

Abstract
Continuous rhythmic neuronal oscillations underpin local and regional cortical communication. The impact of the motor system neurodegenerative syndrome amyotrophic lateral sclerosis (ALS) on the neuronal oscillations subserving movement might therefore serve as a sensitive marker of disease activity. Movement preparation and execution are consistently associated with modulations to neuronal oscillation beta (15–30 Hz) power. Cortical beta‐band oscillations were measured using magnetoencephalography (MEG) during preparation for, execution, and completion of a visually cued, lateralized motor task that included movement inhibition trials. Eleven “classical” ALS patients, 9 with the primary lateral sclerosis (PLS) phenotype, and 12 asymptomatic carriers of ALS‐associated gene mutations were compared with age‐similar healthy control groups. Augmented beta desynchronization was observed in both contra‐ and ipsilateral motor cortices of ALS patients during motor preparation. Movement execution coincided with excess beta desynchronization in asymptomatic mutation carriers. Movement completion was followed by a slowed rebound of beta power in all symptomatic patients, further reflected in delayed hemispheric lateralization for beta rebound in the PLS group. This may correspond to the particular involvement of interhemispheric fibers of the corpus callosum previously demonstrated in diffusion tensor imaging studies. We conclude that the ALS spectrum is characterized by intensified cortical beta desynchronization followed by delayed rebound, concordant with a broader concept of cortical hyperexcitability, possibly through loss of inhibitory interneuronal influences. MEG may potentially detect cortical dysfunction prior to the development of overt symptoms, and thus be able to contribute to the assessment of future neuroprotective strategies. Hum Brain Mapp 38:237–254, 2017. © 2016 Wiley Periodicals, Inc.
Keywords: motor neurone disease, magnetoencephalography, neurophysiology, neuroimaging, inhibition, biomarker
INTRODUCTION
The neurodegenerative disorder amyotrophic lateral sclerosis (ALS) is characterized clinically by progressive motor neuronal system degeneration from cortex to muscle. It is now understood to have multiple aetiologies [Turner and Swash, 2015] and shares clinical, pathological, and genetic overlap with frontotemporal dementia (FTD) [Phukan et al., 2012]. The term primary lateral sclerosis (PLS) encompasses a very slowly progressive phenotype with pure upper motor neuron (UMN) degeneration [Pringle et al., 1992]. The improved characterization of the genetic substrate for familial ALS now enables the study of asymptomatic mutation carriers predisposed to developing ALS [Benatar et al., 2013]. Identification of the earliest pathological events might inform future therapeutic efforts to slow disease progression prior to irreversible neuronal injury.
Neuroimaging has been at the forefront of the drive to explore in vivo cerebral pathology in ALS, concurrently developing candidate biomarkers [Turner and Verstraete, 2015]. Abnormal cortical functional connectivity, typically appraised by coherent fluctuations in the task‐free blood oxygen level‐dependent (BOLD) functional MRI signal, is linked to structural cerebral pathology in ALS [Douaud et al., 2011; Schmidt et al., 2014]. Reflecting the pathological overlap between ALS and FTD, some network specific connectivity changes are shared across these highly related diseases [Trojsi et al., 2015]. Abnormal increased functional connectivity has been demonstrated within specific cortical subregions [Agosta et al., 2013; Douaud et al., 2011; Zhou et al., 2014], perhaps underpinned by cortical hyperexcitability. This potentially pathogenic mechanism is also implicated by abnormal responses to transcranial magnetic stimulation (TMS) [Vucic et al., 2013b], which may reflect loss of cortical inhibitory interneuronal influences [Turner and Kiernan, 2012; Turner et al., 2005a, 2005b].
Direct noninvasive recording of cortical neurophysiology supplements existing functional MRI findings by harnessing millisecond temporal precision at the expense of reduced spatial resolution. The cortical neuronal dynamics underlying motor performance can be ascertained with particularly high sensitivity using magnetoencephalography (MEG) [Proudfoot et al., 2014a, 2014b]. Movement preparation and execution are consistently associated with modulations to motor and premotor neuronal oscillation power, particularly within the beta‐band (15–30 Hz) [Pfurtscheller and Lopes Da Silva, 1999]. Beta‐band limited power is initially reduced (alternatively described as desynchronization) but after movement termination a relative increase (or synchronization) in power follows, accompanied by fluctuation in corticospinal excitability [Chen et al., 1998; Fry et al., 2016; Kilavik et al., 2013]. MEG allows neural signal analysis from anatomically precise cortical structures. As well as their potential as biomarkers, such signals are of heightened relevance in ALS given their proposed use as a user input to brain‐computer interfaces in the advanced stages of disability [Grosse‐Wentrup and Schölkopf, 2014; Kasahara et al., 2012].
Two electroencephalographic (EEG) studies directly considered the effect of ALS on these important neurophysiological markers of cortical activation. Reduction in cortical postmovement synchronization (termed postmovement beta rebound, PMBR) was identified in both studies, but conflicting conclusions drawn regarding the integrity of peri‐movement event‐related desynchronization (ERD) [Bizovičar et al., 2014; Riva et al., 2012]. This study applied the combined temporal and spatial resolution uniquely afforded by MEG to a group of both typical ALS and PLS patients. The central hypothesis was that alterations to cortical beta‐band oscillations may reflect pathological cortical hyperexcitability, which is an early and distinguishing feature of ALS and related phenotypes (Geevasinga et al., 2015a, 2015b]. A group of asymptomatic ALS gene mutation carriers (AGCs) was included to appraise the sensitivity of MEG in the detection of cortical pathology prior to the development of overt symptoms.
METHODS
Participants
Apparently sporadic ALS and PLS patients (i.e., without a family history of ALS or FTD), both prevalent and incident cases, were recruited from a tertiary referral clinic as a component of the Oxford Study for Biomarkers in Motor Neurone Disease (“BioMOx”). Diagnosis was confirmed by one of two experienced neurologists (MRT, KT) according to consensus criteria [Brooks et al., 2000; Gordon et al., 2006]. Six of the ALS patients were taking Riluzole at the time of study. Healthy controls, typically spouses of patients, were similar in age, handedness, and level of education. AGCs included were recruited locally, and through collaboration with the presymptomatic Familial ALS (Pre‐FALS) study (MB, JW) [Benatar and Wuu, 2012], participants in which travelled to Oxford for both MEG and MRI. Demographics for all participant groups are detailed in Table I.
Clinical and cognitive assessments (MRT and MP) were performed on the same day as MEG acquisition (MRT, GR, and MP). Contemporaneous T1‐weighted structural MRI scans were acquired for coregistration with the MEG data (3T Siemens Trio, MPRAGE sequence). Three ALS patients were unable to tolerate MRI, and a standard Montreal Neurological Institute (MNI) template was instead used for MEG coregistration. An additional healthy control was excluded due to incidental white matter changes. Disability was assessed using the revised ALS Functional Rating Score (ALSFRS‐R, range 0–48, lower scores reflecting greater disability). Cognitive function was predominantly assessed using the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) [Abrahams et al., 2013], or the revised Addenbrooke's Cognitive Examination (ACE‐R) [Mioshi et al., 2006] for those cases studied prior to development of the ECAS. Rate of disability progression (ΔALSFRS‐R) was calculated as the decrease in ALSFRS‐R from a presumed baseline score of 48, divided by the disease duration in months from reported symptom onset. A measure of the burden of clinical UMN signs was based upon a pathological reflex sum score [Menke et al., 2014; Turner et al., 2004]
All participants provided written informed consent. The study was approved by the National Research Ethics Service South Central Oxford Research Ethics Committee B (08/H0605/85), and South Central Berkshire Committee (14/SC/0083).
Task Design
A Go‐NoGo task was designed (ACN and GR) to investigate neural activity related to spatially selective motor preparation. Monochromatic visual cues, with one side shaded indicating the hand to be moved, were presented foveally for 200 ms, followed by an interstimulus interval (ISI) of either 1 or 2 s, during which a central fixation cross remained. After the variable time period of lateralized motor preparation, the fixation cross was replaced by either a green “Go” circular target in 80% of trials, or a red “NoGo” target in 20% (randomly distributed; Fig. 1). Participants were instructed to make rapid responses to just the “Go” targets by lifting and replacing the index finger of only the prepared hand. Trials were randomly distributed in laterality and ISI duration. A maximum of three blocks of 100 trials (median intertrial interval 6.25 s) were acquired per participant. Fatigue limited acquisition to two blocks in one PLS patient. Each block lasted ∼12 min. Stimuli were created on Matlab and presented via the Psychtoolbox package [Brainard, 1997].
Figure 1.
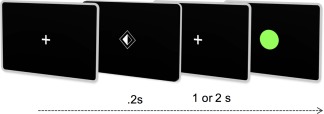
Task design schematic demonstrating visual cues instructing lateralized motor preparation for 1 or 2 s (dependent on cue style) followed by response only to green Go targets. [Color figure can be viewed at http://wileyonlinelibrary.com.]
MEG Data Acquisition
Behavioral responses were measured using a fiber‐optic sensing device, and confirmed by off‐line inspection of surface electromyography (EMG) traces recorded from the extensor digitorum communis. To ensure that participants were engaged in motor preparation, only those trials in which correct responses were confirmed were included in subsequent analyses. Visual fixation and blinks were monitored using an infrared eyetracker (Eyelink 1000) as well as vertical and horizontal EOGs, and subsequently checked off‐line.
MEG data were acquired at the Oxford Centre for Human Brain Activity (OHBA) on a passively shielded Elekta Neuromag system comprising 204 orthogonally oriented planar gradiometers and 102 magnetometers. Participants were seated comfortably, 90 cm away from a back‐projected screen (Panasonic PT D77OOE). Continuous adjustment was made for head position within the MEG helmet using four emitting coils secured to the participant's scalp. A Polhemus 3D tracking system recorded coil positions relative to nasion and preauricular fiducial landmarks, alongside distributed points covering the scalp surface. MEG data were digitized at 1000 Hz with a 0.03 Hz highpass filter.
MEG Data Preprocessing
A locally developed analysis pipeline, OHBA Software Library (MW), was used for analysis, incorporating Matlab toolboxes from SPM12 (UCL, London, UK), FSL (FMRIB, Oxford, UK), and FieldTrip (Donders, Nijmegen, NL). Continuous MEG data were initially inspected to identify noise‐corrupted channels. Maxfilter software (Version 2.2, Elekta) was then applied to the remaining channels for Signal Source Separation and head position adjustment, transforming the data into a set of virtual sensors. Data were then downsampled to 250 Hz, and physiological artefacts were identified and removed using an independent component analysis classification tool based on visual inspection of component spatial topography and timecourse [Baker et al., 2014]. Exactly two independent components pertaining to (1) cardiac pulse and (2) eye‐blink were removed from each data set. Data were then epoched according to the time‐period of interest (including a one second pre‐cue baseline period) for subsequent analysis. Alongside behaviorally incorrect trials and those with premature responses identified by surface EMG, epochs contaminated by artefact were identified by an automated rejection tool, and removed from the analysis. This comprised 1.3% of trials, on average, over subjects. The tool used a robust bisquare linear regression to fit for the mean standard deviation in the data over trials. Any trials that were down weighted by more than 99% points during the regression were classed as outliers. Epochs were centred on either (1) the preparatory cue, to investigate neural correlates of motor preparation, (2) response time (RT), to investigate motor execution while accounting for differences in reaction time, (3) movement termination time, to investigate PMBR, or (4) target appearance, to investigate successfully withheld “NoGo” trials.
Source Analysis
A linearly constrained minimum variance scalar beamformer [Robinson and Vrba, 1999; Van Veen et al., 1997] was used via a single‐shell forward model [Sarvas, 1987] to project preprocessed MEG data, bandpass filtered between 15 and 30 Hz, onto a regular 3D grid spanning the entire brain. Estimates of the data covariance matrix were regularized by removing the weakest PCA components [Woolrich et al., 2011] to leave only 61 (the approximate rank of the data after the use of Maxfilter and removal of two ICA components pertaining to (a) ECG and (b) eye‐movement artefacts). Cue, response and movement termination locked epochs were then inspected across 8‐mm whole‐brain grids to locate subject specific, functionally defined regions of interest (ROIs) corresponding to cortical motor regions: for each subject, the MNI coordinates separately pertaining to either the maximal preparatory ERD, response ERD or PMBR were selected for each hemisphere (locations depicted in Supporting Information Figure S1). Neural signals from these pairs of voxels were carried forward from each subject for comparison of timecourse dynamics. Note that an alternative analysis using the first principal component of the signals from the surrounding voxels within an 8 mm radius, yielded similar results (data not shown).
Time‐frequency transformations as implemented in the FieldTrip toolbox (http://www.fieltriptoolbox.org) were then applied. Hanning tapers of length 300 ms at 50 ms intervals within a frequency range 4–45 Hz were used to reveal the spectral distribution of ERD/S, and multitapers centered at 21.5 Hz with 8.5‐Hz frequency smoothing were used to display the timecourse of beta‐band power. Summary values for preparatory beta ERD (baselined mean value over 0–1.2 s post‐cue), response ERD (±200 ms around response), baselined PMBR (0.15–1.5 s from response completion) were generated per participant from the respective ERD or PMBR ROI data for subsequent clinical correlation. Cluster‐based permutation statistics as implemented in FieldTrip [Maris and Oostenveld, 2007] were used to compare the main task effects between participant groups. For exploration of non‐normal outcome measures, Kendall's Tau was chosen to assess the (uncorrected) 2‐tailed significance of correlations (SPSS, IBM).
Whole‐brain source‐space data (beta‐band, 15–30 Hz) were also compared across groups using a mass univariate trial‐wise general linear model (GLM) approach [Hunt et al., 2012; Woolrich et al., 2009]. The trial‐wise GLM consisted of different regressors that picked out the trials that corresponded to left and right lateralization, and “NoGo” and “Go” conditions. First‐level (within‐subject) contrasts of parameter estimates (COPEs), were then calculated comparing either left or right lateralized effect against baseline, or successfully withheld “NoGo” effect either against baseline or against “Go” effect. The first‐level COPES were averaged over repeated sessions to compute subject‐level means, which were in turn passed into the group‐level subject‐wise GLM. Group‐level regressors were set to model the group means and group contrasts set to detect group differences using unpaired t‐tests. Prior to passing subject‐level COPEs to the group‐level analysis, they were transformed such that only left‐hand preparation/response source‐space COPEs were flipped around the medial surface (i.e., around the x‐axis in MNI coordinates). This enabled merging of both response lateralities into contra‐lateral and ipsi‐lateral hemispheres, relative to the effector limb. 4D spatiotemporal maps were averaged across selected time windows of interest, corresponding to the all‐subjects combined group's maximal ERD following appearance of cue/target, or corresponding to (individual trial specific) response onset/offset. The resulting 3D maps were compared between groups using cluster‐based permutation statistics (Randomize, FSL), with a pre‐defined cluster‐forming threshold (t‐stat = 2.4) and 10,000 permutations with 100‐mm of spatial smoothing applied to the variance of the COPE to improve the effective degrees of freedom. Given the strong a priori expectation that group differences would be prominent within the motor cortices, group comparison was restricted to an anatomically confined binary mask encompassing bilateral motor areas.
The extent to which PMBR was confined to the contra‐lateral motor cortex was investigated using three independent measures of “degree of lateralization” (DoL). Firstly, dynamic beta power timecourses from each subject's motor ROI (as defined by trough beta ERD and subsequently by peak PMBR) were contrasted as a percentage ((contra‐ipsi)/(contra + ipsi)] prior to group comparison over time. Secondly, a 3D voxel‐wise lateralization effect was calculated over the time‐averaged period of maximal PMBR across all subjects (1:2.5s post movement), and over a 400 ms window centered on each group's peak PMBR time‐point. The resultant subject specific DoL spatial map was contrasted between groups using cluster‐permutation tests as above.
Lastly, the correlation between fluctuations of beta power in the same PMBR time‐period were assessed. Using anatomical parcels (from the Harvard‐Oxford cortical structural atlas), time‐courses for each motor cortex were extracted by taking the first principal component over the enclosed voxels, using an orthogonalization procedure to correct for spatial leakage from surrounding anatomical parcels [Colclough et al., 2015]. Correlations were taken between the Hilbert power envelopes of these corrected signals and converted to normal variates with Fisher's transformation. These correlations were averaged within subjects and contrasted between groups, performing inference with 5000 permutations of the subject labels.
The expected oscillatory signature of response inhibition has been shown to involve an augmentation of beta power relative to unsuccessful inhibition [Huster et al., 2013]. The parcellation, orthogonalization and correlation method was additionally applied to an exploratory analysis of “NoGo” trial epochs, assessing the functional connectivity between the motor cortices and two anatomical regions commonly implicated in response inhibition: the supplementary motor area (SMA) and right inferior frontal cortex (rIFC) [Aron et al., 2014].
RESULTS
Demographic and Behavioral
Twelve ALS and 10 PLS patients underwent MEG acquisition. One ALS participant was excluded from analysis after failing to recognize the laterality of the cue. MEG source space variance maps for one PLS participant revealed heavy artefact contamination. Therefore MEG data from the remaining 11 ALS patients (9 male, mean age 63.5 ± 7.6 years) were contrasted against nine patients with PLS (2 male, 59.6 ± 8.0 years) and 10 age‐matched healthy controls (4 male, 61.7 ± 9.3 years). Twelve asymptomatic gene carriers (AGCs; 2 male, 51.7 ± 9.9 years, 10 SOD1, 2 C9orf72) were contrasted against another age‐matched selection of 10 healthy controls (3 male, 51.0 ± 9.5 years). Six healthy controls were included in both age comparators. Full details are given in Table 1.
Table 1.
Demographic and clinical data
| Mean ± SD (range) | ALS (n = 11) | PLS (n = 9) | Asymptomatic genetic carriers (n = 12) | Controls old (n = 10) | Controls young (n = 10) |
|---|---|---|---|---|---|
| Age (years) | 63.5 ± 7.6 (48:74) | 59.6 ± 8.0 (44:70) | 51.7 ± 9.9 (36:66) | 61.7 ± 9.3 (45:75) | 51.0 ± 9.5 (37:64) |
| Gender | 9 M: 2 F | 2 M: 7 F | 2 M: 10 F | 4 M: 6 F | 3 M: 7 F |
| Handedness | 11 R | 9 R | 12 R | 9 R: 1 L | 8 R: 2 L |
| Site of onset or genetics | 1 bulbar | 1 bulbar | 10 SOD1 | N/A | N/A |
| 2 respiratory | 2 both legs | 2 C9orf72 | |||
| 1 RUL, 3 LUL | 1 RLL, 5 LLL | ||||
| 2 RLL, 2 LLL | |||||
| ALSFRS‐r | 34.8 ± 8.8 (21:48) | 35.1 ± 6.3 (24:43) | N/A | N/A | N/A |
| Disease Duration from Symptom Onset (months) | 23.7 ± 18.9 (5:72) | 121.0 ± 57.2 (47:283) | N/A | N/A | N/A |
| Progression Rate (48 – ALSFRS‐R/duration in months) | 0.79 ± 0.69 (0:2.4) | 0.12 ± 0.06 (0.05:0.46) | N/A | N/A | N/A |
| Cognition score (% correct) Intact/Borderline/Impaired | 84.8% ± 8.5 (69:95) | 84.6% ± 9.4 (73:95) | 91% ± 4.5 (80:97) | 96 .1% ± 3.1 (91:100) | 96.3% ± 2.4 (92:99) |
| (10 ECAS, 2 ACE‐r) 8/1/2 | (8 ECAS, 1 ACE‐r) 5/1/3 | (11 ACE‐r) | (9 ACE‐r) | (8 ACE‐r) |
Mean followed by SD (range). Genetic data only available for AGCs.
ALS patients tended to complete fewer correct “Go” responses than other groups; mean ± standard deviation 196 ± 66 from a maximum possible 240 (healthy controls 228 ± 18, P = 0.14). An effect of group membership was noted on RT, (F(4,42) = 5.44, P = 0.001, Fig. 2A), with PLS patients responding the slowest (623 ms, significantly slower than older controls, 440 ms, P = 0.016) and AGCs the fastest (377 ms, not significantly faster than younger controls, 434 ms, P = 0.17). Throughout the groups there was a significant effect of ISI duration on RT, such that participants were on average 35 ms faster following the longer ISI (F(1) = 19.0, P < 0.001). There was no group by ISI duration interaction.
Figure 2.
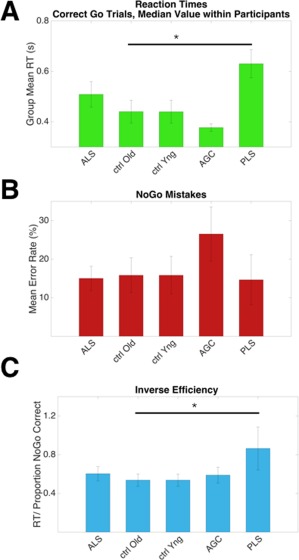
Behavioral data detailing task performance across groups. A: PLS patients responded slower than older controls, P = 0.016. B: A trend towards more NoGo errors was made by pre symptomatic carriers. C: Inverse efficiency, a global measure of task performance (RT/NoGo proportion correct) was impaired in the PLS group (P = 0.022). Median values within participants, mean values within group, error bars = SEM. Uncorrected for multiple comparisons. [Color figure can be viewed at http://wileyonlinelibrary.com.]
The AGC group mean for erroneous responses on “NoGo”' trials was the highest at 26.5%, although not significantly more so than healthy controls (10.6%, P = 0.08, uncorrected, Fig. 2B). A global measure of task performance, inverse efficiency, was calculated as RT/NoGo” proportion correct. PLS patients performed worse relative to older controls (P = 0.022, uncorrected, Fig. 2C).
An increase in RT in the “Go” trials following a “NoGo” trial by 20 ms on average was consistently seen across all groups (Supporting Information Figure S2). This was an expected behavioral effect, equivalent to motor slowing induced by novel stimuli [Wessel and Aron, 2013].
Motor Preparation
Lateralized cortical changes in preparation for movement occurred shortly after cue processing in the form of regional beta‐band desynchronization. All groups demonstrated this preparatory beta‐desychronization, which was maximal at around 600 ms following cue onset (healthy control data from motor ROIs shown in Fig. 3A), and was more prominent in the motor cortex contralateral to the prepared limb. No significant group differences were immediately apparent in the intensity of the preparatory beta desychronization from subject‐specific functionally defined ROIs (Fig. 3B,C). However, analysis of whole‐brain MEG data, time‐averaged around the maximal beta desychronization (t 0 + 500 ms: t 0 + 700 ms. t 0 = cue onset), revealed ALS patients to exhibit regions with significantly increased beta desychronization relative to controls, within (but not restricted to) both the contralateral and ipsilateral precentral gyrus (P = 0.031, Fig. 3D). Directionally similar trends were noted in both PLS and AGC groups.
Figure 3.
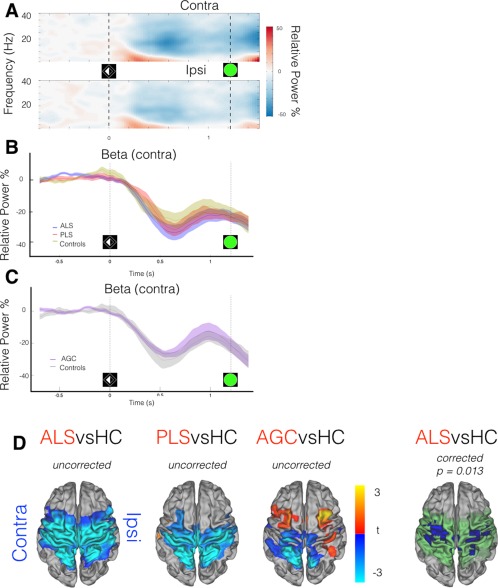
MEG data epoched around cue (t 0 = laterality cue appearance). A: Neural correlate of task performance across frequency range demonstrated in TFR (averaged across healthy controls only) from motor cortex ROIs, contra/ipsilateral relative to effector limb. Beta‐band (15:30 Hz) power decrease (desynchronization) from baseline (more intense desynchronization = deeper blue) occurs during motor preparation, particularly in the contralateral hemisphere, maximally 600 ms post‐cue. B,C: Beta‐band power (relative to baseline) within contralateral motor cortex ROI, group comparisons. D: ALS patients show deeper beta desynchronization. t 0 + 500 ms: t 0 + 700 ms. t stats in red/blue, motor cortex mask used for statistics in green. Cluster correction within motor cortex mask, not across groups. Vertical lines denote appearance of visual laterality cue and go/Nogo target. [Color figure can be viewed at http://wileyonlinelibrary.com.]
Motor Execution
As a unilateral limb movement was executed, beta desychronization became more focal within the motor cortices, yet also more bilateral [control time‐frequency representation (TFR)] (Fig. 4A). The timecourse of the peri‐response beta desychronization from the contralateral motor cortex ROI revealed it to be preserved in both ALS and PLS patient groups (Fig. 4B). The peri‐response beta desychronization extracted from the ROI appeared deeper within the AGC group although not significantly so (P = 0.590; Fig. 4C). Group contrasts were then statistically appraised in whole‐brain data (t 0: t 0 + 200 ms. t 0 = response). Although ALS patients exhibited only a trend towards deeper beta‐desychronization, AGCs demonstrated regions with significantly deeper beta desychronization than controls over a large cortical area encompassing both motor cortices (P = 0.013, Fig. 4D). This result was qualitatively similar after exclusion of the two C9orf72 AGCs. No significant differences were found between PLS patients and controls.
Figure 4.
MEG data epoched around movement initiation ( t 0 = response execution). A: TFR (averaged across healthy controls) demonstrates movement ERD (in blue) as a bilateral motor cortical event. B,C: Beta‐band power (relative to baseline) within contralateral motor cortex ROI; group comparison. Vertical lines indicate cue onset prior to median RT (distribution overlaid). D: AGCs show deeper beta desynchronization [t 0: t 0 + 200 ms] relative to controls, with a directionally similar trend in ALS patients. t stats in red/blue, motor cortex mask used for statistics in green. Vertical lines denote appearance of Go target and timing of response. [Color figure can be viewed at http://wileyonlinelibrary.com.]
Motor Rebound
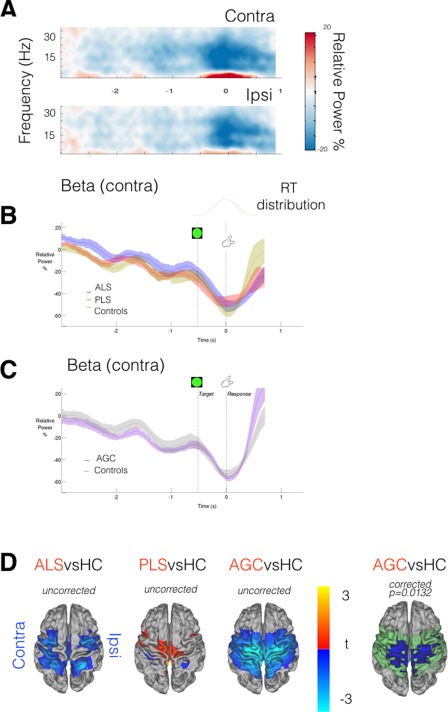
Postmovement beta rebound was expected to be observed from 500 ms after termination of EMG activity, and typically lateralized to the contralateral precentral gyrus [Cheyne, 2013] (control group TFR Fig. 5A, other groups Supporting Information Figure S3, each individual displayed in Supporting Information Figure S4). Group differences in movement duration (F = 2.3, P = 0.07) were accommodated by again realigning data epochs (t 0 = movement completion, as indicated by the fiber‐optic trigger). An expected correlation was found between RT and both EMG peak amplitudes (P < 0.001), and speed of transition in the contralateral motor cortex from “beta desychronization” (ERD) to PMBR (P = 0.0045) [Erbil and Ungan, 2007; Stancák and Pfurtscheller, 1996]. Inspection of beta power timecourses from the contralateral motor cortex confirmed significantly delayed beta rebound in both ALS (P = 0.03) and PLS (P = 0.011) patients (Fig. 5B).
Figure 5.

MEG data epoched around movement completion (t 0 = finger replaced). A: TFR (averaged across healthy controls only) from motor cortex ROIs relative to effector limb. Post Movement Beta Rebound (PMBR, in red) is predominantly contralateral in healthy controls. B,C: Beta‐band power (relative to baseline) within contralateral motor cortex ROI; group comparison. Black bar = timespan of significant group difference via cluster permutation testing, P < 0.05. Vertical lines denote appearance of Go target, timing of response initiation and completion. D: ALS patients demonstrate lower beta power in early transition to PMBR [t 0 + 500 ms: t 0 + 700 ms]. t‐stats in blue, motor cortex mask used for statistics in green. E: Beta‐band power lateralization evolves over time around a response, PLS patients show a diminished DoL during PMBR (black bar, P < 0.05). F: PMBR DoL appraised in whole‐brain data [t 0 + 1 s: t 0 + 2.5 s]. PLS patients again show significantly reduced DoL. t‐stats in ROY‐BIG‐BL. [Color figure can be viewed at http://wileyonlinelibrary.com.]
AGCs showed a more rapid and increased amplitude beta rebound, though not significantly so (P = 0.062, Fig. 5C). Given this unexpected result in the AGC group, averaged rectified surface EMG timecourses were inspected, and confirmed faster EMG onset/offset relative to controls (Supporting Information Figure S5). Given that the transition from beta desychronization to beta rebound is a dynamic process, whole‐brain analysis was focused on the initial time‐period of beta rebound (t 0 + 500 ms: t 0 + 700 ms). During this period, ALS patients were confirmed to exhibit regions with significantly reduced beta power (P = 0.028) relative to controls (Fig. 5D), with a directionally similar trend in PLS patients.
Beta rebound in both patient groups was additionally suspected to be less lateralized, most marked in the PLS patients. This was confirmed only in beta power timecourses from the subject‐specific ERD defined motor ROIs (P = 0.017, Fig. 5E) but not in the PMBR defined ROIs. In the voxel‐wise spatial maps of DoL (Fig. 5F), a significant difference in lateralization was noted across a broad time‐period (1–2.5 s following movement completion) but group differences were not preserved in the 400 ms time‐period centered on each group's peak PMBR. This finding suggests a delay in PMBR, in terms of both intensity and lateralization. Finally, the interhemispheric correlation between beta power timecourses (extracted from the anatomically defined motor cortex parcels, t 0 + 1 s: t 0 + 2.5 s) revealed significantly higher correlation in both PLS patients (P = 0.0054) and ALS patients (P = 0.0086) relative to healthy controls. The implication of this finding is that beta power changes following completion of movement are abnormally coherent between hemispheres in these patient groups, but this may simply again reflect delayed PMBR. Beta rebound lateralization was unaffected in AGCs.
Movement Inhibition
Available trials in which participants successfully withheld a preeminent motor response after appearance of the “NoGo” target were examined to probe integrity of the inhibitory cortical network. Faster responders made more false alarms on “NoGo” trials, as did those with deeper preparatory beta desychronization (P = 0.015). Contrasting correct “NoGo” trials with “Go” trials within the healthy control group, a relative increase in beta power was revealed in the frontal lobe and premotor regions. Data was re‐epoched around t 0 = target presentation. The time period (t 0 + 100 ms: t 0 + 300 ms) was appraised in whole‐brain data, revealing a beta power difference (“NoGo” versus “Go” trials, healthy controls only) in clusters overlying the right frontal lobe and left motor cortex (P = 0.0028, Fig. 6A).
Figure 6.

MEG data epoched around NoGo target presentation (t 0 = target appearance). Beta‐band power increases over a diffuse cortical network in successfully inhibited NoGo trials, demonstrated in (A) healthy controls, beta power NoGo > Go. [t 0 + 100 ms: t 0 + 300 ms]. B: AGCs contrasted against HC, NoGo trials only, reveals increased beta power in posterior cortical regions. No significant differences are noted in ALS or PLS patients. t‐stats in ROY‐BIG‐BL. Whole brain cluster‐permutation correction. [Color figure can be viewed at http://wileyonlinelibrary.com.]
Group contrasts were restricted to successful “NoGo” trials during which the prepared response was inhibited. Within the same time‐period, no significant differences between ALS or PLS patients against controls were apparent in the degree to which beta power increased after “NoGo” targets. AGCs however demonstrated a relative increase in beta power in the right lateral occipital cortex, extending to the precuneus (P = 0.0466, Fig. 6B). Correlation analysis of beta power dynamics between selected cortical parcels (t 0: t 0 + 1 s) also suggested reduced functional connectivity between rIFC and SMA in PLS patients only (FWE P = 0.042). No group differences were noted in the correlation between motor cortex parcels, nor between ALS or AGC groups against controls.
Clinical Correlations
Correlations between relevant behavioral measures, summary MEG statistics, and clinical metrics are presented in Table 2. These were exploratory given the small group numbers, and not corrected for multiple comparisons. Across all patients, slower responses were correlated with increasing ALSFRS‐R disability (global P = 0.005, upper limb P = 0.02), longer disease duration (P = 0.038) and higher UMN score (P = 0.019). Within the ALS group, impaired cognition (% correct ECAS or ACEr) tended to correlate with noGo mistakes (P = 0.073) as did UMN score (P = 0.057). Baselined PMBR correlated negatively with disease duration (P = 0.024) and positively with rate of progression (P = 0.036).
Table 2.
Correlations within the ALS patients (Kendall's Tau, uncorrected) between clinical measures, behavioral data, and summary MEG statistics extracted from per‐subject ROI timecourses [Color table can be viewed at http://wileyonlinelibrary.com]
| ALSFRSr Sum | ALFRSr UL | UMN Score | Disease duration | Progression rate | Cognition | Median RT | noGo mistakes | Preparatory ERD | Execution ERD | ERD lateralization | PMBR | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALSFRSr Sum | x | |||||||||||
| ALFRSr UL | 0.577** | x | ||||||||||
| UMN Score | −0.333 | −0.216 | x | |||||||||
| Disease Duration | 0.187 | −0.112 | 0.000 | x | ||||||||
| Progression Rate | −0.561* | −0.187 | 0.191 | −0.552** | x | |||||||
| Cognition | 0.374 | 0.299 | −0.191 | −0.139 | 0.127 | x | ||||||
| Median RT | −0.524* | −0.785** | 0.343 | 0.406* | 0.164 | −0.345 | x | |||||
| noGo Mistakes | −0.449 | −0.299 | 0.458 | −0.011 | 0.164 | −0.418 | 0.418 | x | ||||
| Preparatory ERD | 0.075 | 0.262 | 0.114 | −0.177 | −0.127 | −0.345 | −0.091 | −0.018 | x | |||
| Execution ERD | −0.224 | 0.000 | 0.381 | −0.368* | −0.018 | −0.309 | 0.164 | 0.236 | 0.673** | x | ||
| PMBR | −0.449 | −0.150 | 0.000 | 0.014 | 0.491* | −0.091 | 0.091 | 0.091 | −0.345 | −0.236 | 0.345 | X |
*P < 0.05, **P < 0.01.
DISCUSSION
This study used MEG to investigate the neural basis of prepared voluntary movement generation, typified by modulation in beta‐band power, revealing abnormalities across the ALS phenotypic spectrum.
Cortical Neurophysiological Dysfunction in ALS and PLS
Previous EEG investigations in ALS largely focused on evoked movement‐related cortical potentials (MRCPs), time‐locked to movement onset and averaged over many trials. The impact on these measures appears most prominent in patients with a high burden of UMN dysfunction [Bizovičar et al., 2013; Inuggi et al., 2011; Westphal et al., 1998], with MRCPs particularly reduced in patients with PLS [Bai et al., 2006]. Event‐related potentials (ERPs) have also revealed abnormal neural correlates of attention control [Pinkhardt et al., 2008], particularly within a bulbar‐onset sub‐group [Mannarelli et al., 2013]. This experimental design permitted assessment of neuronal function during a period of motor preparation, independent of and temporally distinct to subsequent motor performance, thus minimizing the potentially significant confound of group differences in motoric activity.
The precise physiological functions of beta oscillations and their task‐related modulations remain uncertain. More intense beta desychronization is induced as movements require more force [Stančák et al., 1997], speed [Toma et al., 2002], or complexity [Hummel et al., 2003; Manganotti et al., 1998]. Conversely, exaggerated and overly consistent beta synchrony is antikinetic, whether achieved though Parkinson's pathology [Little et al., 2012] or direct stimulation [Pogosyan et al., 2009]. Physiological beta synchrony is promoted in expectation of impending perturbation to a desired posture [Androulidakis et al., 2007] but is also sensitive to the uncertainty of motor outcome estimation [Tan et al., 2016]. Beta oscillations may also contribute to long‐range communication across cortical regions [Engel and Fries, 2010; Kopell et al., 2000] and can facilitate modulation of selective attention in support of action selection [Grent‐'t‐Jong et al., 2013, 2014; Tzagarakis et al., 2010], beyond simple correlation with reaction times [van Ede et al., 2012]. The abnormalities in this characteristic motor system rhythm displayed by ALS patients (amplified beta desychronization and attenuated beta rebound) may reflect or even contribute to an excitotoxic degeneration of neural microcircuitry, particularly given the apparent correlation with rate of disease progression. A more simplistic explanation of the beta desychronization difference in ALS patients might be the relative cognitive demands and task difficulty in comparison to healthy controls [Schoenfeld et al., 2005]. However, this fails to account for the corresponding abnormalities seen in high‐performing AGCs and the lack of excess beta desychronization within equally disabled PLS patients.
Cortical Hyperexcitability in ALS
Although not unique to ALS [Di Lazzaro et al., 2004], converging strands of evidence suggest hyperexcitability as a key mechanism in the pathogenesis of ALS, possibly accompanied by a relative failure of inhibitory cortical interneuronal function [Turner and Kiernan, 2012]. Pathological studies demonstrate a particular vulnerability of parvalbumin‐positive interneurons [Maekawa et al., 2004; Nihei et al., 1993], later shown to contribute to reduced inhibitory GABA‐ergic tone in animal models [McGown et al., 2013; Nieto‐Gonzalez et al., 2011]. Neuroimaging studies across multiple modalities have supported the concept of reduced cortical inhibition. Initial observations were of a “boundary shift” in regional cerebral blood flow measured using positron emission tomography (PET) in ALS patients during a joystick task [Kew et al., 1993]. Further support stems from the observation of widened cortical BOLD activation in ALS patients during functional MRI (fMRI)‐based motor tasks [Mohammadi et al., 2011; Stanton et al., 2007b]; reduced binding of the PET GABAA ligand [11C]‐fluamzenil [Lloyd et al., 2000; Turner et al., 2005a]; reduced GABA MR spectroscopy peak in the motor cortex [Foerster et al., 2012], and elevated Glx (glutamate and glutamine) peak in the medulla [Pioro et al., 1999].
Direct evidence of cortical hyperexcitability in ALS was provided by TMS [Vucic et al., 2013b]. Conditioning pulse protocols have reliably demonstrated reduced intracortical inhibition as a characteristic feature of ALS [Menon et al., 2015; Yokota et al., 1996; Ziemann et al., 1997], which may be partly ameliorated by reducing glutamatergic influence through administration of Riluzole—the only licensed disease‐modifying therapy in ALS [Stefan et al., 2001; Vucic et al., 2013a]. Postmovement beta rebound corresponds well to a period of reduced cortico‐spinal tract excitability, and the present data suggest that ALS patients differ in the speed of transition to this state after movement.
The Endophenotype of PLS
The nosology of PLS continues to be debated [Le Forestier et al., 2001; Singer et al., 2007], although some clinical [Gordon et al., 2009; Pringle et al., 1992], neuroimaging [Agosta et al., 2014; Kolind et al., 2013; Kwan et al., 2013; Müller et al., 2012; Turner et al., 2007] and saccadic [Proudfoot et al., 2015] features support an apparent distinction from ALS. Involvement of the corpus callosum (CC) is a consistent feature of ALS [Filippini et al., 2010], thought to contribute to a functional impairment of interhemispheric inhibition, as evidenced by both TMS [Karandreas et al., 2007; Wittstock et al., 2007] and clinically evident mirror movements [Wittstock et al., 2011]. The CC appears to be a particularly vulnerable structure in PLS [Agosta et al., 2014; Ciccarelli et al., 2009; Iwata et al., 2011; Kolind et al., 2013] and a finding of delayed lateralization of PMBR is in keeping with this observation. Preserved hemispheric autonomy is still demonstrated by the time that PMBR peaks, suggesting still adequate CC functionality, although partial mirror movements during response epochs were also noted in some patients' EMG. Previous EEG‐based investigation of MRCPs also noted ipsilateral premotor recruitment among ALS patients with high UMN burden [Inuggi et al., 2011]. PLS patients by contrast demonstrated diminished preparatory MRCPs during a self‐paced EEG motor task [Bai et al., 2006]. These findings were not in this instance accompanied by any obvious alteration to MRCP topography and beta desychronization appeared preserved, highlighting that distinct neural generators underpin each phenomenon [Toro et al., 1994]. This study did not detect any significant excess in beta desychronization intensity between PLS patients and age‐matched controls in whole brain data. In contrast to the hyperexcitability typically detected in ALS, the motor cortex of PLS patients is often strikingly resistant to TMS stimulation [Brown et al., 1992; Kuipers‐Upmeijer et al., 2001; Zhai et al., 2003], in keeping with the present PLS data failing to demonstrate additional beta‐desychronization.
Presymptomatic Cortical Dysfunction?
Characterization of any presymptomatic phase is a priority for all neurodegenerative conditions if preventative strategies are envisaged. Assessment of asymptomatic familial Alzheimer gene carriers has highlighted the added sensitivity of functional neuroimaging [Chhatwal et al., 2013] in keeping with the age‐dependent impact of APOE ε4 status on both fMRI [Filippini et al., 2011] and MEG [Cuesta et al., 2015] in the task‐free state. It remains an open question to what extent symptoms of ALS are preceded by temporally remote cellular abnormalities [Eisen et al., 2014]. Although animal models of ALS demonstrate abnormal neural architecture and function during embryonic stages [Martin et al., 2013; Vinsant et al., 2013], human epidemiological [Byrne et al., 2013; Schoder et al., 2010] and pathological [Proudfoot et al., 2014a] links to neurodevelopmental disorders remain sparse. Suggestions that ALS (or FTD) pathology might manifest in a behavioural prodrome long before diagnostic symptoms remain speculative [Eisen et al., 2014; Lule et al., 2008].
There are inconsistent reports of structural and functional MRI abnormalities prior to symptom onset in those at high genetic risk of ALS [Carew et al., 2011; Menke et al., 2016; Ng et al., 2008; Vucic et al., 2008; Walhout et al., 2015]. Evidence of cortical hyperexcitability in asymptomatic carriers of genetic mutations who are “at risk” of ALS is restricted to a very limited finding of reduced ligand [11C]‐flumazenil binding in two individuals with the D90A SOD1 mutation [Turner et al., 2005a], and three SOD1 mutation carriers who demonstrated reduced or absent intra‐cortical inhibition within three months of symptom onset [Vucic et al., 2008]. Further studies on seven asymptomatic SOD1 mutation carriers (mean age 33 years) and 11 C9orf72 carriers (mean age 49 years) failed to differentiate them from controls on TMS measures [Geevasinga et al., 2015a; Vucic et al., 2010]. The older age of the SOD1 mutation carrier participants in the current study may contribute to the apparent gain in sensitivity offered by MEG.
Both patients with ALS and AGCs differ from controls in beta rebound latency, but the group effect directions are divergent. Review of the averaged EMG timecourses revealed that AGC participants completed their responses faster than controls, functionally corresponding to a more rapid transition from beta desychronization to beta rebound. However, across all groups EMG metrics and behavioral measures correlated poorly with beta rebound latency and intensity. Conflicting abnormalities between pre‐symptomatic carriers and manifest patients have previously been noted on fMRI studies of both Huntington's [Kloppel et al., 2009] and monogenetic Alzheimer's [Quiroz et al., 2010], therefore compensatory or pathological neural abnormalities may still underpin these MEG group differences. Slight differences in task performance pose a challenge in the interpretation of functional data from particularly motivated asymptomatic AGC. Blinded genetic testing of at‐risk individuals is a solution that raises novel ethical considerations as well as practical complexities, especially with regards to future therapeutic trials [Kim et al., 2015].
Does Motoric Inhibition Reflect Executive Dysfunction?
Although cognitively impaired ALS patients are predictably under‐represented in demanding functional neuroimaging tasks, comparatively reduced activation in the dorsolateral prefrontal cortex has been demonstrated [Witiuk et al., 2014], while other frontal regions reveal increased activation during inhibition of prepared manual movements [Mohammadi et al., 2015]. The present finding of abnormal beta power in AGCs during successful inhibition was limited to posterior cortical regions in the context of task‐induced inferior and pre‐frontal activity. Despite previous findings of reduced stop‐signal ERPs [Thorns et al., 2010], no abnormalities in beta‐band dynamics were noted in ALS patients, so that the changes in AGCs might represent very early compensatory neural changes. In contrast to an ERP study utilising the Stroop task [Amato et al., 2013], but in keeping with eye‐tracking findings [Proudfoot et al., 2015], the present data suggest that executive control dysfunction may not spare PLS patients, given that task relevant functional connectivity was diminished between rIFC and SMA. Multiple nonexclusive biological and methodological restrictions may underlie the lack of correlation between the MEG data and cognitive profiles of the ALS participants [Verstraete et al., 2015], not least the brevity of the ECAS test as a opposed to more comprehensive and granular neuropsychological assessment.
Study Limitations
These results complement existing EEG based investigations of sensorimotor oscillations in ALS which also found abnormally attenuated beta rebound but did not report increased beta desychronization [Bizovičar et al., 2014; Riva et al., 2012]. These previous studies differed from this investigation not only in acquisition technologies, but also in protocol design by requiring self‐paced movements of the right hand only, whereas the present study was a laterally cued task and thus facilitated analysis of a specific preparatory period. Additional analysis variables pertinent to neural signal interpretation include the selection of baseline period and frequency band of interest. Furthermore patient groups may differ in the degree to which they actively participate in a task, and this severely limits the conclusions that can be drawn from tasks of motor imagery (Kasahara et al., 2012; Lulé et al., 2007; Stanton et al., 2007a, 2007b). Riluzole administration could possibly dilute the reported ALS group effects, although analysis of the five remaining patients revealed a directionally similar trend. Unique challenges arise in the investigation of a genetically and phenotypically heterogenous condition such as ALS. Comparative studies against other neurological conditions with significant UMN burden of disease remain valuable. Furthermore, longitudinal study, particularly of a precisely defined group of AGCs before and after conversion to symptomatic ALS could pinpoint important aetiological pathways but might have restricted relevance to the wider sporadic ALS population.
MEG benefits from improved spatial resolution over EEG, as neuromagnetic signals pass through skull structures without spatial smearing. Reconstruction of the MEG signal into source‐space components overcomes the ambiguity inherent in selection of specific EEG sensors, but coregistration errors are still likely to limit precise anatomical conclusions regarding the current group differences. This methodology included selection of an ROI‐based purely on subject‐specific functional data, in addition to anatomically defined sources perhaps more vulnerable to coregistration error.
CONCLUSIONS
Motor system cortical function assessed by beta‐band oscillations revealed abnormalities across the syndrome of ALS. MEG affords a useful contribution to the non‐invasive investigation of ALS pathology that complements more established techniques. The present results provide further distinguishing features between motor neurodegenerative phenotypes and supporting evidence of a detectable pre‐symptomatic phase to ALS pathophysiology. The current study has not exhausted the analytic options available from high‐dimensional MEG data, in particular further analysis of coherence measures may confirm and extend the existing literature concerning functional connectivity in ALS. Resting‐state networks similar in topography to those delineated by fMRI have also been decomposed from fluctuations in band‐limited MEG power [Brookes et al., 2011; Hipp et al., 2012] and the precise temporal sensitivity afforded by MEG has also enabled discovery of more rapidly cycling brain states [Baker et al., 2014] that remain unexplored across disease states. MEG was a well‐tolerated investigation for functionally disabled patients and could serve as a platform for the appraisal of novel therapeutic agents [Suntrup et al., 2013], as well as providing unique “real time” mechanistic insights into cortical dysfunction that will guide the emerging era of targeted therapeutics in ALS.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
ACKNOWLEDGMENTS
The authors would like to thank all study participants for their enthusiastic contribution to clinical research; Eliana Reyes and Sumaira Hussain (University of Miami, USA) for the Pre‐fALS study coordination; Dr F van Ede (University of Oxford Centre for Human Brain Activity, UK) for analysis advice; and Prof PM Andersen (Umeå University, Sweden) for the genetic analysis.
Contributor Information
Anna C. Nobre, Email: martin.turner@ndcn.ox.ac.uk.
Martin R. Turner, Email: kia.nobre@ohba.ox.ac.uk.
REFERENCES
- Abrahams S, Newton J, Niven E, Foley J, Bak TH (2013): Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 1–6. [DOI] [PubMed] [Google Scholar]
- Agosta F, Canu E, Valsasina P, Riva N, Prelle A, Comi G, Filippi M (2013): Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging 34:419–427. [DOI] [PubMed] [Google Scholar]
- Agosta F, Galantucci S, Riva N, Chiò A, Messina S, Iannaccone S, Calvo A, Silani V, Copetti M, Falini A, Comi G, Filippi M (2014): Intrahemispheric and interhemispheric structural network abnormalities in PLS and ALS. Hum Brain Mapp 35:1710–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato N, Riva N, Cursi M, Martins‐Silva A, Martinelli V, Comola M, Fazio R, Comi G, Leocani L (2013): Different frontal involvement in ALS and PLS revealed by Stroop event‐related potentials and reaction times. Front Aging Neurosci 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androulidakis AG, Doyle LMF, Yarrow K, Litvak V, Gilbertson TP, Brown P (2007): Anticipatory changes in beta synchrony in the human corticospinal system and associated improvements in task performance. Eur J Neurosci 25:3758–3765. [DOI] [PubMed] [Google Scholar]
- Aron AR, Robbins TW, Poldrack RA (2014): Inhibition and the right inferior frontal cortex: one decade on. Trends Cogn Sci 18:177–185. [DOI] [PubMed] [Google Scholar]
- Bai O, Vorbach S, Hallett M, Floeter MK (2006): Movement‐related cortical potentials in primary lateral sclerosis. Ann Neurol 59:682–690. [DOI] [PubMed] [Google Scholar]
- Baker AP, Brookes MJ, Rezek IA, Smith SM, Behrens T, Smith PJP, Woolrich M (2014): Fast transient networks in spontaneous human brain activity. Elife 2014:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatar M, Wuu J (2012): Presymptomatic studies in ALS: Rationale, challenges, and approach. Neurology 79:1732–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatar M, Wuu J, Ravits J (2013): Opportunity and innovation in studying pre‐symptomatic amyotrophic lateral sclerosis. Muscle Nerve 47:629–631. [DOI] [PubMed] [Google Scholar]
- Bizovičar N, Koritnik B, Zidar I, Dreo J, Zidar J (2013): Movement‐related cortical potentials in ALS increase at lower and decrease at higher upper motor neuron burden scores. Amyotroph Lateral Scler Frontotemporal Degener 14:380–389. [DOI] [PubMed] [Google Scholar]
- Bizovičar N, Dreo J, Koritnik B, Zidar J (2014): Decreased movement‐related beta desynchronization and impaired post‐movement beta rebound in amyotrophic lateral sclerosis. Clin Neurophysiol 125:1689–1699. [DOI] [PubMed] [Google Scholar]
- Brainard DH (1997): The Psychophysics Toolbox. Spat Vis 10:433–436. [PubMed] [Google Scholar]
- Brookes MJ, Woolrich M, Luckhoo H, Price D, Hale JR, Stephenson MC, Barnes GR, Smith SM, Morris PG (2011): Investigating the electrophysiological basis of resting state networks using magnetoencephalography. Proc Natl Acad Sci U S A 108:16783–16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL (2000): El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord 1:293–299. [DOI] [PubMed] [Google Scholar]
- Brown WF, Ebers GC, Hudson AJ, Pringle CE, Veitch J (1992): Motor‐evoked responses in primary lateral sclerosis. Muscle Nerve 15:626–629. [DOI] [PubMed] [Google Scholar]
- Byrne S, Heverin M, Elamin M, Bede P, Lynch C, Kenna K, Maclaughlin R, Walsh C, Al Chalabi A, Hardiman O (2013): Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: A population‐based case‐control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann Neurol 74:699–708. [DOI] [PubMed] [Google Scholar]
- Carew JD, Nair G, Andersen PM, Wuu J, Gronka S, Hu X, Benatar M (2011): Presymptomatic spinal cord neurometabolic findings in SOD1‐positive people at risk for familial ALS. Neurology 77:1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Yaseen Z, Cohen LG, Hallett M (1998): Time course of corticospinal excitability in reaction time and self‐paced movements. Ann Neurol 44:317–325. [DOI] [PubMed] [Google Scholar]
- Cheyne DO (2013): MEG studies of sensorimotor rhythms: A review. Exp Neurol 245:27–39. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Schultz AP, Johnson K, Benzinger TLS, Jack C, Ances BM, Sullivan C. a, Salloway SP, Ringman JM, Koeppe RA, Marcus DS, Thompson P, Saykin AJ, Correia S, Schofield PR, Rowe CC, Fox NC, Brickman AM, Mayeux R, McDade E, Bateman R, Fagan AM, Goate AM, Xiong C, Buckles VD, Morris JC, Sperling RA (2013): Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81:736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli O, Behrens TE, Johansen‐Berg H, Talbot K, Orrell RW, Howard RS, Nunes RG, Miller DH, Matthews PM, Thompson AJ, Smith SM (2009): Investigation of white matter pathology in ALS and PLS using tract‐based spatial statistics. Hum Brain Mapp 30:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colclough GL, Brookes MJ, Smith SM, Woolrich MW (2015): A symmetric multivariate leakage correction for MEG connectomes. Neuroimage 117:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta P, Garcés P, Castellanos NP, López ME, Aurtenetxe S, Bajo R, Pineda‐Pardo JA, Bruña R, Marín AG, Delgado M, Barabash A, Ancín I, Cabranes JA, Fernandez A, Del Pozo F, Sancho M, Marcos A, Nakamura A, Maestú F (2015): Influence of the APOE ε4 allele and mild cognitive impairment diagnosis in the disruption of the MEG resting state functional connectivity in sources space. J Alzheimers Dis 44:493–505. [DOI] [PubMed] [Google Scholar]
- Douaud G, Filippini N, Knight S, Talbot K, Turner MR (2011): Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain 134:3470–3479. [DOI] [PubMed] [Google Scholar]
- van Ede F, de Lange FP, Maris E (2012): Attentional cues affect accuracy and reaction time via different cognitive and neural processes. J Neurosci 32:10408–10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A, Kiernan M, Mitsumoto H, Swash M (2014): Amyotrophic lateral sclerosis: A long preclinical period? J Neurol Neurosurg Psychiatry 85:1232–1238. [DOI] [PubMed] [Google Scholar]
- Engel AK, Fries P (2010): Beta‐band oscillations‐signalling the status quo? Curr Opin Neurobiol 20:156–165. [DOI] [PubMed] [Google Scholar]
- Erbil N, Ungan P (2007): Changes in the alpha and beta amplitudes of the central EEG during the onset, continuation, and offset of long‐duration repetitive hand movements. Brain Res 1169:44–56. [DOI] [PubMed] [Google Scholar]
- Filippini N, Douaud G, Mackay CE, Knight S, Talbot K, Turner MR (2010): Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology 75:1645–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N, Ebmeier KP, MacIntosh BJ, Trachtenberg AJ, Frisoni GB, Wilcock GK, Beckmann CF, Smith SM, Matthews PM, Mackay CE (2011): Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage 54:602–610. [DOI] [PubMed] [Google Scholar]
- Foerster BR, Callaghan BC, Petrou M, Edden RAE, Chenevert TL, Feldman EL (2012): Decreased motor cortex γ‐aminobutyric acid in amyotrophic lateral sclerosis. Neurology 78:1596–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Forestier N, Maisonobe T, Piquard a, Rivaud S, Crevier‐Buchman L, Salachas F, Pradat PF, Lacomblez L, Meininger V (2001): Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain 124:1989–1999. [DOI] [PubMed] [Google Scholar]
- Fry A, Mullinger KJ, O'Neill GC, Barratt EL, Morris PG, Bauer M, Folland JP, Brookes MJ (2016): Modulation of post‐movement beta rebound by contraction force and rate of force development. Hum Brain Mapp 37:2493–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, Yiannikas C, Kiernan MC, Vucic S (2015a): Cortical Function in Asymptomatic Carriers and Patients With C9orf72 Amyotrophic Lateral Sclerosis. JAMA Neurol 2145:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geevasinga N, Menon P, Sue CM, Kumar KR, Ng K, Yiannikas C, Kiernan MC, Vucic S (2015b): Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol 22:826–e58. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP (2006): The natural history of primary lateral sclerosis. Neurology. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Cheng B, Katz IB, Mitsumoto H, Rowland LP (2009): Clinical features that distinguish PLS, upper motor neuron‐dominant ALS, and typical ALS. Neurology 72:1948–1952. [DOI] [PubMed] [Google Scholar]
- Grent‐'t‐Jong T, Oostenveld R, Jensen O, Medendorp WP, Praamstra P (2013): Oscillatory dynamics of response competition in human sensorimotor cortex. Neuroimage 83:27–34. [DOI] [PubMed] [Google Scholar]
- Grent‐'t‐Jong T, Oostenveld R, Jensen O, Medendorp WP, Praamstra P (2014): Competitive interactions in sensorimotor cortex: Oscillations express separation between alternative movement targets. J Neurophysiol 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse‐Wentrup M, Schölkopf B (2014): A brain‐computer interface based on self‐regulation of gamma‐oscillations in the superior parietal cortex. J Neural Eng 11:056015. [DOI] [PubMed] [Google Scholar]
- Hipp JF, Hawellek DJ, Corbetta M, Siegel M, Engel AK (2012): Large‐scale cortical correlation structure of spontaneous oscillatory activity. Nat Neurosci 15:884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel F, Kirsammer R, Gerloff C (2003): Ipsilateral cortical activation during finger sequences of increasing complexity: Representation of movement difficulty or memory load? Clin Neurophysiol 114:605–613. [DOI] [PubMed] [Google Scholar]
- Hunt LT, Kolling N, Soltani A, Woolrich MW, Rushworth MFS, Behrens TEJ (2012): Mechanisms underlying cortical activity during value‐guided choice. Nat Neurosci 15:470–476. S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster RJ, Enriquez‐Geppert S, Lavallee CF, Falkenstein M, Herrmann CS (2013): Electroencephalography of response inhibition tasks: Functional networks and cognitive contributions. Int J Psychophysiol 87:217–233. [DOI] [PubMed] [Google Scholar]
- Inuggi A, Riva N, González‐Rosa JJ, Amadio S, Amato N, Fazio R, Del Carro U, Comi G, Leocani L (2011): Compensatory movement‐related recruitment in amyotrophic lateral sclerosis patients with dominant upper motor neuron signs: An EEG source analysis study. Brain Res 1425:37–46. [DOI] [PubMed] [Google Scholar]
- Iwata N, Kwan J, Danielian L (2011): White matter alterations differ in primary lateral sclerosis and amyotrophic lateral sclerosis. Brain 134:2642–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karandreas N, Papadopoulou M, Kokotis P, Papapostolou A, Tsivgoulis G, Zambelis T (2007): Impaired interhemispheric inhibition in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 8:112–118. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Terasaki K, Ogawa Y, Ushiba J, Aramaki H, Masakado Y (2012): The correlation between motor impairments and event‐related desynchronization during motor imagery in ALS patients. BMC Neurosci 13:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JJ, Leigh PN, Playford ED, Passingham RE, Goldstein LH, Frackowiak RS, Brooks DJ (1993): Cortical function in amyotrophic lateral sclerosis. A positron emission tomography study. Brain 116:655–680. [DOI] [PubMed] [Google Scholar]
- Kilavik BE, Zaepffel M, Brovelli A, MacKay W. a, Riehle A (2013): The ups and downs of β oscillations in sensorimotor cortex. Exp Neurol 245:15–26. [DOI] [PubMed] [Google Scholar]
- Kim SYH, Karlawish J, Berkman BE (2015): Ethics of genetic and biomarker test disclosures in neurodegenerative disease prevention trials. Neurology 84:1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppel S, Draganski B, Siebner HR, Tabrizi SJ, Weiller C, Frackowiak RSJ (2009): Functional compensation of motor function in pre‐symptomatic Huntington's disease. Brain 132:1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolind S, Sharma R, Knight S, Johansen‐Berg H, Talbot K, Turner MR (2013): Myelin imaging in amyotrophic and primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 14:562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopell N, Ermentrout GB, Whittington MA, Traub RD (2000): Gamma rhythms and beta rhythms have different synchronization properties. Proc Natl Acad Sci U S A 97:1867–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers‐Upmeijer J, de Jager AE, Hew JM, Snoek JW, van Weerden TW (2001): Primary lateral sclerosis: clinical, neurophysiological, and magnetic resonance findings. J Neurol Neurosurg Psychiatry 71:615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JY, Meoded A, Danielian LE, Wu T, Floeter MK (2013): Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. Neuroimage Clin 2:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero a, Pilato F, Saturno E, Dileone M, Marra C, Daniele a, Ghirlanda S, Gainotti G, Tonali PA (2004): Motor cortex hyperexcitability to transcranial magnetic stimulation in Alzheimer's disease. J Neurol Neurosurg Psychiatry 75:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S, Pogosyan A, Kuhn AA, Brown P (2012): Beta band stability over time correlates with Parkinsonian rigidity and bradykinesia. Exp Neurol 236:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd CM, Richardson MP, Brooks DJ, Al‐Chalabi A, Leigh PN (2000): Extramotor involvement in ALS: PET studies with the GABA(A) ligand [(11)C]flumazenil. Brain 123:2289–2296. [DOI] [PubMed] [Google Scholar]
- Lule D, Ludolph AC, Ludolph AG (2008): Neurodevelopmental and neurodegenerative diseases ‐ Is there a pathophysiological link? Attention‐deficit/hyperactivity disorder and amyotrophic lateral sclerosis as examples. Med Hypotheses 70:1133–1138. [DOI] [PubMed] [Google Scholar]
- Lulé D, Diekmann V, Kassubek J, Kurt A, Birbaumer N, Ludolph AC, Kraft E (2007): Cortical plasticity in amyotrophic lateral sclerosis: motor imagery and function. Neurorehabil Neural Repair 21:518–526. [DOI] [PubMed] [Google Scholar]
- Maekawa S, Al‐Sarraj S, Kibble M, Landau S, Parnavelas J, Cotter D, Everall I, Leigh PN (2004): Cortical selective vulnerability in motor neuron disease: A morphometric study. Brain 127:1237–1251. [DOI] [PubMed] [Google Scholar]
- Manganotti P, Gerloff C, Toro C, Katsuta H, Sadato N, Zhuang P, Leocani L, Hallett M (1998): Task‐related coherence and task‐related spectral power changes during sequential finger movements. Electroencephalogr Clin Neurophysiol 109:50–62. [DOI] [PubMed] [Google Scholar]
- Mannarelli D, Pauletti C, Locuratolo N, Vanacore N, Frasca V, Trebbastoni A, Inghilleri M, Fattapposta F (2013): Attentional processing in bulbar‐ and spinal‐onset amyotrophic lateral sclerosis: Insights from event‐related potentials. Amyotroph Lateral Scler Frontotemporal Degener 1–9. [DOI] [PubMed] [Google Scholar]
- Maris E, Oostenveld R (2007): Nonparametric statistical testing of EEG‐ and MEG‐data. J Neurosci Methods 164:177–190. [DOI] [PubMed] [Google Scholar]
- Martin E, Cazenave W, Cattaert D, Branchereau P (2013): Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54:116–126. [DOI] [PubMed] [Google Scholar]
- McGown A, McDearmid JR, Panagiotaki N, Tong H, Al Mashhadi S, Redhead N, Lyon AN, Beattie CE, Shaw PJ, Ramesh TM (2013): Early interneuron dysfunction in ALS: Insights from a mutant sod1 zebrafish model. Ann Neurol 73:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke RAL, Körner S, Filippini N, Douaud G, Knight S, Talbot K, Turner MR (2014): Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain 137:2546–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke RAL, Proudfoot M, Wuu J, Andersen PM, Talbot K, Benatar M, Turner MR (2016): Increased functional connectivity common to symptomatic amyotrophic lateral sclerosis and those at genetic risk. J Neurol Neurosurg Psychiatry 87:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S (2015): Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol 14:478–484. [DOI] [PubMed] [Google Scholar]
- Mioshi E, Dawson K, Mitchell J, Arnold R, Hodges JR (2006): The Addenbrooke's Cognitive Examination Revised (ACE‐R): A brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 21:1078–1085. [DOI] [PubMed] [Google Scholar]
- Mohammadi B, Kollewe K, Samii A, Dengler R, Münte TF (2011): Functional neuroimaging at different disease stages reveals distinct phases of neuroplastic changes in amyotrophic lateral sclerosis. Hum Brain Mapp 32:750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi B, Kollewe K, Cole DM, Fellbrich A, Heldmann M, Samii A, Dengler R, Petri S, Münte TF, Krämer UM (2015): Amyotrophic lateral sclerosis affects cortical and subcortical activity underlying motor inhibition and action monitoring. Hum Brain Mapp 36:2878–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H‐P, Unrath A, Huppertz H‐J, Ludolph AC, Kassubek J (2012): Neuroanatomical patterns of cerebral white matter involvement in different motor neuron diseases as studied by diffusion tensor imaging analysis. Amyotroph Lateral Scler 13:254–264. [DOI] [PubMed] [Google Scholar]
- Ng M‐C, Ho JT, Ho S‐L, Lee R, Li G, Cheng T‐S, Song Y‐Q, Ho PW‐L, Fong GC‐Y, Mak W, Chan K‐H, Li LS‐W, Luk KD‐K, Hu Y, Ramsden DB, Leong LL‐Y (2008): Abnormal diffusion tensor in nonsymptomatic familial amyotrophic lateral sclerosis with a causative superoxide dismutase 1 mutation. J Magn Reson Imaging 27:8–13. [DOI] [PubMed] [Google Scholar]
- Nieto‐Gonzalez JL, Moser J, Lauritzen M, Schmitt‐John T, Jensen K (2011): Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb Cortex 21:625–635. [DOI] [PubMed] [Google Scholar]
- Nihei K, McKee AC, Kowall NW (1993): Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol 86:55–64. [DOI] [PubMed] [Google Scholar]
- Pfurtscheller G, Lopes Da Silva FH (1999): Event‐related EEG/MEG synchronization and desynchronization: Basic principles. Clin Neurophysiol 110:1842–1857. [DOI] [PubMed] [Google Scholar]
- Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, Lynch C, Pender N, Hardiman O (2012): The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population‐based study. J Neurol Neurosurg Psychiatry 83:102–108. [DOI] [PubMed] [Google Scholar]
- Pinkhardt EH, Jürgens R, Becker W, Mölle M, Born J, Ludolph AC, Schreiber H (2008): Signs of impaired selective attention in patients with amyotrophic lateral sclerosis. J Neurol 255:532–538. [DOI] [PubMed] [Google Scholar]
- Pioro EP, Majors AW, Mitsumoto H, Nelson DR, Ng TC (1999): 1H‐MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology 53:71–79. [DOI] [PubMed] [Google Scholar]
- Pogosyan A, Gaynor LD, Eusebio A, Brown P (2009): Boosting Cortical Activity at Beta‐Band Frequencies Slows Movement in Humans. Curr Biol 19:1637–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC (1992): Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain 115:495–520. [DOI] [PubMed] [Google Scholar]
- Proudfoot M, Gutowski NJ, Edbauer D, Hilton DA, Stephens M, Rankin J, Mackenzie IR. a (2014a): Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol 127:451–458. [DOI] [PubMed] [Google Scholar]
- Proudfoot M, Menke RALL, Sharma R, Berna CM, Hicks SL, Kennard C, Talbot K, Turner MR (2015): Eye‐tracking in amyotrophic lateral sclerosis: A longitudinal study of saccadic and cognitive tasks. Amyotroph Lateral Scler Front Degener 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot M, Woolrich MW, Nobre AC, Turner MR (2014b): Magnetoencephalography. Pract Neurol 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, Lopera F, Stern CE (2010): Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann Neurol 68:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva N, Falini a, Inuggi a, Gonzalez‐Rosa JJ, Amadio S, Cerri F, Fazio R, Del Carro U, Comola M, Comi G, Leocani L (2012): Cortical activation to voluntary movement in amyotrophic lateral sclerosis is related to corticospinal damage: Electrophysiological evidence. Clin Neurophysiol 123:1586–1592. [DOI] [PubMed] [Google Scholar]
- Robinson S, Vrba J (1999): Functional neuroimaging by synthetic aperture magnetometry (SAM). Recent Adv 2–5. [Google Scholar]
- Sarvas J (1987): Basic mathematical and electromagnetic concepts of the biomagnetic inverse problem. Phys Med Biol 32:11–22. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Verstraete E, de Reus MA, Veldink JH, van den Berg LH, van den Heuvel MP (2014): Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum Brain Mapp 35:4386–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoder D, Hannequin D, Martinaud O, Opolczynski G, Guyant‐Maréchal L, Le Ber I, Campion D (2010): Morbid risk for schizophrenia in first‐degree relatives of people with frontotemporal dementia. Br J Psychiatry 197:28–35. [DOI] [PubMed] [Google Scholar]
- Schoenfeld MA, Tempelmann C, Gaul C, Kühnel GR, Düzel E, Hopf J‐M, Feistner H, Zierz S, Heinze H‐J, Vielhaber S (2005): Functional motor compensation in amyotrophic lateral sclerosis. J Neurol 252:944–952. [DOI] [PubMed] [Google Scholar]
- Singer MA, Statland JM, Wolfe GI, Barohn RJ (2007): Primary lateral sclerosis. Muscle Nerve 35:291–302. [DOI] [PubMed] [Google Scholar]
- Stancák A, Pfurtscheller G (1996): Event‐related desynchronisation of central beta‐rhythms during brisk and slow self‐paced finger movements of dominant and nondominant hand. Cogn Brain Res 4:171–183. [DOI] [PubMed] [Google Scholar]
- Stančák A, Riml A, Pfurtscheller G (1997): The effects of external load on movement‐related changes of the sensorimotor EEG rhythms. Electroencephalogr Clin Neurophysiol 102:495–504. [DOI] [PubMed] [Google Scholar]
- Stanton BR, Williams VC, Leigh PN, Williams SCR, Blain CRV, Giampietro VP, Simmons A (2007a): Cortical activation during motor imagery is reduced in Amyotrophic Lateral Sclerosis. Brain Res 1172:145–151. [DOI] [PubMed] [Google Scholar]
- Stanton BR, Williams VC, Leigh PN, Williams SCR, Blain CRV, Jarosz JM, Simmons A (2007b): Altered cortical activation during a motor task in ALS. Evidence for involvement of central pathways. J Neurol 254:1260–1267. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Benecke R, Classen J (2001): Effects of riluzole on cortical excitability in patients with amyotrophic lateral sclerosis. Ann Neurol 49:518–521. [PubMed] [Google Scholar]
- Suntrup S, Teismann I, Wollbrink A, Winkels M, Warnecke T, Flöel A, Pantev C, Dziewas R (2013): Magnetoencephalographic evidence for the modulation of cortical swallowing processing by transcranial direct current stimulation. Neuroimage 83C:346–354. [DOI] [PubMed] [Google Scholar]
- Tan H, Wade C, Brown P (2016): Post‐Movement Beta Activity in Sensorimotor Cortex Indexes Confidence in the Estimations from Internal Models. J Neurosci 36:1516–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorns J, Wieringa BM, Mohammadi B, Hammer A, Dengler R, Münte TF (2010): Movement initiation and inhibition are impaired in amyotrophic lateral sclerosis. Exp Neurol 224:389–394. [DOI] [PubMed] [Google Scholar]
- Toma K, Mima T, Matsuoka T, Gerloff C, Ohnishi T, Koshy B, Andres F, Hallett M (2002): Movement rate effect on activation and functional coupling of motor cortical areas. J Neurophysiol 88:3377–3385. [DOI] [PubMed] [Google Scholar]
- Toro C, Deuschl G, Thatcher R, Sato S, Kufta C, Hallett M (1994): Event‐related desynchronization and movement‐related cortical potentials on the ECoG and EEG. Electroencephalogr Clin Neurophysiol Potentials Sect 93:380–389. [DOI] [PubMed] [Google Scholar]
- Trojsi F, Esposito F, de Stefano M, Buonanno D, Conforti FL, Corbo D, Piccirillo G, Cirillo M, Monsurrò MR, Montella P, Tedeschi G (2015): Functional overlap and divergence between ALS and bvFTD. Neurobiol Aging 36:413–423. [DOI] [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CCJ, Shaw CE, Brooks DJ, Leigh PN, Banati RB (2004): Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)‐PK11195 positron emission tomography study. Neurobiol Dis 15:601–609. [DOI] [PubMed] [Google Scholar]
- Turner MR, Hammers A, Al‐Chalabi A, Shaw CE, Andersen PM, Brooks DJ, Leigh PN (2005a): Distinct cerebral lesions in sporadic and “D90A” SOD1 ALS: Studies with [11C]flumazenil PET. Brain 128:1323–1329. [DOI] [PubMed] [Google Scholar]
- Turner MR, Osei‐Lah a. D, Hammers A, Al‐Chalabi A, Shaw CE, Andersen PM, Brooks DJ, Leigh PN, Mills KR (2005b): Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J Neurol Neurosurg Psychiatry 76:1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Hammers A, Al‐Chalabi A, Shaw CE, Andersen PM, Brooks DJ, Leigh PN (2007): Cortical involvement in four cases of primary lateral sclerosis using [(11)C]‐flumazenil PET. J Neurol 254:1033–1036. [DOI] [PubMed] [Google Scholar]
- Turner MR, Kiernan MC (2012): Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler 13:245–250. [DOI] [PubMed] [Google Scholar]
- Turner MR, Swash M (2015): The expanding syndrome of amyotrophic lateral sclerosis: A clinical and molecular odyssey. J Neurol Neurosurg Psychiatry 86:667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Verstraete E (2015): What Does Imaging Reveal About the Pathology of Amyotrophic Lateral Sclerosis? Curr Neurol Neurosci Rep 15: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagarakis C, Ince NF, Leuthold AC, Pellizzer G (2010): Beta‐band activity during motor planning reflects response uncertainty. J Neurosci 30:11270–11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veen BD, van Drongelen W, Yuchtman M, Suzuki A (1997): Localization of brain electrical activity via linearly constrained minimum variance spatial filtering. IEEE Trans Biomed Eng 44:867–880. [DOI] [PubMed] [Google Scholar]
- Verstraete E, Turner MR, Grosskreutz J, Filippi M, Benatar M (2015): Mind the gap: The mismatch between clinical and imaging metrics in ALS. Amyotroph Lateral Scler Front Degener 16:524–529. [DOI] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez‐Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C (2013): Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II, results and discussion. Brain Behav 3:431–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Nicholson G. a, Kiernan MC (2008): Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131:1540–1550. [DOI] [PubMed] [Google Scholar]
- Vucic S, Winhammar JMC, Rowe DB, Kiernan MC (2010): Corticomotoneuronal function in asymptomatic SOD‐1 mutation carriers. Clin Neurophysiol 121:1781–1785. [DOI] [PubMed] [Google Scholar]
- Vucic S, Lin CSY, Cheah BC, Murray J, Menon P, Krishnan AV, Kiernan MC (2013a): Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 136:1361–1370. [DOI] [PubMed] [Google Scholar]
- Vucic S, Ziemann U, Eisen A, Hallett M, Kiernan MC (2013b): Transcranial magnetic stimulation and amyotrophic lateral sclerosis: Pathophysiological insights. J Neurol Neurosurg Psychiatry 84:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout R, Schmidt R, Westeneng H‐J, Verstraete E, Seelen M, van Rheenen W, de Reus MA, van Es MA, Hendrikse J, Veldink JH, van den Heuvel MP, van den Berg LH (2015): Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology 85:1780–1788. [DOI] [PubMed] [Google Scholar]
- Wessel JR, Aron AR (2013): Unexpected events induce motor slowing via a brain mechanism for action‐stopping with global suppressive effects. J Neurosci 33:18481–18491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal KP, Heinemann HA, Grözinger B, Kotchoubey BJ, Diekmann V, Becker W, Kornhuber HH (1998): Bereitschaftspotential in amyotrophic lateral sclerosis (ALS): lower amplitudes in patients with hyperreflexia (spasticity). Acta Neurol Scand 98:15–21. [DOI] [PubMed] [Google Scholar]
- Witiuk K, Fernandez‐Ruiz J, McKee R, Alahyane N, Coe BC, Melanson M, Munoz DP (2014): Cognitive Deterioration and Functional Compensation in ALS Measured with fMRI Using an Inhibitory Task. J Neurosci 34:14260–14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittstock M, Wolters A, Benecke R (2007): Transcallosal inhibition in amyotrophic lateral sclerosis. Clin Neurophysiol 118:301–307. [DOI] [PubMed] [Google Scholar]
- Wittstock M, Meister S, Walter U, Benecke R, Wolters A (2011): Mirror movements in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 12:393–397. [DOI] [PubMed] [Google Scholar]
- Woolrich M, Hunt L, Groves A, Barnes G (2011): MEG beamforming using Bayesian PCA for adaptive data covariance matrix regularization. Neuroimage 57:1466–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, Beckmann C, Jenkinson M, Smith SM (2009): Bayesian analysis of neuroimaging data in FSL. Neuroimage 45:S173–S186. [DOI] [PubMed] [Google Scholar]
- Yokota T, Yoshino A, Inaba A, Saito Y (1996): Double cortical stimulation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 61:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai P, Pagan F, Statland J, Butman JA, Floeter MK (2003): Primary lateral sclerosis: A heterogeneous disorder composed of different subtypes? Neurology 60:1258–1265. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xu R, Dowd E, Zang Y, Gong H, Wang Z (2014): Alterations in regional functional coherence within the sensory‐motor network in amyotrophic lateral sclerosis. Neurosci Lett 558:192–196. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W (1997): Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology 49:1292–1298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.