected using Integrative Genomics Viewer (IGV)(30) (Supplementary Table S3).
Significant changes in VR between paired samples (i.e. subclonal expansion) was defined using a similar approach to a previously published criteria,(8) based on changes in the cancer cell fraction (CCF) greater than or equal to 20% between time points. The lack of copy number and loss of heterozygosity data has impeded us to precisely calculate the CCF of each mutation. Instead, we defined a threshold of 10% change in VRs between time points, which is equivalent to 20% of CCF for heterozygous mutations in a diploid karyotype.
Statistical Analysis
We employed univariable Cox proportional hazards regression models and Kaplan Meier survival analysis to test statistical associations between the genetic findings and two survival outcomes: TTT and progression free survival (PFS). TTT is defined as time from MBL diagnosis to date of first-line treatment for CLL. PFS was defined as time from MBL diagnosis to progression to intermediate (Rai I/II) or advanced (Rai III/IV) CLL. Three individuals with MBLs who progressed to CLL and participated in early intervention trials before meeting the NCI/IWCLL criteria(31) to initiate therapy were excluded from the TTT statistical analyses. First, we compared the TTT and PFS between those MBLs harboring mutations in any of the 21 genes at the time of MBL diagnosis to those without any mutations. Next, we refined the analysis by dividing the cohort into three groups: a) MBLs harboring mutations in NOTCH1, BIRC3, or SF3B1 (genes previously associated with poor outcome in CLL; N=12); b) MBLs with mutations in one or more of the remaining 18 genes in the panel (N=11); and c) MBLs without mutations in any gene (N=22). We further evaluated whether subclonal expansion was associated with TTT and PFS. Subclonal expansion was defined as a time dependent covariate in the Cox proportional hazards regression analysis. Statistical calculations were performed using SPSS 17.0 (SPSS Inc, Chicago). P values ≤ 0.05 were considered significant.
Results
Characterization of the mutation profile of driver genes in MBL
We studied a total of 48 individuals with high-count MBL, 47 of who had two or more longitudinal blood samples available. The median age at MBL diagnosis was 66.5 years (range: 44-80). The median time between the first and last analyzed samples in these individuals was 55 months (range: 10-119 months). At the time the last sample was collected, 30 cases (64%) remained as MBL or Rai 0 CLL, whereas 17 (36%) progressed to intermediate/advanced stage CLL (Rai stage I-IV), including five individuals (10%) who required treatment. Individuals were also followed clinically for a median of 19 months after the last sample (range: 0-85 months), providing a total median follow-up of 82 months (range: 33-137). At the last follow-up, 22 MBLs (46%) progressed to intermediate/advanced stage CLL, 10 (21%) required treatment, and three (6%) died with disease progression.
The samples were sequenced with an average coverage depth of 727x. At the initial time point analyzed, we identified somatic non-synonymous mutations in 25 of 48 MBL individuals (52%). Recurrent mutations were found in SF3B1 (5 cases, 10%), BIRC3, DDX3X, CHD2 (4 cases each, 8%), NOTCH1 (3 cases, 6%), and ATM, BRAF, EGR2, FBXW7, KRAS, MED12, MYD88, and ZMYM3 (2 cases each, 4%). Furthermore, BCOR, ITPKB, POT1, RIPK1 and SAMHD1 were each mutated in one case. No mutations were found in HIST1H1E, TP53 and XPO1 (Figure 1A-B and Supplementary Table S1). Collectively, 27% of MBLs had more than one gene mutated (range 2-7), and 8% showed multiple mutations in the same gene (Figure 1A). By comparing the mutation profile of MBLs to a similar sized cohort of CLL patients (N=48) analyzed with the same gene panel and with similar coverage depth,(13) we found that the mutation frequency was comparable for most genes; with the exception that individuals with MBL had an increased mutation rate in CHD2, a decreased rate in NOTCH1, and a lack of mutations in TP53 and XPO1 (Figure 1B). Although the difference on TP53 and XPO1 mutation rate between MBL and CLL showed marginal statistical significance (p=0.12 and p=0.06, respectively), the small number of cases precluded reaching definitive conclusions about these differences.
Figure 1. MBL has a mutation profile similar to CLL years before disease progression.

A) Heatmap of mutation distributions in the MBL cohort. Rows and columns represent genes and individuals, respectively. Black boxes indicate mutations (white numbers inside the black boxes indicate the number of mutations found in cases with more than one mutation per gene). IGHV mutation status and FISH data are shown below the mutation profiles. B) Mutation prevalence comparison between 48 MBL and 48 CLL with progressive disease analyzed using the same gene platform and similar sequencing depth coverage (CLL data published by Ojha et al. (13)).
Association between driver mutations and time to first treatment
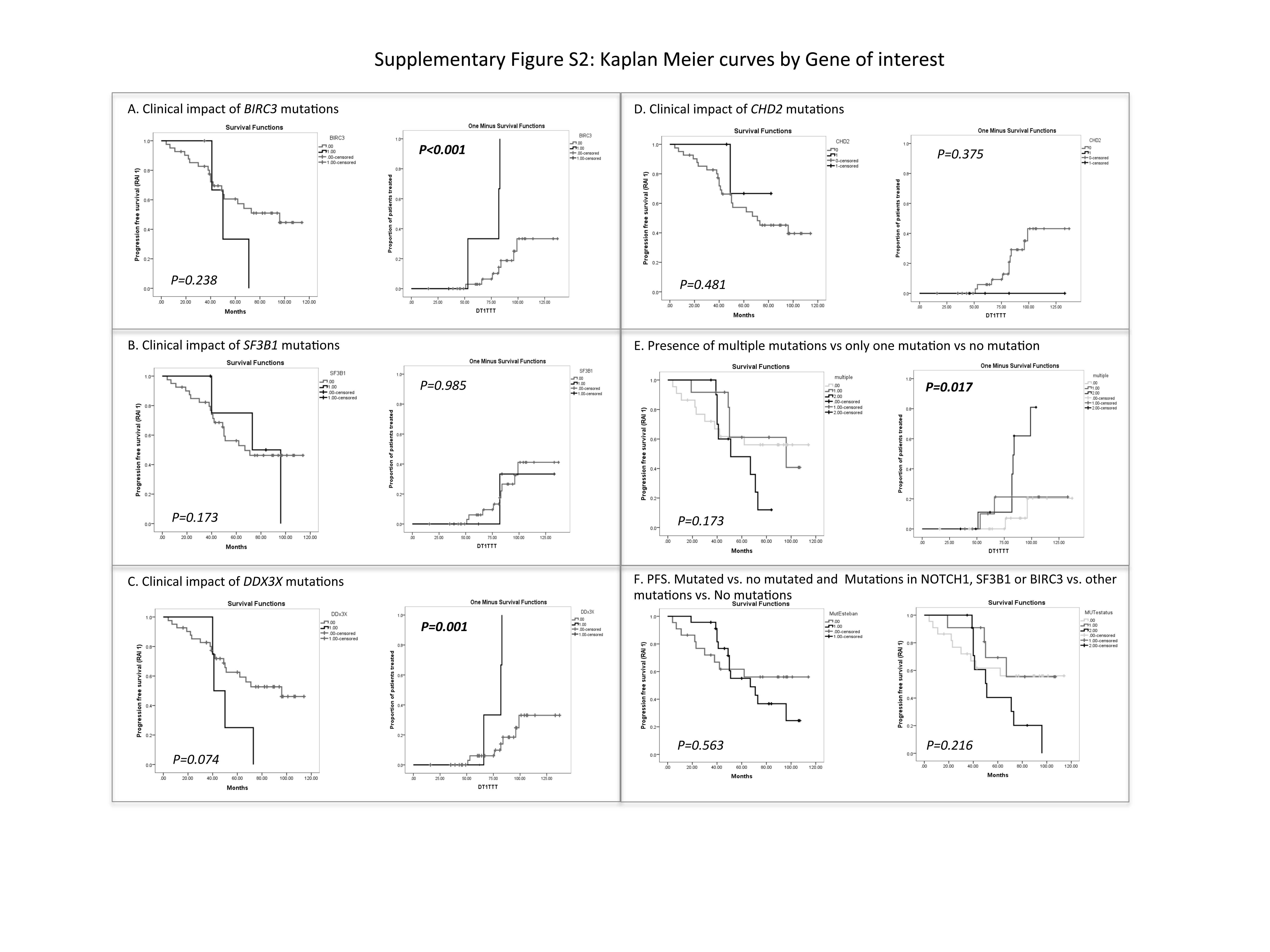
Due to the low mutation incidence for each gene analyzed individually, we first pooled all MBLs harboring any mutations and compared their TTT to that of individuals without mutations in any of the 21 genes. MBL individuals with any mutation showed a shorter TTT (median: 99 months vs. not reached [NR]; p=0.04) (Figure 2A). Next, we explored the clinical significance of specific mutations by dividing the cohort into three groups: a) MBLs harboring mutations in NOTCH1, BIRC3, or SF3B1 (genes previously associated with poor outcome in CLL,(3-7) N=12); b) MBLs with mutations in one or more of the remaining genes in the panel (N=11); and c) MBLs without mutations in any gene (N=22). The subgroup with NOTCH1, BIRC3 or SF3B1 mutations had the shortest median TTT among these subgroups (mutation in driver genes: 84 months; mutation in other genes: NR; no mutation: NR; p=0.02) (Figure 2B). Finally, we analyzed the association between individual genes that were mutated in at least four cases (SF3B1, DDX3X, BIRC3 and CHD2) and TTT. Statistically significant correlation with shorter TTT was found for individuals with MBL who had mutations in BIRC3 (p<0.001) and DDX3X (p=0.001)(Supplementary Figure S2). These associations, although obtained from a small number of cases, are promising and should be explored in larger cohorts.
Figure 2. Time to Treatment and presence of mutations.

A) Comparison of TTT in cases with mutations in any of the 21 genes versus cases without mutations. B) Comparison of TTT between cases with mutations in BIRC3, NOTCH1 or SF3B1 vs. other mutations vs. no mutations.
Clonal dynamics in the MBL-CLL continuum
The high coverage depth achieved with the targeted sequencing approach we employed, allowed us to evaluate for small subclones with markedly higher sensitivity than obtained using WES. A total of 13 cases (29%) with small subclonal mutations (defined as VR lower than 5%) were identified. Mutations found in VR <5%, were confirmed on an independent sequencing run. Notably, in four MBL cases, we detected two or more mutations in the same genes. In most of these cases, one or more mutations were identified at the initial time point, and additional mutations were identified at subsequent time points (Figure 3 and Supplementary Figures S3 and S4). Thus, one individual (MBL08) was characterized by the presence of two distinct SAMHD1 mutations, one detected at both time points (p.I136S) and the other found only at the second time point (p.Q105*). Case MBL10 had two mutations in DDX3X (p.D164G and p.I415V) as well as two mutations in SF3B1 (p.Y623C; p.K700E) at the initial time point, and acquired a third mutation in SF3B1 (p.K666Q) at the second time point. MBL38 initially had three distinct mutations in BRAF mutations (p.D594G; p.G466E; and p.G469V) in small subclones (2-18% VR) and acquired a fourth BRAF activating mutation (p.K601E) at the second time point (Figure 3B). Finally, MBL51 showed an exceptionally complex pattern, initially with mutations in KRAS (p.G12D), DDX3X (p.I522Nfs1*), BIRC3 (p.E553Efs14*), and subsequently acquired distinct additional mutations in all three genes at the second time point (KRAS p.K117N; DDX3X p.Y163fs*; and BIRC3 p.Q547Nfs21*) (Figure 3D). This patient was analyzed at two additional time points, confirming the co-existence of all mutations with slight changes in mutation predominance between the sequential time points. The complete list of mutations and VR for each time point is shown in Supplementary Table S3.
Figure 3. Examples of different clonal dynamics in paired MBL samples.

X-axis shows the variant reads (VRs) of each mutation at the first time point analyzed, whereas the y-axis shows the VRs at the last time point. A) Example harboring a near clonal (DDX3X) and 2 subclonal mutations (SF3B1 and NOTCH1) that remain stable between time points analyzed. B) Example of an individual showing 4 independent subclonal mutations affecting BRAF. C) Example of clonal tides, where an initial clone with a NOTCH1 mutation is replaced with a second clone harboring a MED12 mutation. D) Example of a MBL individual showing more than one mutation in multiple genes. The individual shows a single mutation in KRAS, DDX3X and BIRC3 at the initial time point, with the subsequent identification of a second mutation in all 3 genes and a mutation in BCOR at the last time point analyzed.
Subclonal expansion as a predictive marker of earlier need of therapy
We then analyzed whether the detection of subclonal expansion over time could predict which patients would require earlier treatment. To investigate the clinical implications of clonal dynamics over time, we divided the individuals in three groups based on whether or not longitudinal changes in the observed mutation profile were identified: a) 22 cases with no mutations at any of the time points analyzed; b) 12 cases with stable mutations that did not show changes in VR between time points (Figure 3A-B and Supplementary Figures S3), and c) 11 cases with mutations that showed an increase of >10% on VR between analyzed time points (Figure 3C-D and Supplementary Figure S4).
Subclonal expansion of mutated drivers was found in 6 out of 10 (60%) cases who progressed to require treatment, compared with only 5 of 35 (14%) non-treated cases. Of the four treated cases who did not undergo subclone expansion, two had a constant VR between samples, and two had no mutations in the tested genes. Subclonal expansion was found, on average, 24 months (range 3-42 months) before initiation of first-line treatment (Figure 4). Finally, we evaluated whether subclonal expansion was associated with TTT and/or PFS. Individuals with clonal expansion had both significantly shorter PFS (HR: 5.3 95% CI: 1.3-20.9; p=0.02), as well as significantly shorter TTT (HR: 6.4 95% CI: 1.8-23.2; p=0.005).
Figure 4. Subclonal expansion was found in most MBLs that required treatment, years before the beginning of therapy.

Timeline analysis of MBL individuals that subsequently received treatment. In each case we plot time (months) before treatment (X axis) and Variant Reads (VR) frequency of the different mutations detected in each time point (Y axis). Genes with mutations below 5% are listed but there is no graph associated. Left boxes: Clinical and biological parameters. Prog: Progression to CLL Rai> 0. Grey: Mutations without subclonal expansion; Black: Mutations with subclonal expansion; Arrows: Time (months) from detection of subclonal expansion to initial treatment.
Discussion
Little is known regarding the molecular, biologic, and genetic events that cause high-count MBLs to progress to CLL. Here, we performed a longitudinal analysis, evaluating mutation status and clonal dynamics of 21 putative CLL driver genes in high-count MBLs. We have confirmed the existence of mutations in CLL driver genes in over 50% of cases at the premalignant MBL stage and in 60% of MBLs that ultimately progressed to intermediate/advanced Rai stage. Among cases that progressed, mutations were detected 41 months prior to progression indicating their early origin in the MBL/CLL continuum. Furthermore, we also showed that most of these genes are mutated in MBL at a similar prevalence to CLL.
The current observations regarding MBL should be considered in the context of prior studies characterizing the mutation profile, clonal heterogeneity and evolution in patients with CLL.(8, 10, 15-17, 32, 33) Despite the limited size of our cohort, we found strong evidence that the tumor mutation profile is present in the pre-malignant MBL stage and predicts progression to clinically significant CLL. This observation suggests that genetic characteristics can be used to identify MBL cases who will progress to advanced stage CLL and require treatment. In longitudinal analysis of our cohort, 60% of MBL cases that progressed to require treatment for CLL exhibited subclonal expansion compared with only 14% of untreated cases suggesting its potential use as a biomarker predicting the likelihood of earlier disease progression.
The results obtained in this study are based on a TDS approach including an array of potential CLL driver genes. By obtaining a sequencing mean coverage depth of ~750x, we found a large reservoir of small subclonal mutations in driver genes, including several cases with multiple mutations in the same genes.
Some limitations of our study should be noted. First, although this study is the largest longitudinal genomic analysis of MBL reported to date, our sample size still remains limited. Second, although our TDS approach evaluated the most relevant recurrent mutations reported in CLL patients, this strategy would miss mutations in genes not included in the panel. That is evident in the study of cases such as MBL29, where a subclone harboring a NOTCH1 mutation is replaced by another having a MED12 mutation, either implying that the subclone with the not so well characterized MED12 mutation has a selective advantage and/or the rising subclone has additional, high-fitness, mutations not included in the sequencing panel. Further studies are needed to better dissect the effect of each mutation, as was previously done with TP53.(34) On the other hand, this limitation is in part offset by the fact that the deep coverage of the TDS strategy provides more detailed analysis of clonal evolution and sub-clone dynamics, which was one of the primary goals of the analysis. Although longer follow-up of this cohort will continue to provide new insights, the current 84-month median follow-up is long enough to reach sound conclusions. This long follow-up also allowed us to use a high threshold for determining clinically significant progression defined as requiring treatment or progression to intermediate/advanced Rai stage rather than simply a higher lymphocyte count which has been used in many studies as an expedient parameter but which does not have clear clinical relevance.
In summary, mutations in driver genes are an early event in the MBL-CLL continuum and sequential genetic evaluation of individuals with MBL provides insights that add to our understanding of the events that contribute to progression from MBL to CLL and have potentially profound clinical implications.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the Henry Predolin Foundation and NIH grant CA197120.
Footnotes
Authorship
S.B., J.O., C.S., K.M.K., S.P., E.B. performed experiments; S.B., K.G.C., S.L.S., E.B. analyzed results; S.B., T.S., D.L.V., R.F., S.L.S., N.E.K, E.B. designed the research; S.B., E.B. wrote the paper.
Conflict of Interest: Dr. Fonseca has received a patent for the prognostication of MM based on genetic categorization of the disease. He has received consulting fees from Celgene, Genzyme, BMS, Bayer, Lilly, Onyx, Binding Site, Novartis, Sanofi, Millennium and AMGEN. He also has sponsored research from Onyx. He is also a member of the Scientific Advisory Board of Applied Biosciences.
References
- 1.Call TG, Phyliky RL, Noel P, Habermann TM, Beard CM, O'Fallon WM, et al. Incidence of chronic lymphocytic leukemia in Olmsted County, Minnesota, 1935 through 1989, with emphasis on changes in initial stage at diagnosis. Mayo Clin Proc. 1994;69(4):323–8. doi: 10.1016/s0025-6196(12)62215-0. [DOI] [PubMed] [Google Scholar]
- 2.Diehl LF, Karnell LH, Menck HR, The American College of Surgeons Commission on Cancer and the American Cancer Society The National Cancer Data Base report on age, gender, treatment, and outcomes of patients with chronic lymphocytic leukemia. Cancer. 1999;86(12):2684–92. [PubMed] [Google Scholar]
- 3.Baliakas P, Hadzidimitriou A, Sutton LA, Rossi D, Minga E, Villamor N, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29(2):329–36. doi: 10.1038/leu.2014.196. [DOI] [PubMed] [Google Scholar]
- 4.Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Dohner K, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247–54. doi: 10.1182/blood-2014-01-546150. [DOI] [PubMed] [Google Scholar]
- 5.Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–8. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108–17. doi: 10.1038/leu.2013.263. [DOI] [PubMed] [Google Scholar]
- 7.Pflug N, Bahlo J, Shanafelt TD, Eichhorst BF, Bergmann MA, Elter T, et al. Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood. 2014;124(1):49–62. doi: 10.1182/blood-2014-02-556399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 10.Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191–6. doi: 10.1182/blood-2012-05-433540. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015 doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 13.Ojha J, Secreto CR, Rabe KG, Van Dyke DL, Kortum KM, Slager SL, et al. Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br J Haematol. 2015;169(3):445–8. doi: 10.1111/bjh.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–30. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knight SJ, Yau C, Clifford R, Timbs AT, Sadighi Akha E, Dreau HM, et al. Quantification of subclonal distributions of recurrent genomic aberrations in paired pre-treatment and relapse samples from patients with B-cell chronic lymphocytic leukemia. Leukemia. 2012;26(7):1564–75. doi: 10.1038/leu.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braggio E, Kay NE, Vanwier S, Tschumper RC, Smoley S, Eckel-Passow JE, et al. Longitudinal genome-wide analysis of patients with chronic lymphocytic leukemia reveals complex evolution of clonal architecture at disease progression and at the time of relapse. Leukemia. 2012;26(7):1698–701. doi: 10.1038/leu.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ojha J, Ayres J, Secreto C, Tschumper R, Rabe K, Van Dyke D, et al. Deep sequencing identifies genetic heterogeneity and recurrent convergent evolution in chronic lymphocytic leukemia. Blood. 2015;125(3):492–8. doi: 10.1182/blood-2014-06-580563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marti GE, Rawstron AC, Ghia P, Hillmen P, Houlston RS, Kay N, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. British journal of haematology. 2005;130(3):325–32. doi: 10.1111/j.1365-2141.2005.05550.x. [DOI] [PubMed] [Google Scholar]
- 19.Shanafelt TD, Ghia P, Lanasa MC, Landgren O, Rawstron AC. Monoclonal Bcell lymphocytosis (MBL): biology, natural history and clinical management. Leukemia. 2010;24(3):512–20. doi: 10.1038/leu.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawstron AC, Bennett FL, O'Connor SJ, Kwok M, Fenton JA, Plummer M, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575–83. doi: 10.1056/NEJMoa075290. [DOI] [PubMed] [Google Scholar]
- 21.Greco M, Capello D, Bruscaggin A, Spina V, Rasi S, Monti S, et al. Analysis of SF3B1 mutations in monoclonal B-cell lymphocytosis. Hematol Oncol. 2013;31(1):54–5. doi: 10.1002/hon.2013. [DOI] [PubMed] [Google Scholar]
- 22.Rasi S, Monti S, Spina V, Foa R, Gaidano G, Rossi D. Analysis of NOTCH1 mutations in monoclonal B-cell lymphocytosis. Haematologica. 2012;97(1):153–4. doi: 10.3324/haematol.2011.053090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ojha J, Secreto C, Rabe K, Ayres-Silva J, Tschumper R, Dyke DV, et al. Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression. Leukemia. 2014;28(12):2395–8. doi: 10.1038/leu.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lionetti M, Fabris S, Cutrona G, Agnelli L, Ciardullo C, Matis S, et al. High-throughput sequencing for the identification of NOTCH1 mutations in early stage chronic lymphocytic leukaemia: biological and clinical implications. Br J Haematol. 2014;165(5):629–39. doi: 10.1111/bjh.12800. [DOI] [PubMed] [Google Scholar]
- 25.Winkelmann N, Rose-Zerilli M, Forster J, Parry M, Parker A, Gardiner A, et al. Low frequency mutations independently predict poor treatment free survival in early stage chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis. Haematologica. 2015 doi: 10.3324/haematol.2014.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shanafelt TD, Kay NE, Rabe KG, Call TG, Zent CS, Maddocks K, et al. Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(24):3959–63. doi: 10.1200/JCO.2008.21.2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 28.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kocher JP, Quest DJ, Duffy P, Meiners MA, Moore RM, Rider D, et al. The Biological Reference Repository (BioR): a rapid and flexible system for genomics annotation. Bioinformatics. 2014;30(13):1920–2. doi: 10.1093/bioinformatics/btu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouillette P, Saiya-Cork K, Seymour E, Li C, Shedden K, Malek SN. Clonal evolution, genomic drivers, and effects of therapy in chronic lymphocytic leukemia. Clin Cancer Res. 2013;19(11):2893–904. doi: 10.1158/1078-0432.CCR-13-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guieze R, Robbe P, Clifford R, de Guibert S, Pereira B, Timbs A, et al. Presence of multiple recurrent mutations revealed by targeted NGS confers poor trial outcome of relapsed/refractory CLL. Blood. 2015 doi: 10.1182/blood-2015-05-647578. [DOI] [PubMed] [Google Scholar]
- 34.Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Fama R, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–47. doi: 10.1182/blood-2013-11-539726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.