

Abstract
Purpose
To assess clinical characteristics, foveal structure, mutation spectrum, and prevalence rate of Åland eye disease (AED)/incomplete congenital stationary night blindness (iCSNB).
Methods
A retrospective survey included individuals diagnosed with AED at a national low-vision center from 1980 to 2014. A subset of affected males underwent ophthalmologic examinations including psychophysical tests, full-field electroretinography, and spectral-domain optical coherence tomography.
Results
Over the 34-year period, 74 individuals from 35 families were diagnosed with AED. Sixty individuals from 29 families participated in a follow-up study of whom 59 harbored a CACNA1F mutation and 1 harbored a CABP4 mutation. Among the subjects with a CACNA1F mutation, subnormal visual acuity was present in all, nystagmus was present in 63%, and foveal hypoplasia was observed in 25/43 subjects. Foveal pit volume was significantly reduced as compared to normal (P < 0.0001). Additionally, outer segment length at the fovea was measured in 46 subjects and found to be significantly reduced as compared to normal (P < 0.001). Twenty-nine CACNA1F variations were detected among 34 families in the total cohort, and a novel CABP4 variation was identified in one family. The estimated mean birth prevalence rate was 1 per 22,000 live-born males.
Conclusions
Our data support the viewpoint that AED, iCSNB, and X-linked cone–rod dystrophy 3 are designations that refer to a broad, continuous spectrum of clinical appearances caused in the majority by a variety of mutations in CACNA1F. We argue that the original designation AED should be used for this entity.
Keywords: Åland eye disease, iCSNB, CSNB2A, CACNA1F, CABP4, prevalence, OCT
Aland eye disease (AED) was first described as an X-linked disorder in a family from the archipelago of Åland in the Baltic Sea.1 The disorder was initially categorized as a type of X-linked ocular albinism, Forsius-Eriksson type (gene/locus Online Mendelian Inheritance in Man [OMIM] No. 300600).2 However, the demonstration of normal retinocortical projections by visual evoked potential recording definitely excluded the concept of AED being a type of ocular albinism.3 A similar disorder was later categorized as incomplete congenital stationary night blindness (iCSNB) type 2A (OMIM No. 300071), and in a separate report of a single family a comparable entity was referred to as X-linked cone–rod dystrophy 3 (OMIM No. 300476).4–6 The three entities were subsequently shown to be caused by mutations in the CACNA1F gene.7–9 The phenotype of this clinical entity is highly variable and may comprise subnormal best-corrected visual acuity, nystagmus, a broad refractive span from high myopia to moderate hyperopia, photophobia, color vision deficiency, and/or foveal hypoplasia with an otherwise normal, sometimes sparsely pigmented fundus.1,10,11 Mild or moderately impaired dark adaptation is often present, but subjective complaints are reported by a part of the patients only. None of the clinical symptoms are specific and at least one major feature (impaired night vision, nystagmus, or myopia) was absent in 75% of the subjects in a previous study.12 The full-field electroretinography (ffERG) is diagnostic in most cases showing a negative configuration of the dark-adapted cone–rod response and a severely reduced photopic 30-Hz flicker a-wave with a conspicuous double configuration.13–15 Electrophysiologically, AED/iCSNB is a cone–rod synaptic disorder with predominantly cone affliction, and the confusion with X-linked and autosomal recessive complete CSNB mainly reflects that both disorders show negative ERG. We recognize that AED, CSNB2A, and cone–rod dystrophy 3, in the absence of distinct discriminating features, represent a broad overlap of symptoms within a single entity, which historically was named Åland eye disease. We convened a Danish national study of phenotypes with emphasis on the foveal structure, mutation spectrum, and prevalence of AED of a sizeable cohort of clinically and molecularly diagnosed AED patients. New information was added on prevalence and from optical coherence tomography (OCT) imaging, which among other things showed that this disorder also involves an effect on photoreceptor outer segments.
Methods
Subjects
This single-center study included a retrospective review of the files on all subjects diagnosed with AED at the National Eye Clinic for the Visually Impaired in Denmark from 1980–2014. The study followed the tenets of the Declaration of Helsinki and informed written consent was obtained from all subjects or from the parents of subjects younger than 18 years. The criteria for referral to this clinic were subnormal visual function or otherwise unexplained visual difficulties in daily life. The subjects underwent a routine clinical evaluation including fundus photography, ffERG, and Goldmann-Weekers dark adaptometry with integral technique. All affected males were invited to participate in a follow-up study consisting of a reexamination at the National Eye Clinic for the Visually Impaired. The diagnosis was based on clinical evaluation and confirmed by molecular genetic analysis, as described below.
Clinical Assessment
The study protocol included assessment of refraction, best-corrected visual acuity (Snellen and ETDRS), biomicroscopy with fundus examination, motility, Goldman manual kinetic perimetry (III/4e), color vision testing (Ishihara, Farnsworth panels D15 standard and D15 desaturé), Goldman-Weekers dark adaptometry (normal median threshold: 4 × 10−3 cd/m2 after 30 minutes), color fundus photography, and ffERG according to the standard protocol of the International Society for Clinical Electrophysiology of Vision.16
Optical Coherence Tomography
Imaging of foveal structure was attempted by nonmydriatic spectral-domain OCT (SD-OCT) in both eyes of all subjects with one or both of two different instruments (Spectralis SD-OCT, Heidelberg Engineering, Heidelberg, Germany; Cirrus SD-OCT, Carl Zeiss Meditec, Humphrey Division, Dublin, CA, USA). The respective ability of these two instruments to overcome the effects of nystagmus differed from subject to subject as reflected in the success rates described below. A combination of fast and detailed protocols was used to mitigate the effect of nystagmus during SD-OCT scanning. If fixation was deemed by the operator to be more than 500 μm off the anatomic center of the fovea, the scan was repeated at least once (though this did not always result in an acceptable scan). Morphologic measurements were made on scans where one or more of the following findings suggested that they were made through the anatomic center of the macula: the presence of a reflex at the bottom of a depression on the inside of the retina, a point of maximal depth in a foveal depression, a peak point of attenuation of the middle layers of the retina, or scan line that transected the center of the foveal avascular zone. Not all subjects had scans that could be analysed, though when possible the following measurements were made: (1) The SD-OCT scans were qualitatively assessed for the presence of hypoplasia (persistence of one or more inner retinal layers at the foveal center)17,18; (2) The foveal pit volume was measured and compared to normative values by using the method described by Wilk et al.19; (3) The length of the photoreceptor outer segments at the fovea was measured, taken as the distance between the second (inner segment/outer segment junction or ellipsoid zone) and third (interdigitation zone or cone outer segment tips) hyperreflective bands; and (4) The subfoveal choroidal thickness was measured, taken as the distance between Bruch's membrane and the chorioscleral interface.
Molecular Genetics and Genealogic Studies
Venous blood samples were collected from all subjects and available family members. Total genomic DNA was extracted according to standard procedures. Mutation screening of CACNA1F was performed by PCR amplification of genomic DNA and Sanger sequencing of both DNA strands of the entire coding region and the flanking intron–exon splice junctions. Two samples were sequenced by next-generation sequencing using a targeted approach with a gene panel of 124 genes, among these CACNA1F, CABP4, and CACNA2D4. Target regions were enriched by using Agilent SureSelect (Agilent Technologies, Inc., Santa Clara, CA, USA) and sequenced by using Illumina HiSeq2000 platform (Illumina, Inc., San Diego, CA, USA). Raw image files were processed by Illumina HCS software. Alignment to the human reference genome GRCh37/hg19 was performed by using Burrows-Wheeler Aligner and genotypes were called by using SOAPsnp software. Called variants were filtered, leaving only coding nonsynonymous and splice-site variants with a frequency in publicly available databases under 1%. Reported variants were verified by Sanger sequencing. Sequence variations were assigned into five classes: pathogenic, likely pathogenic, variant of uncertain significance, likely benign, or benign based on an in-house classification system including type of variation, frequency in databases, segregation analysis, previously reported in affected individuals, functional assays, and in silico analyses using prediction.
Genealogic investigation involved at least two generations of ancestors in all families and was performed to identify possible relations to the other families. The prevalence rate of AED was expressed as the proportion of all live-born males that were affected in the population during 3 decades.
Results
Clinical Phenotyping Results
Retrospectively, we ascertained data on 74 males diagnosed with AED in 35 families, including a previously described large multigenerational family with 11 affected members.20 Reexamination according to the current institutional routine was made in 60 affected subjects, 59 with a CACNA1F mutation from 29 families. One individual, No. 59, with a probably pathogenetic CABP4 variant was excluded from all calculations and presented separately. Most of the families were residing in Denmark as far as traceable, while three families originated from Germany (subject No. 55), Norway (subject No. 53), and Pakistan (subject No. 26). The reexamined subjects had a median age of 22 years, range 4 to 76 years, and showed a median best-corrected binocular visual acuity (BCVA) of 0.33, range 0.05 to 0.8 (Fig. 1; Supplementary Table S1). None of the participants, except No. 7 and No. 53, showed progression between the initial examination and the follow-up (median 9 years, range 0–40 years). Nystagmus was present in 63% (23/55) of subjects and this subgroup had distinctly lower visual acuity. Ten subjects showed color vision deficiency, six of whom belonged to a family segregating deuteranomaly. The retina appeared to be normally pigmented with normal retinal vessel diameters in all subjects except in subjects with high myopia (≤−6.00 diopters [D]), which was present in 31% of the cases. Seventy-six percent of the cohort had some degree of myopia in at least one eye. The median refraction was −3 D, range from −18 D to +4.75 D. Cross-tabulation between nystagmus (n = 34) and high myopia ≤ −6 D (n = 18) among 59 subjects showed no significant association (two-tailed Fisher exact probability test, P = 0.16). Dark adaptation was normal or only moderately deficient (<2 log elevation) in all subjects, except in subject No. 7. Goldmann perimetry (object III/4e) was normal in all subjects. Full-field electroretinography was performed in 54 subjects. Dark-adapted 3-ERG recordings (combined rod–cone standard flash ERG) showed a-wave amplitudes between 58 and 384 μV (median 202 μV), which did not deviate markedly from controls (N = 80, 63–387 μV, median 205 μV) (Fig. 1D). The b-wave amplitudes were distinctly subnormal resulting in an electronegative waveform of the scotopic bright flash ERG (Fig. 2). Scotopic oscillatory potentials were present but reduced in number and amplitudes. The photopic bright flash 30-Hz flicker recordings showed severe reduction in amplitude and a distinct b-wave separation resulting in a double-peak configuration (Fig. 2).
Figure 1.

Distribution of four clinical characteristics in the Danish AED cohort. Age distribution among 59 Danish AED patients with a CACNA1F mutation. (A) Refractive values in diopters (equivalent spherical values) in the right eye of 58 Danish AED patients. (B) Best-corrected visual acuity (logMAR) of 58 Danish AED patients. (C) a-Wave amplitude of 51 Danish AED patients in response to a combined rod–cone standard flash ERG (dark-adapted 3 ERG) (D).
Figure 2.

Full-field electroretinography recordings from the right eyes of three subjects with AED and a normal control (right column). (A–E) Scotopic recordings. (F–G) Photopic recordings. (A) Rod ERG (dark-adapted 0.01 ERG). (B) Rod–cone response to single flash 1.22 cd-s/m2. (C) Combined rod–cone standard flash ERG (3.0 cd-s/m2). (D) Strong flash ERG (10.0 cd-s/m2). (E) Dark-adapted 3 oscillatory potentials. (F) Standard flash “cone” ERG (light-adapted 3 ERG). (G) Light-adapted flicker ERG (3.0 cd-s/m2).
Disrupted Foveal Morphology in AED
Spectral-domain OCT scans (Cirrus and/or Spectralis) of sufficient quality to judge the presence or absence of foveal hypoplasia were available for 43 subjects, with the observed incidence of hypoplasia being 58% (n = 25). For the 48 subjects in whom both eyes were assessed on at least one device, we observed 100% interocular symmetry. Cross-tabulation between foveal hypoplasia (n = 25) and nystagmus (n = 27) in 46 individuals with CACNA1F mutations showed no significant association (two-tailed Fisher exact probability test, P = 0.77). Cross-tabulation between foveal hypoplasia and high myopia (≤−6, n = 19) versus low myopia/hypermetropia (n = 19) in the right eye also showed no significant association (two-tailed Fisher exact probability test, P = 0.32). To further assess foveal morphology, we quantified the volume of the foveal pit by using Cirrus SD-OCT scans and a previously described algorithm.19 Scans were available for 22 subjects, 19 images from right eyes (mean ± SD: 0.0328 ± 0.0163 mm3), and 19 from left eyes (mean ± SD: 0.0326 ± 0.0153 mm3). Note that subject 59 (CAPB4 mutation) was not included in the calculation of mean values, though this patient's foveal pit volume was similarly shallow. These values were significantly different from the normal values previously reported19 (mean ± SD: 0.0864 ± 0.0377 mm3) (P < 0.0001, Mann-Whitney test). Of note, we used the right eye foveal volume from each subject unless a right eye image was not available or could not be processed because of its quality; if that occurred, the left foveal volume was used, if available (Fig. 3; Supplementary Table S2).
Figure 3.

Foveal hypoplasia in AED. Foveal hypoplasia was assessed by the presence of inner retinal layers at the foveal center. The subject in (A) (No. 26) shows persistence of the inner plexiform layer (IPL) and ganglion cell layer (GCL), while the subject in (B) (No. 24) shows persistence of the IPL, GCL, inner nuclear layer (INL), and outer plexiform layer (OPL). In our subjects, 53% showed some degree of foveal hypoplasia. Retinal thickness maps from two normal controls (C, D) show foveal pits with volumes of 0.104 and 0.033 mm3, respectively. The AED patient in (E) has an estimated foveal pit volume of 0.013 mm3. While there is some uncertainty as to the absolute accuracy of these measurements owing to the unavailability of axial length measurements to correct for differences in the lateral magnification, the retinal thinning and smaller pit profiles are readily visible and consistent with the high incidence of foveal hypoplasia in the AED population.
Abnormal Cone Development in AED
Elongation of the cone outer segments represents one aspect of normal foveal development.22,23 Therefore, we measured the outer segment length in subjects with AED from all available scans. Foveal outer segment length (mean ± SD) was 41.65 ± 4.99 μm for OD and 42.07 ± 5.65 μm for OS, based on measurements from 48 subjects (OU = 45, OS = 3, OD = 3) (Fig. 4; Supplementary Table S3). Normal values of 46.04 ± 4.03 μm were derived from the study of Wilk et al.19 Comparing these distributions revealed significantly shorter foveal cone outer segment lengths in patients with AED (Z score test; P < 0.001).
Figure 4.

Outer segment length in AED. Images from four patients with AED demonstrating the variation in foveal outer segment length (A = 35 μm, B = 37 μm, C = 43 μm, D = 51 μm). On average, the outer segment length in AED was significantly shorter than normal, consistent with reduced foveal cone packing in these patients (see text).
Subfoveal Choroidal Thinning in AED
Among 36 subjects for whom subfoveal choroidal thickness (micrometers) could be assessed in both eyes with Spectralis SD-OCT, comparable thickness values in right and left eyes (paired t-test, mean ± SD: 192.0 ± 79.3 and 190.7 ± 83.1, respectively, P = 0.830) was found. The range of subfoveal choroidal thickness in our AED patients is shown in Figure 5 and Supplementary Table S4.
Figure 5.

Variability in choroidal thickness in AED. Shown are subjects with (A) normal subfoveal choroidal thickness (subject No. 9; 341.8 μm), (B) moderate thinning (subject No. 17; 92.2 μm), and (C) severe thinning (subject No. 30; 19.2 μm). On average, the AED patients had significantly reduced subfoveal choroidal thickness (see text).
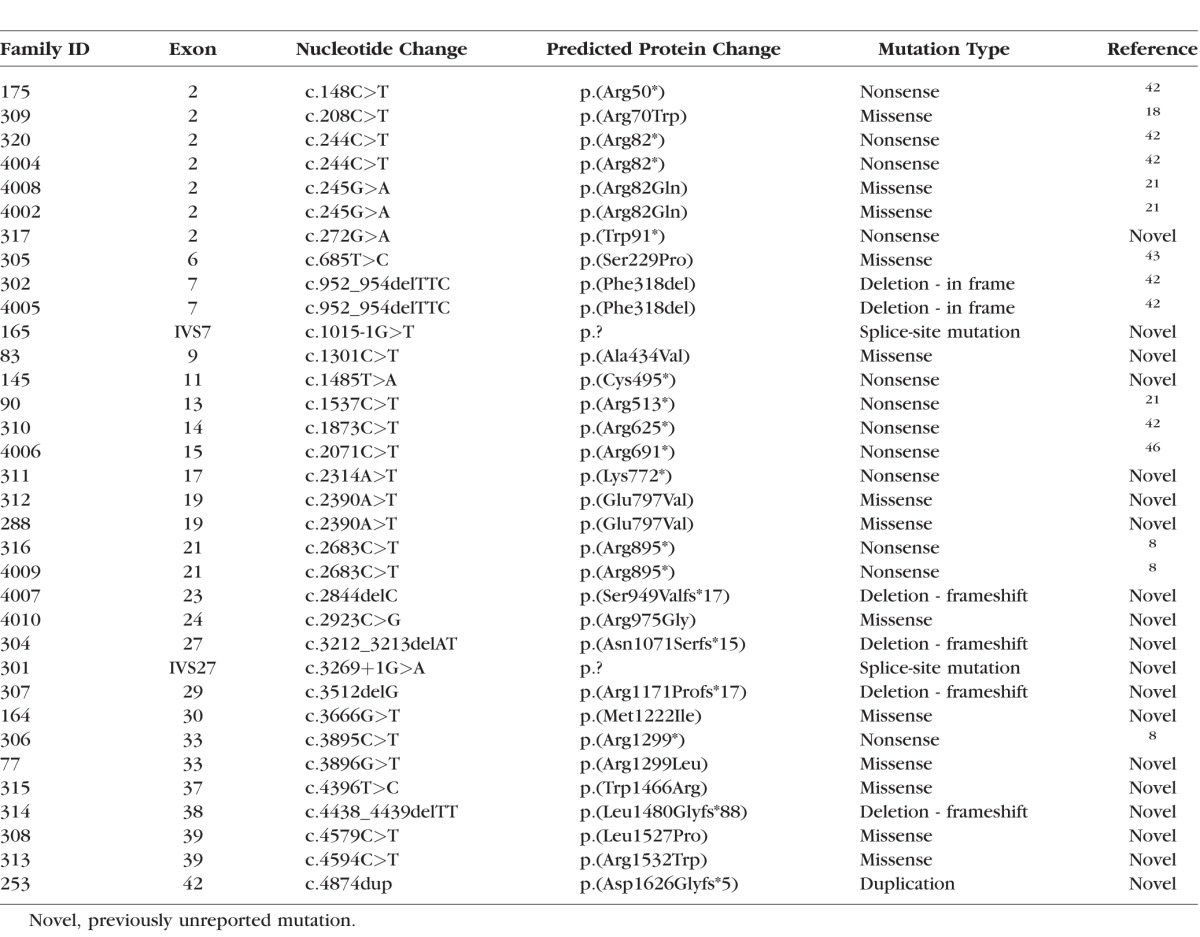
Molecular Genetics
In 34 families, a total of 29 variations in CACNA1F were identified, among which 18 have not been reported previously (Table 1). The variations were located in 21 of the 48 exons in CACNA1F and included 11 missense and 10 nonsense mutations, four frame-shift deletions, two splice-site deletions, one in-frame deletion, and one duplication (Table 1). Furthermore, an apparently homozygous variant, c.773A>T, p.(Asn258Ile), in CABP4 was detected in individual No. 59, though a deletion of one allele could not be excluded.
Table 1.
Overview of CACNA1F Mutations in 34 Danish Families With Åland Eye Disease
Prevalence
The number of subjects born in the decades 1980–1989, 1990–1999, and 2000–2009 were 8, 14, and 22, respectively, corresponding to a mean birth prevalence of at least 4.6 per 100,000 live-born males (Table 2).
Table 2.
Male-Specific Prevalence Rates of Subjects With AED

Discussion
CACNA1F codes for the α1F subunit of the Cav1.4 calcium channel. Cav1.4 maintains a continuous or tonic calcium-dependent neurotransmitter release from the photoreceptors and, with the αβ2 subunit and the α2δ4 auxiliary subunit, participates in the formation and function of presynaptic cone and rod photoreceptor ribbon synapses.24–28 Rod signal transmission primarily occurs through depolarizing ON-bipolar cells, whereas cones connect and transmit with both ON- and hyperpolarizing OFF-bipolar cells. Therefore, in AED rod and cone signal transmission from photoreceptors to bipolar cells involves both ON and OFF pathway activity.
A second gene, CABP4, encoding the calcium-binding protein 4 has been shown to be involved in a minor portion of the patients and families diagnosed with AED or autosomal recessive CSNB2B (OMIM No. 610427). The CABP4 protein interacts with the C-terminal domain of the Cav 1.4α calcium channel.12,29 The phenotype of this disorder is not yet fully elucidated; however, the same discussion about nomenclature as in AED was raised and the name “congenital cone–rod synaptic disorder” proposed.30,31 It is generally accepted that this disorder of AED/iCSNB-like patients with mutations in CACNA1F, is also not clinically a type of night blindness. Historically, the original description of AED in 1974 has been neglected since the publications by Miyake and coworkers4 and, despite the recent use of the designation of CSNB2, we consider it appropriate to consider returning to the original name of AED for this subgroup of vision disorders. Mutations in a third gene, CACNA2D4, in the photoreceptor voltage-gated calcium channel family have been associated with a rare retinal dysfunction, retinal cone dystrophy 4 (RCD4) (OMIM No. 610478) with an overlapping phenotype including reduced cone responses and negative or normal rod signals.32–34
The phenotypes in our cohort are in accordance with existing knowledge. With regard to night blindness the term “incomplete” refers to a slight to moderate elevation of the threshold after 30 minutes of dark adaptation, which is stated to lie between 0.3 and 2.5 log units (median 1.8 log unit) above normal threshold.11,13 Reports on the frequency of subjective complaints of impaired night vision are scarce. Bijveld et al.11 have reported a frequency of 54%, reporting signs of night blindness at medical history taking. The present investigation did not address this issue; however, in an earlier publication20 we have reported subjective night blindness in only 2 of 13 patients with AED.
The anatomic observations reported here are consistent with a generalized underdevelopment of the foveal pit, with hypoplasia similar to that seen in conditions such as albinism and achromatopsia.17–19 Segmentation of SD-OCTs of the retinal layers in five individuals with X-linked and autosomal recessive CSNB2 patients has shown thinning of both inner and outer retinal layers, compared with those of myopic controls.35 While we could not combine the data from the Spectralis and Cirrus instruments owing to different segmentation methods, the qualitative retinal thinning was visible in many subjects (Fig. 3E). Furthermore, we found that AED patients have, on average, reduced length of foveal cone outer segments, which has been observed in some patients with albinism.19 A scatterplot of foveal cone outer segment length against dark-adapted 3-ERG a-wave amplitude showed no correlation, while a nonparametric test found a weak trend toward decreasing a-wave amplitudes with decreasing height of the foveal cone outer segment layer (P = 0.082). The lacking correlation was unexpected but may be due to the stimulus intensity, which predominantly reflects rod function. No other remarkable trends or associations were found. Based on mouse models of Cav channelopathies, the ribbon synapse structure maturation is disturbed or essentially absent and ectopic synapses develop by sprouting from bipolar and horizontal cells.25,28 Thus, the shortening of foveal outer segments observed here may indicate still other direct or indirect impact on the cone photoreceptors.27,36 Moreover, the disruption of the dendritic branching pattern of horizontal cells in a murine model of CSNB2A, which has a loss-of-function mutation in Cacna1f, suggests that afferent signaling from photoreceptors has downstream effects on retinal development.37
One limitation of our analysis was that axial length measurements were not available. While this would affect OCT measurements made laterally, the axial scale of the OCT images is not affected by individual differences in axial length. Thus, the only measurement that may be compromised is the foveal volume measures. However, our quantitative findings of smaller foveal pits in AED were supported by the qualitative observations of a high incidence of foveal hypoplasia in this group (58%). Thus, the difference reported between AED and normal is likely correct, though the exact magnitude of the difference may be more or less. Future analyses looking at foveal pit dimensions should aim to integrate axial length measurements in order to remove the possible confound of differences in axial length. In a group of 54 healthy subjects—mean age 31.8 years, BCVA 20/25–20/10, spherical equivalent refraction −0.5 D, range −7.1 to 3.8 D—examined at our institution (Rönnbäck et al., 2015), the subfoveal choroidal thickness is 368.4 ± 179.2 μm. The thickness of the subfoveal choroid in patients with AED is substantially lower than in healthy subjects, and the difference exceeds by far the effect of myopia.35,38
All previously reported sequence variations that had been detected in our AED patients were classified as either pathogenic or likely pathogenic, including the three previously reported missense mutations (Supplementary Table S5). Of the 18 novel sequence variations, 10 cause premature truncation of the protein (nonsense, deletion-frameshift, or duplication-frameshift) or are located in splice-site consensus sequences and are thus classified as either pathogenic or likely pathogenic, further supported by these variants being absent in any population databases. None of the novel eight missense mutations are present in 2000 Danish individuals and all but one (p.Met1222Ile) are reported in the Exome Sequencing Project, demonstrating that the missense mutations found among our AED patients are not common polymorphisms.39 From our in-house classification system modified from Richards and coworkers,40 three missense variations were classified as likely pathogenic, while the remaining five were all classified as variants of unknown significance.
In two individuals with CACNA1F mutations, the phenotypes differed substantially from the rest, in particular as their clinical presentation was progressive in contrast with the ordinary nonprogressive course in AED. Subject No. 7 was one of four examined males in family 320. He was reexamined at the age of 76 years, 26 years after the initial examination at which time he was highly myopic (−17.5/−20.0 D). At the first visit the ERG was characteristic of AED and both color vision and visual fields were normal. At reexamination, however, his visual acuity had decreased, color vision was affected, both retinas showed large peripapillary and upper temporal atrophic areas (Fig. 6), and the visual fields were concentrically restricted. No measurable ERG responses were recordable and the dark-adaptation threshold was substantially elevated. In this patient who had a known pathogenetic CACNA1F nonsense mutation (c.244C>T, p.[Arg82*]; Table 1), an additional potentially pathogenic mutation in the ROM1 gene (c.47G>A) was identified. The variant is known in the Single Nucleotide Polymorphism Database (dbSNP; https://www.ncbi.nlm.nih.gov/SNP/, available in the public domain as rs143166696). Exome sequencing of 2000 individuals residing in Denmark has shown that six individuals out of 1965 are heterozygous for c.47G>A.39 Three in silico programs (Align GVGD, MutationTaster, and Polyphen2 [HumVar model]) predict the change as benign, while one (SIFT) predicts deleterious. The variant is thus classified as a variant of uncertain clinical significance. It can be speculated that the missense mutation in ROM1 contributes to the atypical phenotype in subject No. 7; however, not many sequence variations in ROM1 have been reported in subjects with retinal dystrophies and very often the pathogenicity is questionable. Digenic inheritance has been reported for PRPH2 and ROM1, and the gene products have been shown to form heterotetramers.41 No physical interaction between CACNA1F and ROM1 gene products are known. We therefore find it unlikely that the ROM1 sequence change contributes to the phenotype. Moreover, it cannot be excluded that there is some other variant/mutation present in another gene not represented among the 124 sequenced genes, which has a more direct impact on the phenotype in this subject.
Figure 6.

Fundus photographs of both eyes taken at an interval of 26 years, from subject No. 7 with high myopia and a mutation in both CACNA1F and ROM1. The interpretation of this is treated in the Discussion. (A) Fundus pictures when the subject was aged 50 years. (B) Fundus pictures at the age of 76 years show distinct progression of the dystrophy.
The second subject (ID No. 53), who had a novel missense CACNA1F mutation (c.3666G<T, p.[Met1222Ile]) (Table 1), was the only subject who at reexamination 6 years after the initial examination showed normal scotopic ERG responses, nonmeasurable photopic responses, macular OCT abnormalities, and acquired-type dyschromatopsia. These findings were in accordance with a cone dystrophy rather than AED, and a sequence variation in GUCA1A was also found to be present (c.463G>A, p.[Glu155Lys]) in subject No. 53. This variation is predicted pathogenic by four prediction programs (Align GVGD, MutationTaster, Polyphen2 [HumVar model], and SIFT) and is not reported in dbSNP or other population databases. The variation is located in a functional domain (EF-hand domain involved in Ca2+ ion binding). Mutations in GUCA1A are known to cause autosomal dominant cone dystrophy 3 (OMIM No. 602093) and we find it likely that the variation found in GUCA1A is the main reason for the phenotype seen in this patient. He was the only one affected in the family. Generally, a clinical assessment including ffERG is sufficient for a clinical diagnosis of AED. However, the case stories of patients No. 7 and No. 53 show that even in the presence of a pathogenic CACNA1F variation, an unusual course should lead to reevaluation of the diagnosis and a search for additional genetic variations. A retinal dystrophy resembling sector retinopathy has been reported by Nakamura and coworkers42 in a patient with a stop mutation in CACNA1F, yet, without a molecular explanation of the deviating phenotype.
An apparently homozygous missense variation in CABP4 (c.773A>T, p.[Asn258Ile]) was detected in a third subject, No. 59, diagnosed with AED and in whom no CACNA1F variation was identified. The variant is classified of uncertain significance; however, since it is present with a very low frequency in the ExAC database (<0.002), it is located in a functional domain (EF-hand domain), and all four in silico prediction programs (SIFT, Polyphen2, MutationTaster, and AlignGVGD) score the variant pathogenic and everything points in the direction of a pathogenic missense mutation. This individual was examined from the age of 10 years and reexamination as described was performed at age 47 years. A right convergent squint was noticed from early infancy. He had nystagmus and was photophobic. He experienced no problems in the dark. The visual acuity on right and left eye was +0.8 and +0.5 logMar and stationary, and the refractive values were +2.50 and +2.00 D. The color discrimination was reduced with a score of 248 and blue–yellow axis in Farnsworth-Munsell 100 hue test. Nagel anomaloscope showed a deuteranomaly. Dark adaptation had a normal final threshold, and dark-adapted ffERG with standard flash was negative with a-wave amplitude of 138 μV. The light-adapted cone response was severely reduced. Optical coherence tomography exhibited foveal hypoplasia with foveal volumes of 0.018 and 0.026 mm3, outer segments length were normal with 38.94 and 33.41 μm on right and left eye. Choroidal thickness was analyzable on left eye only and was normal, 360.96 μm. The electroretinographic similarity with the AED phenotype has been described earlier.11,31
The prevalence calculations for AED (Table 2) were based on referrals to a national specialized low-vision eye clinic covering the whole population of 5.5 million inhabitants in Denmark. Electroretinography was routinely performed in most children with impaired vision, including individuals with congenital nystagmus, unexplained low visual acuity, and/or high myopia. Åland eye disease was recognized as a relatively common cause of visual impairment in such subjects. Table 2 demonstrates an increase in the number of diagnosed cases over 3 recent decades of birth, which reflects a growing awareness of the diagnosis of AED and the improved accessibility to mutation analysis. A supplementary search in the Danish National Database for visually impaired children with visual acuity 6/18 or lower, based on mandatory notifications, showed that ERG examination was missing in a considerable number of recorded children who had not been referred to the National Eye Clinic. This finding suggests that AED may be underdiagnosed in Denmark, not unlike the prevalence of this condition in other countries.43–45 This postulated underestimation may be due to limited use of ERG in cases with nystagmus, low vision with or without high myopia. Thus, our frequency estimate of 4.6:100,000 live-born males in Denmark likely represent an absolute minimum.
Since the first description more than 40 years ago, AED has been classified as a type of ocular albinism, a cone–rod dystrophy, and a type of congenital stationary night blindness. All the proposed classifications are inappropriate and we argue for a return to the original designation, which does not indicate any specific category. In this article we presented data on foveal morphology and prevalence in a fairly large sample of cases with mutations in either CACNA1F or CABP4 genes.
Supplementary Material
Acknowledgments
The authors thank BGI for next-generation sequencing, and Erik Kann for pertinent genealogic studies.
Supported in part by grants from The Support Foundation of the Blind (Blindes Støttefond) (MNH), Foundation Fighting Blindness (TB-H) (Canada), and The Velux Foundation (KG), with additional support from the National Eye Institute of the National Institutes of Health under Award No. P30EY001931 (JC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure: M.N. Hove, None; K.Z. Kilic-Biyik, None; A. Trotter, None; K. Grønskov, None; B. Sander, None; M. Larsen, None; J. Carroll, None; T. Bech-Hansen, None; T. Rosenberg, None
References
- 1. Forsius H,, Eriksson AW. A new eye syndrome with X-chromosomal transmission: a family clan with fundus albinism, fovea hypoplasi, nystagmus, myopia, astigmatism and dyschromatosia [in German]. Klin Monatsbl Augenheilk. 1964; 144: 447–457. [PubMed] [Google Scholar]
- 2. Waardenburg PJ. Some notes on publications of Professor Arnold Sorsby and on Aland eye disease (Forsius-Erikssonsyndrome). J Med Genet. 1970; 7: 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Dorph DB,, Eriksson AW,, Delleman JW,, et al. Åland eye disease: no albino misrouting. Clin Genet. 1985; 28: 526–531. [DOI] [PubMed] [Google Scholar]
- 4. Miyake Y,, Yagasaki K,, Horiguchi M. Congenital stationary night blindness with negative electroretinogram: a new classification. Arch Ophthalmol. 1986; 104: 1013–1020. [DOI] [PubMed] [Google Scholar]
- 5. Miyake Y,, Yagasaki K,, Horiguchi M. A rod-cone dysfunction syndrome with separate clinical entity: incomplete-type congenital stationary night blindness (Miyake). : Hollyfield JG,, Anderson RE,, LaVail MM, Degenerative Retinal Disorders: Clinical and Laboratory Investigations. New York: Alan R. Liss; 1987: 137–145. [PubMed] [Google Scholar]
- 6. Jalkanen R,, Mantyjarvi M,, Tobias R,, et al. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. Med Genet. 2006; 43: 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strom TM,, Nyakatura G,, Apfelstedt-Sylla E,, et al. An L-type calcium-channel gene mutated in incomplete stationary night blindness. Nat Genet. 1998; 19: 260–263. [DOI] [PubMed] [Google Scholar]
- 8. Bech-Hansen NT,, Naylor MJ,, Maybaum TA,, et al. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998; 19: 264–267. [DOI] [PubMed] [Google Scholar]
- 9. Jalkanen R,, Bech-Hansen NT,, Tobias R,, et al. A novel CACNA1F gene mutation causes Aland Island eye disease. Invest Ophthalmol Vis Sci. 2007; 48: 2498–2502. [DOI] [PubMed] [Google Scholar]
- 10. Boycott KM,, Pearce WG,, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can J Ophthalmol. 2000; 35: 204–213. [DOI] [PubMed] [Google Scholar]
- 11. Bijveld MMC,, Florijn RJ,, Bergen AAB,, et al. Genotype and phenotype of 101 Dutch patients with congenital stationary night blindness. Ophthalmology. 2013; 120: 2072–2081. [DOI] [PubMed] [Google Scholar]
- 12. Pearce WG,, Reedyk M,, Coupland SG. Variable expressivity in X-linked congenital stationary night blindness. Can J Opthalmol. 1990; 25: 3–10. [PubMed] [Google Scholar]
- 13. Miyake Y,, Horiguchi M,, Ota I,, Shiroyama N. Characteristic ERG flicker anomaly in incomplete congenital stationary night blindness. Invest Ophthalmol Vis Sci. 1987; 28: 1816–1823. [PubMed] [Google Scholar]
- 14. Tremblay F,, Laroche RG,, De Becker I. The electroretinographic diagnosis of the incomplete form of congenital stationary night blindness. Vision Res. 1995; 35: 2383–2393. [DOI] [PubMed] [Google Scholar]
- 15. Langrová H,, Gamer D,, Friedburg C,, Besch D,, Zrenner E,, Apfelstedt-Sylla E. Abnormalities of the long flash ERG in congenital stationary night blindness of the Schubert–Bornschein type. Vision Res. 2002; 42: 1475–1483. [DOI] [PubMed] [Google Scholar]
- 16. McCulloch DL,, Marmor MF,, Brigell MG,, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015; 130: 1–12. [DOI] [PubMed] [Google Scholar]
- 17. Thomas MG,, Kumar A,, Mohammad S,, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography: a predictor of visual acuity? Ophthalmology. 2011; 118: 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sundaram V,, Wilde C,, Aboshiha J,, et al. Retinal structure and function in achromatopsia:Implications for gene therapy. Ophthalmology. 2014; 121: 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilk MA,, McAllister JT,, Cooper RF,, et al. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014; 55: 4186–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenberg T,, Schwartz M,, Simonsen SE. Åland eye disease (Forsius-Eriksson-Miyake syndrome) with probability established in a Danish family. Acta Ophthalmol (Copenh). 1990; 68: 281–291. [DOI] [PubMed] [Google Scholar]
- 21. Zeitz C,, Robson AG,, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res. 2015; 45: 58–110. [DOI] [PubMed] [Google Scholar]
- 22. Hendrickson A,, Youdelis C. The morphological development of the human fovea. Ophthalmology. 1984; 91: 603–612. [DOI] [PubMed] [Google Scholar]
- 23. Youdelis C,, Hendrickson A. A qualitative and quantitative analysis of the human fovea during development. Vision Res. 1986; 26: 847–855. [DOI] [PubMed] [Google Scholar]
- 24. McRory JE,, Hamid J,, Doering CJ,, et al. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004; 24: 1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mansergh F,, Orton NC,, Vessey JP,, et al. Mutation of the calcium channel gene CACNA1F disrupts calcium signalling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005; 14: 3035–3046. [DOI] [PubMed] [Google Scholar]
- 26. Hoda JC,, Zaghetto F,, Koschak A,, Striessnig J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels. J Neurosci. 2005; 25: 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mercer AJ,, Thoreson WB. The dynamic architecture of photoreceptor ribbon synapses: cytoskeletal, extracellular matrix, and intramembrane proteins. Vis Neurosci. 2011; 28: 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu X,, Kerov V,, Haeseleer F,, et al. Dysregulation of Cav 1.4 channels disrupts the maturation of photoreceptor synaptic ribbons in congenital stationary night blindness type 2. Channels. 2013; 7: 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeitz C,, Kloeckener-Gruissem B,, Forster U,, et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006; 79: 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Littink KW,, van Genderen MM,, Collin RWJ,, et al. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009; 50: 2344–2350. [DOI] [PubMed] [Google Scholar]
- 31. Khan AO. CABP4 mutations do not cause congenital stationary night blindness. Ophthalmology. 2014; 121: e15. [DOI] [PubMed] [Google Scholar]
- 32. Wycisk KA,, Zeitz C,, Feil S,, et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006; 79: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang X-F,, Huang F,, Wu K-C,, et al. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med. 2015; 17: 271–278. [DOI] [PubMed] [Google Scholar]
- 34. Ba-Abbad R,, Arno G,, Carss K,, et al. Mutations in CACNA2D4 cause distinctive retinal dysfunction in humans. Ophthalmology. 2016; 123: 668–671. [DOI] [PubMed] [Google Scholar]
- 35. Chen RW,, Greenberg JP,, Lazow MA,, et al. Autofluorescence imaging and spectral-domain optical coherence tomography in incomplete congenital stationary night blindness and comparison with retinitis pigmentosa. Am J Ophthalmol. 2012; 153: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McAllister JT,, Dubis AM,, Tait DM,, et al. Arrested development: high-resolution imaging of foveal morphology in albinism. Vision Res. 2010; 50: 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raven MA,, Orton NC,, Nassar H,, et al. Early afferent signaling in the outer plexiform layer regulates development of horizontal cell morphology. J Comp Neurol. 2008; 506: 745–758. [DOI] [PubMed] [Google Scholar]
- 38. Li XQ,, Jeppesen P,, Larsen M,, Munch IC. Subfoveal choroidal thickness in 1323 children aged 11 to 12 years and association with puberty: the Copenhagen Child Cohort 2000 Eye Study. Invest Ophthalmol Vis Sci. 2014; 55: 550–555. [DOI] [PubMed] [Google Scholar]
- 39. Lohmueller KE,, Sparsø T,, Li Q,, et al. Whole-exome sequencing of 2000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013; 93: 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Richards S,, Aziz N,, Bale S,, Bick D,, Das S,, Gastier-Forster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kajiwara K,, Berson EL,, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1loci. Science. 1994; 264: 1604–1608. [DOI] [PubMed] [Google Scholar]
- 42. Nakamura M,, Ito S,, Terasaki H,, Miyake Y. Incomplete congenital stationary night blindness associated with symmetrical retinal atrophy. Am J Ophthalmol. 2002; 134: 463–465. [DOI] [PubMed] [Google Scholar]
- 43. Héon E,, Musarella M. Congenital stationary night blindness: a critical review for molecular approaches. : Wright AF,, Jay B, Molecular Genetics of Inherited Eye Disorders. Chur, Switzerland: Harwood Academics; 1994: 277–301. [Google Scholar]
- 44. Boycott KM,, Maybaum TA,, Naylor MJ,, et al. A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum Genet. 2001; 108: 91–97. [DOI] [PubMed] [Google Scholar]
- 45. Wutz K,, Sauer C,, Zrenner E,, et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur J Hum Genet. 2002; 10: 449–456. [DOI] [PubMed] [Google Scholar]
- 46. Zeitz C,, Minotti R,, Feil S,, et al. Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness. Mol Vis. 2005; 11: 179–183. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.