

Abstract
Research in recent years has focused significantly on the role of selective macroautophagy in targeting intracellular pathogens for lysosomal degradation, a process termed xenophagy. In this review we evaluate the proposed roles for xenophagy in controlling bacterial infection, highlighting the concept that successful pathogens have evolved ways to subvert or exploit this defense, minimizing the actual effectiveness of xenophagy in innate immunity. Instead, studies in animal models have revealed that autophagy-associated proteins often function outside of xenophagy to influence bacterial pathogenesis. In light of current efforts to manipulate autophagy and the development of host-directed therapies to fight bacterial infections, we also discuss the implications stemming from the complicated relationship that exists between autophagy and bacterial pathogens.
Keywords: Autophagy, Xenophagy, bacterial pathogens, Mycobacterium tuberculosis, host-directed therapies
Xenophagy as an Immune Defense against Intracellular Bacteria
The rise in the prevalence of antibiotic-resistant infections and the dearth of new antibiotics in the pipeline has raised significant interest in developing host-directed therapies that harness the immune response to control bacterial infection as an alternative therapeutic strategy[1]. One attractive cellular target for immunotherapy is the stimulation of macroautophagy (herein referred to as autophagy, see Glossary), and more specifically xenophagy. Autophagy is a dynamic process that targets cellular cytoplasmic contents for lysosomal degradation and serves myriad roles in eukaryotic cells. Xenophagy is a type of selective macroautophagy that specifically targets intracellular pathogens to lysosomes, restricting their replication and survival.
Xenophagy involves the same steps as canonical autophagy, which are often delineated as initiation, elongation, substrate targeting, and maturation/lysosomal fusion (resulting in degradation of cargo)(Fig. I, Box 1)[2]. In the case of bacterial infection, substrate targeting involves ubiquitination of bacteria or colocalization of ubiquitin to bacteria, and the recognition of the ubiquitin by autophagy receptors that interact with LC3 (microtubule-associated protein 1 light chain 3to recruit the bacteria to autophagosomes, which then fuse to lysosomes to degrade the cargo [2] (Box 2). The first report indicating that bacteria could be eliminated by xenophagy was published in 1984 when Rikihisa observed autophagosome formation in neutrophils infected by Rickettsia conorii[3]. Since then, the role of xenophagy in cell culture infection experiments has been investigated for all widely studied intracellular bacterial pathogens. Using mammalian cell culture approaches (Box 3), researchers have gained tremendous insight into key players and mechanisms of xenophagy. However, no single experimental approach captures the complete dynamics of xenophagy and, despite the immense focus in this area, these studies often raise uncertainty with regard to the role of xenophagy in controlling intracellular bacterial infections. Therefore, a more critical and comprehensive evaluation of the role of xenophagy and autophagy-related proteins during bacterial infection is necessary to understand the range of effects that might occur while intervening with autophagic flux. In this review we evaluate the data available regarding the interplay between xenophagy and different intracellular bacterial pathogens, highlighting Mycobacterium tuberculosis, which has historically been used as a model of xenophagic control of bacterial replication used as a model of xenophagic control of bacterial replication (for different intracellular bacterial pathogens, see Table S1 in the supplemental information online). Detailed analysis of the data reveals that successful pathogens have evolved ways to subvert or exploit xenophagy, but autophagy-associated proteins can function outside of xenophagy to influence bacterial pathogenesis. Finally, we discuss the implications of the complicated relationship between autophagy and bacterial pathogens which may impact the development of host-directed therapies to treat these infections.
Figure I. Schematic of the Stages of Canonical Macroautophagy.
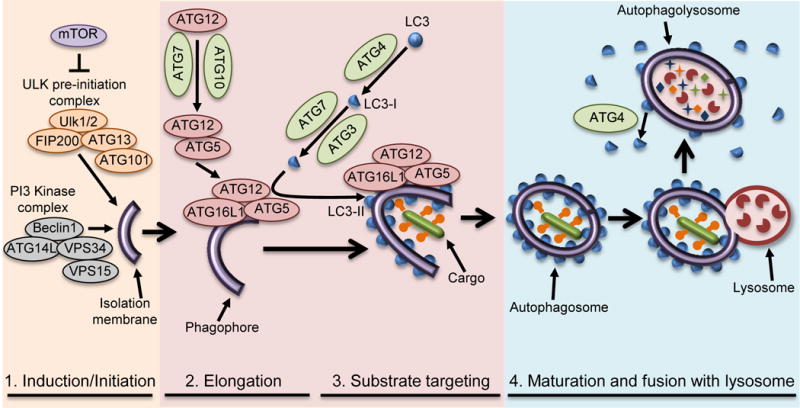
(1) Induction/initiation, (2) elongation, (3) substrate targeting, and (4) maturation/lysosomal fusion and targeting of bacterial pathogens (substrate targeting) as detailed in Box 2.
Box 1. Steps of Canonical Macroautophagy.
Initiation of autophagy involves the sequestration of a small portion of the cytoplasm in a membranous sac referred to as a phagophore (Figure I) [2,114]. Phagophore formation is mediated by translocation of the ULK1 complex (ULK1/ULK2, ATG13, FIP200, ATG101) from the cytoplasm to the endoplasmic reticulum (ER) and, as such, the ER is thought to be the source of the autophagosome membrane. The ULK1 complex functions in the recruitment of the autophagosome specific [phosphatidylinositol3-kinase(PI3K)] complex consisting of ATG14L, BECLIN1, VPS15, and VPS34[2,114]. The PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), which is not normally found in the ER and is essential for canonical autophagy[2,87,115].
Elongation of the autophagosomal double membrane depends on two ubiquitin-like conjugation systems. In the first system, ATG12 (ubiquitin-like protein) is synthesized with an exposed C-terminal glycine, activated by ATG7 (E1-like)[2,87,115], and transferred to ATG10 (E2-like), which facilitates its final ligation to ATG5. ATG5 non-covalently interacts with ATG16L1, resulting in an ATG5-ATG12—ATG16L1 complex that is present on the phagophore and elongating membrane, but dissociates upon completion of the autophagosome[2,116,117]. The second ubiquitin like component is LC3 (microtubule-associated protein 1 light chain 3)[2,87,115]. Upon synthesis, LC3 (pro-LC3) is rapidly processed by the cysteine-protease ATG4 to generate a cytoplasmic form called LC3-I. LC3-I is conjugated to phosphatidylethanolamine, generating the membrane bound form called LC3-II through the actions of ATG7 (E1-like) and ATG3 (E2-like). ATG5-ATG12 facilitates LC3 lipidation through its interactions with ATG3, while ATG16L1 specifies the localization of LC3 conjugation to the autophagosome membrane.
Upon completion of the autophagosome membrane, removal of external LC3 by ATG4 facilitates autophagosome maturation as well as recycling of LC3, while LC3 on the interior of the autophagosome is inaccessible to ATG4 and is degraded. Autophagosome maturation occurs through SNARE-mediated fusion of autophagosomes with lysosomes, degrading the cargo[2].
Box 2. Targeting Bacteria by Xenophagy.
In general, once a bacterial pathogen either escapes into the cytosol or is exposed to the cytosol through damage to the vesicle in which it resides, xenophagy is induced by cytoplasmic recognition of pathogen-associatedmolecular patterns (PAMPs) or damage-associatedmolecularpatterns(DAMPs) [6,15,118–120]. Xenophagy occurs through the same basic steps as canonical macroautophagy (Box 1, Figure I).
Once phagophore formation is induced, bacteria are targeted by the autophagosome via ubiquitination of the bacteria or colocalization of ubiquitin to the bacteria via ubiquitin ligases such as PARKIN[15]. Ubiquitin is then recognized and bound by autophagy adaptors such as p62 (also called Sequestosome-1, SQSTM1), neighbor of BRCA1 gene 1 (NBR1), nuclear dot protein 52 kDa (NDP52) and optineurin (OPTN). These same adaptors interact with LC3 to recruit the bacteria to autophagosomes[87].
Autophagy adaptors are regulated by a variety of mechanisms including expression, spatial organization, cellular localization, and post-translational modifications[121]. In particular, regulation of autophagy adaptors by phosphorylation is emerging as an important theme for control of selective autophagy. For example, p62 can be phosphorylated at S403 by casein kinase 2, and this leads to increased affinity of p62 for polyubiquitin chains and plays a role in regulation of mitophagy[122].
TANK-binding kinase 1 (TBK1) can also phosphorylate p62 on S403, and has been shown to play a role in control of Mycobacterium bovis BCG infection[123]. During S. Typhimurium infection, NDP52 recruits TBK1, which constitutively associates with a second autophagy adaptor OPTN. TBK1-mediated phosphorylation of OPTN on S177 regulates its LC3-binding affinity and subsequent targeting of Salmonella by xenophagy[124]. Following bacterial targeting to the autophagosome, the autophagosome matures and fuses with lysosomes, killing invading bacteria.
Box 3. Approaches for Studying Xenophagy in Mammalian Cell Cultures: Necessary Considerations.
Most studies into the role of xenophagy in controlling bacterial infections have used cell culture models of infection. Broadly, there are two approaches used to investigate whether bacterial pathogens are targeted by xenophagy[125,126]: 1) Observation of hallmarks of autophagy including the colocalization of autophagy factors to the bacteria and 2) Experimental manipulation of autophagy and analysis of the impact on bacterial replication.
To investigate the presence of hallmarks of autophagy, electron microscopy can be used to visualize double membrane autophagosomes around targeted bacteria. Alternatively, fluorescent microscopy can be used to visualize colocalization of autophagy-associated proteins with the bacterium. LC3 colocalization is the most common assay performed because autophagosomes will remain LC3+ until they are degraded in the lysosome. However, a common misconception is that increased numbers of LC3-associated autophagosomes represent an increase in autophagy[126]. Autophagy is a dynamic process and increases in LC3 puncta can be caused by either increased LC3 lipidation (autophagosome biogenesis) or decreased LC3 turnover (autophagosome degradation). Furthermore, because many LC3 molecules are degraded upon lysosomal fusion, extended stimulation of autophagy will result in a decrease in cellular LC3, which could be misinterpreted as decreased autophagy. LC3 is also central to an ever-growing list of processes that are distinct from canonical autophagy, but still result in vacuole-associated LC3-II. Therefore, measurement of LC3 puncta alone is no longer sufficient to demonstrate a role for autophagy[126].
For the second approach, autophagic flux can be manipulated by targeting autophagosome formation or degradation. Chemically, autophagy is most often induced by inhibiting mTOR through nutrient starvation or rapamycin treatment. mTOR is the main negative regulator of autophagy but also regulates other processes including cell growth, proliferation, motility, survival, protein synthesis, and transcription. Autophagosome formation can be inhibited by PI3K inhibitors such as wortmannin or 3-methyladenine (3-MA), which also inhibit kinases involved in diverse cell signaling and membrane-trafficking processes. Drugs such as bafilomycin A1 or chloroquine block autophagosome degradation by inhibiting lysosomal acidification, but also affect processes such as endocytosis and mitosis. Because of such diverse effects, these drugs should be used as complementary approaches to identify if an observed effect is actually the result of interference with autophagy[126]. Autophagy can also be manipulated through genetic approaches by knockdown, knockout, or ectopic expression of autophagy genes. While targeting individual autophagy proteins may have fewer pleiotropic effects than the chemical approaches listed, it is still important to consider the effects of each gene in autophagy-independent pathways as well as dominant negative effects or off-target effects, all of which have been documented[126].
Importantly, when used individually, none of these methods are able to capture the complete dynamics of xenophagy. Therefore, it is critical to employ multiple approaches when measuring autophagic flux and to consider the effects of cell type, culture conditions, and timing of analysis, which can all impact the results.
Xenophagy and Mycobacterium tuberculosis Pathogenesis
The role of xenophagy in controlling M. tuberculosis replication in immune cells has been extensively studied in cell culture by multiple groups. As a result, M. tuberculosis is often cited as an example of a bacterial pathogen controlled by xenophagy. The survival of M. tuberculosis in phagocytes is dependent on its ability to inhibit phagolysosome fusion. Interferon-γ (IFN-γ activation of cultured macrophages results in increased trafficking of M. tuberculosis to the lysosome and subsequent killing[4], and xenophagy is often implicated in this process. Many studies have demonstrated that autophagy factors including LC3 are targeted to a subset of intracellular M. tuberculosis bacteria [4–8]. There are also reports that knockdown (KD) of Unc 51-like kinase 1Ulk1[9], Beclin1[10], Atg5[10], Atg7[11], or p62[6] autophagy genes can result in improved M. tuberculosis survival in cell cultures. It is notable that at most, 30% of M. tuberculosis bacteria appear to be targeted by LC3 at any given appear to be targeted by LC3 at any given timepoint, and only twofold to threefold changes in bacterial survival are observed following modulation of autophagy [4–8].
In 2012, two groups reported that mice lacking Atg5 in myeloid derived cells (Atg5fl/fl-LysM-Cre) succumb to M. tuberculosis infection within the first 40 days of infection[6,12], a phenotype that is similar to mice completely lacking IFN-γ signaling[13] and among the most severe phenotypes described for a knockout (KO) mouse infected with M. tuberculosis. Control of M. tuberculosis is extremely impaired in Atg5fl/fl-LysM-Cre mice, resulting in 1–2 logs higher bacterial burden and more severe inflammation in lungs than control mice. Given the focus of previous cell culture studies, a prevailing theory was that this effect was due to loss of xenophagy-dependent control of bacterial replication. However, a recent study found that while ATG5 is required in LysM+ cells for mice to survive acute M. tuberculosis infection, other essential autophagy factors are not. In fact, mice lacking ATG3, ATG7, ATG12, ATG14L, or ATG16L1 in LysM+ cells or lacking ULK1, ULK2, p62, or ATG4B in all cells effectively control M. tuberculosis burden and survive for more than 80 days post-infection with no overt signs of morbidity[14]. This demonstrates that neither xenophagy nor other autophagy-mediated processes are required in myeloid cells to control M. tuberculosis infection in mice. Instead, ATG5 plays a specific role in limiting neutrophil-mediated inflammation through a mechanism that is independent of all pathways involving autophagy or LC3-lipidation[14]. These data demonstrate a lack of correlation between in vitro and in vivo findings of the roles of autophagy genes in controlling M. tuberculosis replication and highlight the importance of studying the roles for xenophagy in vivo. These in vivo genetic analyses also argue for a shift in focus onto macroautophagy-independent roles of ATG5 in controlling resistance to Mtb infection in vivo.
A separate study investigated the role of PARKIN, a ubiquitin ligase important for mitophagy (autophagic targeting of mitochondria), in controlling M. tuberculosis infection[15]. Similar to other cell culture experiments involving autophagy-associated proteins, the PARKIN protein colocalized to approximately 12% of M. tuberculosis and loss of the gene encoding PARKIN, Park2, resulted in a 2 fold increase in M. tuberculosis survival in murine macrophages. Moreover, Park2−/− mice infected with M. tuberculosis had a 10 fold increase in bacterial titers by 21 days post-infection and succumbed to infection by 85 days post-infection. Given that mice deficient in autophagy survived past this time point[14], PARKIN likely plays an autophagy-independent role in controlling M. tuberculosis infection. Furthermore, as the susceptibility of Park2−/− mice was not as severe as that of Atg5fl/fl-LysM-Cre mice, PARKIN is not required for the autophagy-independent role of ATG5, highlighting that these factors have diverse functions beyond canonical autophagy. Notably, genetic polymorphisms in the Park2 regulatory region are associated with increased susceptibility to Mycobacterium leprae and Salmonella enterica serovar Typhi in humans[16,17] and Park2−/− mice are more susceptible to Listeria monocytogenes infection[15], indicating that PARKIN may function in general antibacterial responses.
Recent mouse studies challenging the paradigm that xenophagy is important in controlling M. tuberculosis infection, along with the modest effects of xenophagy in cell cultures, suggest that M. tuberculosis has evolved ways to evade host defense. In fact, a study characterizing autophagic flux over a time course of M. tuberculosis infection in cell cultures found that although M. tuberculosis was targeted by xenophagy, virulent strains of M. tuberculosis inhibited autophagosome maturation, thereby avoiding fusion with the lysosome and subsequent degradation[18] (Fig. 1). Several M. tuberculosis virulence factors that alter host signaling and vesicle trafficking also decrease autophagy, and this may explain the mechanism by which M. tuberculosis manipulates autophagic flux. Specifically, M. tuberculosis prevents phagolysosome fusion in cultured murine macrophages by interfering with fusion in cultured murine macrophages by interfering with phosphatidylinositol 3-phosphate (PI3P) levels PIP3 levels through the activity of mannose-capped lipoaribomannan (ManLAM)[19]. ManLAM also inhibits of Ca2+ influx, which can alter mammalian target of rapamycin (mTOR) signaling and ULK1-activation[19]. Consistent with this, ManLAM-coated beads have been shown to decrease LC3-targeting in cultured murine macrophages[20], but the role this plays during M. tuberculosis infection remains to be elucidated. M. tuberculosis also recruits the host factor Coronin1a to phagosomes within cultured murine macrophages, which blocks autophagosome formation through an unknown mechanism[21]. M. tuberculosis expresses Eis, an N-acetyltransferase that inactivates JUN N-terminal kinase (JNK), leading to a decrease in reactive oxygen species (ROS) that normally activates autophagy in cultured murine macrophages[22,23]. However, the Δeis mutant does not present a survival defect in cell cultures or mice, suggesting that this is not a critical mechanism of xenophagy evasion, and that M. tuberculosis likely employs multiple factors to inhibit xenophagy.
Figure 1. Intracellular Bacterial Pathogens Avoid or Block Xenophagy-Mediated Clearance.
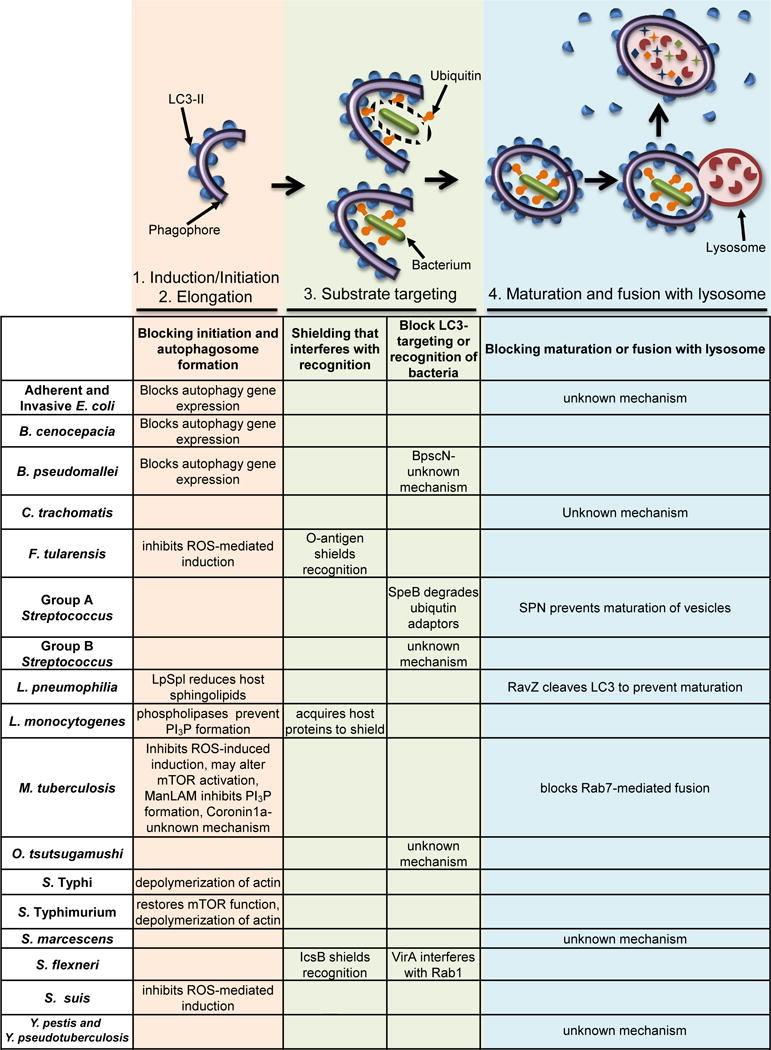
The schematic images above the table depict the relevant stages of xenophagy: 1. Initiation of xenophagy via phagophore formation. 2. Elongation of the autophagosomal double membrane. LC3-II (blue half-circles) is formed and localizes to the inner and outer autophagosome membranes. 3. Bacteria (green rods) within a damaged vesicle (top) or cytosolic bacteria (bottom) colocalize with ubiquitin (orange shapes), which is then recognized by autophagy receptors. The latter also interact with LC3-II, thus targeting bacteria to autophagosomes. 4. Autophagosomes mature and fuse with lysosomes (pink circle with red partial circle shapes inside). The cargo, including the bacteria, is then degraded. The ways in which bacteria interfere with each step of xenophagy are listed in the first column for each step. Many bacterial species use multiple strategies to interfere with autophagic flux at multiple steps. Some of these mechanisms of evasion may be cell type or host species specific. If a cell in the table is blank, there is no current evidence that the pathogen interferes with that stage of xenophagy. References are within the main text.
Xenophagy beyond M. tuberculosis as Revealed by Cell Culture Approaches
M. tuberculosis is not an exception in its evolution of ways to evade xenophagy. Although many cell culture studies have suggested roles for xenophagy in controlling replication of certain pathogenic bacteria, for all intracellular bacterial pathogens that have been investigated, there is evidence of strategies that allow them to subvert or exploit (Figures 1 and 2) autophagy to promote pathogenesis. In fact, one may conclude that in order to become a successful intracellular pathogen, a bacterium must evolve mechanisms to avoid this ancient innate immune defense.
Figure 2. Intracellular Bacterial Pathogens Exploit Autophagy-Associated Proteins for Replication and/or Survival.
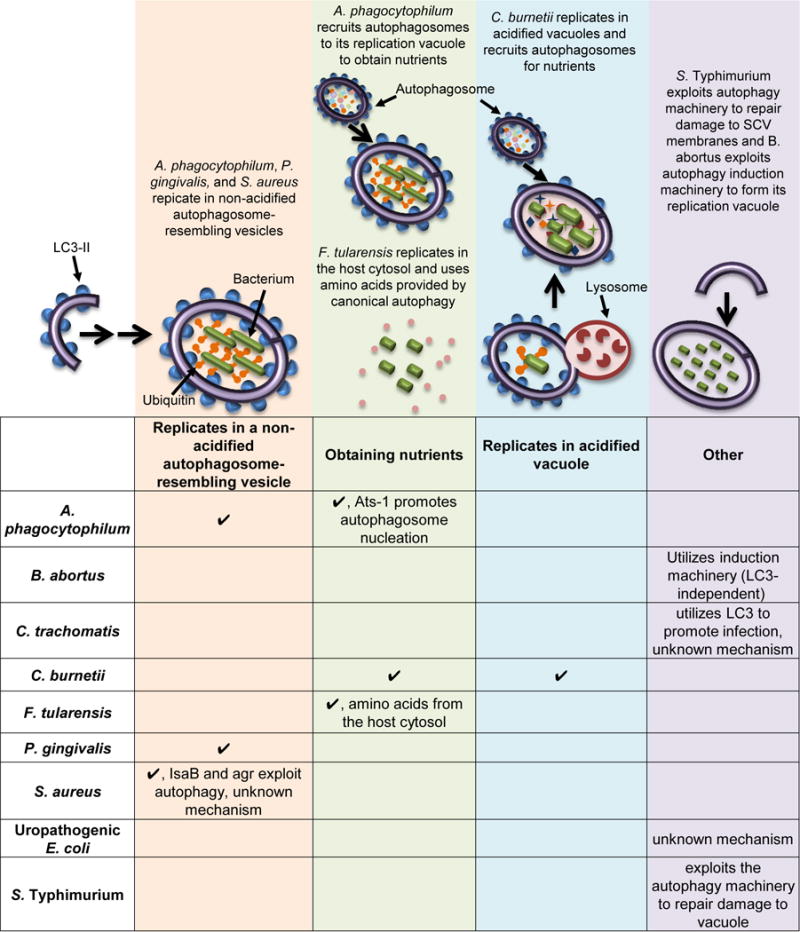
For the pathogens listed in this table, inhibiting autophagy would block infection and inducing autophagy would promote the infection. A check mark signifies that a given pathogen uses the mechanism denoted at the top of that column. If a cell in the table is blank, there is no current evidence that the pathogen exploits that mechanism. The schematic image above depicts each stage exploited by various bacteria and describes how the bacterium uses autophagy, with the shapes designating autophagy factors and bacteria as green rods. Note that A. phagocytophilum both replicates in a non-acidified vesicle and uses nutrients derived from autophagic flux. References are within the main text.
Shielding to Prevent Detection
One strategy used by multiple pathogens to avoid xenophagy is to shield themselves from being recognized by autophagy factors. Francisella tularensis invades macrophages and, after a transient interaction with the endocytic pathway, the bacteria escape into the cytosol, proliferate, and avoid being targeted by LC3 by at least two mechanisms. First, F. tularensis surface polysaccharidic O-antigen acts as a shield against ubiquitinylation and subsequent xenophagic recognition during infection of cultured murine macrophages[24]. Second, antioxidant superoxide dismutase enzymes of F. tularensis inhibit ROS-mediated induction of xenophagy and LC3-associated phagocytosis (LAP) during infection of cultured murine macrophages[25]. In addition, F. tularensis exploits an ATG5-independent autophagy pathway in fibroblasts and macrophages to obtain nutrients released from degraded cellular constituents to promote its replication[26]. Following replication within the cytosol of macrophages, F. tularensis re-enters the endocytic pathway and resides in Francisella-containing vacuoles (FCV) that colocalize with LC3[27]. FCV formation is critical for F. tularensis to complete its replication cycle and 3-methyladenine (3-MA) treatment, which among other effects inhibits autophagy (Box 3), blocks FCV formation and F. tularensis replication. Listeria monocytogenes replicates rapidly in the cytosol of macrophages and epithelial cells and avoids being targeted by xenophagy by decorating its surface with host proteins through the activity of ActA and InlK[28,29]. L. monocytogenes also expresses phospholipases that prevent the formation of PI3P, thus stalling autophagosome formation in macrophages and epithelial cells[30,31]. Shigella flexneri avoids recognition by autophagy factors while it replicates in the cytosol of epithelial cells and fibroblasts by expressing the effector IcsB, which masks a region of the S. flexneri surface protein IcsA that is recognized by ATG5. The effector VirA, a GTPase-activating protein (GAP) for Rab1, also prevents LC3 recruitment to S. flexneri [32–35]. These shielding strategies allow these bacteria to replicate undetected by the autophagy machinery, thus subverting this type of immune defense.
Blocking Induction of Autophagy, Autophagosome Formation, and LC3Targeting
Other bacteria block autophagosome formation or targeting by autophagy factors without known mechanisms of shielding the pathogen. After entry into epithelial, endothelial cells, macrophages, and dendritic cells (DC), Orientia tsutsugamushi escapes from the endosomal pathway and replicates in the cytosol of eukaryotic host cells. O. tsutsugamushi infection induces autophagy, however, most of the bacteria do not colocalize with autophagosomes, even when further stimulated with rapamycin. Therefore, O. tsutsugamushi must block autophagic recognition[36,37]. Another pathogen, Burkholderia pseudomallei promotes its replication in the cytosol of murine macrophages by evading LC3 targeting through an undefined process involving the type III secretion system ATPase BpscN[38]. In addition, infection of macrophages and epithelial cells with B. pseudomallei or Burkholderia cenocepacia leads to downregulation of certain autophagy genes, resulting in decreased levels of autophagy[39,40]. Group B streptococcus (GBS) induces autophagy during infection of EC, but still resides primarily in single membrane-bound compartments, suggesting that GBS evades targeting to autophagosomes[41]. Streptococcus suis blocks xenophagy during infection of macrophages by secreting superoxide dismutase to scavenge cellular ROS[42]. Salmonella enterica serovar Typhimurium uses a type III secretion system (T3SS-1) to invade epithelial cells and form the Salmonella-containing vacuole (SCV). However, the T3SS-1 also damages the SCV, and this induces xenophagy and recruits the autophagic machinery to the SCV. S. Typhimurium exploits the autophagic machinery to repair damage to SCV membranes caused by T3SS-1, thereby allowing compartment maturation and subsequent expression of a second type III secretion system, T3SS-2, which together promote intracellular survival[43]. S. Typhimurium then downregulates LC3 colocalization in epithelial cells, fibroblasts, and macrophages by restoring mTOR activation, which in turn inhibits autophagy[44–47]. Clinically important Salmonella, including Typhi and Typhimurium, also express SpvB from the virulence plasmid pRST98, which can suppress autophagosome formation in macrophages by depolymerizing actin and enhancing bacterial survival[48]. The diverse mechanisms employed by these bacteria to avoid clearance by xenophagy facilitate their ability to replicate in host cells.
Blocking Autophagosome Maturation
Another strategy commonly used by bacteria to evade xenophagy is to block autophagosome maturation and fusion with lysosomes, thus allowing for continued bacterial replication and survival. The obligate intracellular pathogen Chlamydia trachomatis induces autophagy during infection, but then blocks the fusion of its vacuole to the lysosome in human epithelial cells, murine fibroblasts, and murine macrophages[49]. C. trachomatis also co-opts LC3 for an autophagy-independent function that promotes bacterial infection in human epithelial cells[50]. Adherent and invasive Escherichia coli (AIEC) infection in enterocytes leads to up-regulation of microRNAs that reduce the levels of ATG5 and ATG16L1, and inhibit autophagy[51]. AIEC also blocks autophagy at the autolysosomal step during infection of neutrophil-like PLB-985 cells, allowing for intracellular survival of these bacteria[52]. Legionella pneumophila survives within a vacuole that is targeted by LC3 during infection in macrophages, but blocks acidification by cleaving membrane-conjugated LC3 via the effector protein RavZ[53]. In addition, L. pneumophila blocks autophagy during infection of macrophages by translocating the effector protein sphingosine-1 phosphate lyase (LpSpl), and this triggers a reduction of several host sphingolipids critical for autophagy[54]. Serratia marcescens replicates and persists inside large membrane-bound compartments in epithelial cells that colocalize with LC3 but are non-acidic, indicating that the bacteria prevent fusion of the vesicles with lysosomes to generate a suitable niche for proliferation inside the host cell[55]. Similarly, Yersinia pestis and Yersinia pseudotuberculosis replicate in LC3-positive vacuoles within macrophages, but prevent vacuole acidification to allow sustained bacterial replication[56,57]. Therefore, although xenophagy is triggered in infected host cells and bacteria are targeted by autophagy proteins, the ability of these pathogens to block autophagosome maturation allows them to survive and replicate within the cell.
Group A Streptococcus (GAS)- Employing Cell Type-Specific Evasion Mechanisms
Following internalization, GAS escapes into the cytosol, which results in stimulation of autophagy. This leads to co-localization of LC3 to a subset of intracellular bacteria, but the degree of targeting depends on the cell type infected. In HeLa cells, 80% of GAS were shown to colocalize with LC3, correlating with a fivefold decrease in bacterial titers, suggesting that xenophagy might contribute to the control of bacterial killing within epithelial cells. However, by 24 h post-infection, ~50% of the infected cells underwent apoptosis, suggesting that xenophagic killing of GAS is not sufficient to protect host cells[58]. During infection of macrophages, LC3 is only targeted to approximately 26% of GAS; however, proficient replication of GAS continues despite the recruitment of autophagy receptors[59]. In addition, because not all cytosolic bacteria are targeted by LC3, there is likely a subpopulation of GAS within macrophages that evades recognition by autophagy factors[59]. In keratinocytes, 80–90% of GAS was found to localize in LC3 vesicles, but translocation of Streptococcus pyogenes NAD+ glycohydrolase (SPN) into the cytosol prevented maturation of the GAS-containing autophagosome-like vacuoles[60]. Similarly, in endothelial cells, the low pH is not maintained in GAS-containing autophagosomes, and the bacteria replicate efficiently[61]. In addition, the globally disseminated GAS serotype M1T1 secretes SpeB, a protease that degrades ubiquitin adaptors within the host cell cytosol to avoid recognition by LC3 in epithelial cells[62]. Thus, GAS employs multiple mechanisms to evade xenophagy and promote its replication in diverse types of host cells.
Exploiting Autophagy Proteins to Promote Replication
Other bacterial pathogens exploit autophagy or autophagy-associated proteins for intracellular replication. During infection of human promyelocytic leukemia cells, Anaplasma phagocytophilum replicates in membrane-bound compartments devoid of lysosomal markers, indicating that the pathogen inhibits fusion to lysosomes[63]. A. phagocytophilum secretes an effector, Ats-1, to promote autophagosome nucleation and fusion to its vesicle, stimulating its own growth by using the nutrients contained in the autophagosomes[64]. The obligate intracellular bacterium Coxiella burnetii survives and replicates within large, acidified, vacuoles that colocalize with LC3 during infection of epithelial cells and macrophages[65–67]. C. burnetii exploits these vacuoles as a favorable replication niche while also recruiting autophagosomes to deliver nutrients and provide a membrane for the expanding vacuole[68]. C. burnetii also secretes CvpB, which manipulates PI3P metabolism, promoting replication[69]. Porphyromonas gingivalis avoids targeting to lysosomes in human DCs and instead traffics to vesicles resembling autophagosomes where it replicates. Indeed, selective engagement of DC-SIGN by P. gingivalis during uptake promotes evasion of lysosome fusion[70]. Induction of autophagy promotes the replication of A. phagocytophilum, C. burnetii, and P. gingivalis, while inhibition of autophagy blocks the replication of these pathogens in cell culture systems[63,67,71–73].
Staphylococcus aureus exploits autophagy factors to form a niche for replication that resembles an autophagosome but is blocked for maturation and acidification[74,75]. S. aureus depends on this vacuole for replication and, as such, is unable to survive and replicate in Atg5−/− mouse embryonic fibroblasts (MEFs)[74]. Expression of IsaB and agr by S. aureus is important for exploiting the autophagic machinery through undefined mechanisms[74,75]. The intracellular bacterium Brucella abortus also co-opts autophagy factors to support its replication, but is unique in that this process does not involve the LC3-conjugation machinery. After initial entry into fibroblasts or macrophages, B. abortus forms a Brucella-containing vacuole (BCV) that traffics from the endocytic compartment to the endoplasmic reticulum (ER), where the bacterium proliferates[76]. Following B. abortus replication, the BCV is converted into a compartment with autophagic features in a process that is dependent upon the autophagy-initiation proteins, but independent of autophagy-elongation or LC3-conjugation machinery. These autophagosome-like vesicles are required to complete the B. abortus lifecycle[77]. Brucella melitensis also requires autophagic flux for replication in macrophages[78]. Autophagy has also been proposed to be beneficial for the survival of uropathogenic E. coli (UPEC) in macrophages, based on the observation that ATG16L1-deficient macrophages display increased uptake and enhanced killing of UPEC[79].
A handful of studies have also explored the roles of xenophagy in controlling bacterial pathogens that are primarily extracellular but may have intracellular stages during infection, including Helicobacter pylori[80–82], Streptococcus pneumonia[83], Pseudomonas aeruginosa[84], and Citrobacter rodentium[85]. Even for these pathogens that may be less adapted to an intracellular environment, there is evidence for xenophagy evasion strategies[80–85]. However, more studies will be necessary to understand the relevance of xenophagy during infection of these mostly extracellular pathogens.
The reoccurring theme is that xenophagy is insufficient to fully control bacterial replication of successful intracellular bacterial pathogens and most, if not all, intracellular bacterial pathogens employ multiple strategies to avoid clearance by xenophagy (Figures 1 and 2). These studies also highlight that colocalization of autophagy factors does not equate to the role of xenophagy in controlling bacterial replication, and may actually represent evasion strategies instead. Thus, detailed mechanistic studies will be necessary to understand the relationship between autophagy-associated proteins and a particular bacterial pathogen. In addition, roles for autophagy proteins can vary based on cell type and species (i.e., human versus mouse), and it is important to consider these caveats when interpreting the significance of findings.
Autophagy Associated Proteins during Bacterial Infection in Animal Models
Although xenophagy has been proven to be limited in its impact on bacterial pathogenesis, autophagy-associated proteins can still influence bacterial infections in other autophagy-dependent and -independent ways[86–88] (Box 4). Due to the diverse roles for autophagy-associated proteins, detailed in vivo studies are necessary to fully understand how autophagy-related genes and proteins impact on bacterial infection. Work with M. tuberculosis highlights the importance of in vivo studies to delineate how processes observed in cell culture models relate to mechanisms of pathogen control in vivo. Unfortunately, in vivo approaches may be limited by the animal models available for specific pathogens as well as by the degree to which a particular animal model represents human infection. Nonetheless, the generation of genetically engineered mice with mutations in the autophagy pathway has facilitated the discovery and study of myriad roles for autophagy proteins during bacterial pathogenesis. Mice with germline deletions in redundant autophagy factors have been generated, but these are limited by the ability of other factors to compensate for the absence of the targeted gene. Germline deletions of essential autophagy factors are embryonically or perinatally lethal and are often studied using conditional deletion Cre/lox recombination systems. As with all methods for studying autophagy, the use of conditional KOs comes with its own complications. For instance, autophagy-associated proteins also play roles in cell differentiation, proliferation, and survival[89], which may complicate analyses. To reconcile some of these issues and discern the function of specific pathways, different complementary mouse strains and approaches should be used in combination.
Box 4. Additional Roles of Autophagy-Associated Proteins in Host Immunity to Bacteria.
In addition to xenophagy, autophagy-associated proteins can influence bacterial infections in many other autophagy-dependent and -independent ways[86–88]. First, autophagic flux cross-regulates phagocytosis. One mechanism for this occurs when autophagy is downregulated: the adaptor protein p62 accumulates, resulting in increased Nrf2-mediated expression of scavenger receptors and increased phagocytosis of certain bacteria[127]. Many autophagy proteins also participate in LC3-associated phagocytosis (LAP), which also traffics pathogens to the lysosome for degradation. In contrast to canonical macroautophagy, LAP uses RUBICON and UV irradiation resistance-associated gene A (UVRAG) instead of ATG14L in the PI3K complex and does not depend on ULK1[88,128]. During LAP, LC3 is recruited to the phagosome, resulting in LC3+ single-membrane vesicles instead of the double membrane characteristic of autophagosomes. Because LAP was discovered relatively recently, previous studies describing pathogen-associated LC3+ vesicles now require additional investigation to determine if these are generated through xenophagy or LAP.
Once the pathogen invades the cell, autophagy can increase Toll-like receptor (TLR) signaling by delivering nucleic acids to TLRs, activating the production of type I IFN[129]. By contrast, ATG5-ATG12 can negatively regulate RIG-I, and ATG9A can negatively regulate DNA receptor stimulator of interferon genes (STING), leading to a decrease in type I IFN responses[87]. Type I IFN signaling can be detrimental or protective to the host, depending on the bacterial pathogen[130]. Autophagy also affects the production and secretion of IL-1β, a potent pro-inflammatory molecule, typically important for control of bacterial infections, but can also lead to significant immunopathology, and ultimately be detrimental to the host[131–135].
Autophagy can also directly increase host cell survival by acting as a starvation response, as well as decrease cell survival through autophagic cell death or autosis[136]. The mechanism by which an infected cell dies can have a major impact on immune responses since DAMPs released from dying cells will trigger inflammation[137]. Notably, ATG5 also impacts on cell death in neutrophils independently of autophagy when it is cleaved by calpain and translocates to the mitochondria where it exerts pro-apoptotic effects[138]. Autophagy is also important for the removal of apoptotic cells by phagocytic cells (efferocytosis) or endothelial cells (entosis)[139]. During M. tuberculosis infection, efferocytosis facilitates antigen presentation by DCs, and macrophages are better able to kill M. tuberculosis when it is acquired through efferocytosis as opposed to phagocytosis of free bacteria[140,141]. Autophagy-related genes are also associated with the maintenance and function of numerous immune cell populations including DCs, B cells, T cells, and fetal hematopoietic stem cells[87,89,115]. Thus, autophagy-associated proteins have diverse effects on inflammation and immunity that, depending on the pathogen and timing of the response, can exert both positive and negative effects for the host.
In addition to M. tuberculosis, mouse studies with other pathogens have revealed roles for autophagy-associated proteins in controlling bacterial infections by showing that loss of autophagy-associated proteins is detrimental to the host. One study reported that mice lacking ATG5 in myeloid cells presented a modest increase in susceptibility to L. monocytogenes, as measured by bacterial levels in spleen and liver 3 days after infection[90]. Mice in which Atg5 or Atg16l1 were conditionally deleted from the intestinal epithelium, presented abnormalities in granule secretion from Paneth cells and were more susceptible to Salmonella enterica serovar Typhimurium infection[91–93]. Mice with conditionally-deleted Atg7 in the intestinal epithelium also exhibited increased susceptibility to C. rodentium infection[94], possibly due to a similar mechanism as in Salmonella infection. Another study showed that mice, globally hypomorphic for ATG16L1 expression (ATG16L1HM), displayed increased lethality during methicillin-resistant Staphylococcus aureus (MRSA) infection. However, this effect did not seem to be due to the role of ATG16L1 in reducing bacterial burden, but rather to protection of host cells from α-toxin-induced damage[95].
Consistent with these results, induction of autophagy has been shown to be beneficial to the host during some bacterial infections. For instance, deletion of RNF5, which decreases the stability of ATG4B to limit LC3 processing, enhances autophagy and improves survival of mice during GAS infection[96]. Induction of autophagy in myeloid cells during S. Typhimurium infection also appears to be beneficial to the host. Focal adhesion kinase (FAK) is normally recruited to the surface of the Salmonella-containing vacuole (SCV) where it amplifies signaling through the Akt-mTOR axis to inhibit autophagy; when mice do not express FAK in myeloid cells, autophagy is enhanced and S. Typhimurium is cleared faster from infected tissues [45].
In other cases, mouse studies have revealed roles for autophagy-associated proteins in promoting bacterial infections. ATG16L1HM mice infected with UPEC showed more rapid bacterial clearance and epithelial recovery[97], indicating that ATG16L1 usually promotes UPEC infection. Loss of ATG16L1 leads to higher levels of IL-1β production from macrophages in response to UPEC, and this could contribute to the protection of ATG16L1HM mice[79]. These same ATG16L1HM mice are also resistant to intestinal disease induced by C. rodentium, at least in part as a result of enhanced immune responses[98]. Beclin1 heterozygous-deficient mice have been reported to be resistant to A. phagocytophilum infection 64], supporting the requirement for autophagy in A. phagocytophilum replication, as shown in cell cultures.
These animal studies have revealed the impact of autophagy associated proteins beyond xenophagy and highlight the complicated roles of autophagy proteins in regulating bacterial pathogenesis. The animal data further demonstrate how a single autophagy protein can have different effects on bacterial infection depending on the pathogen and the cell in which it functions. For many cases, whether the susceptibility of these mice is due to loss of autophagy, loss of an autophagy-independent role for the particular ATG protein, or effects on a combination of these processes remains unknown since conclusions about the role of autophagy in the immune response to a pathogen cannot be made based on data from a single autophagy protein.
Targeting Autophagy as a Host-Directed Therapy to Treat Bacterial Infections
There has been growing interest in modulating autophagy as a therapeutic intervention for a number of ailments, including bacterial infections (Box 5)[99]. Given the ability of intracellular bacterial pathogens to evade and exploit xenophagy and the impact of autophagy-associated proteins on diverse immune responses, pursuing autophagy as a therapeutic target requires the consideration of a number of factors. For example, some infections will be exacerbated by induction of autophagy (including those discussed in this review (Figure 2)), as well as by co-infections with some viruses or parasites[100,101]. In addition, manipulating the activity of autophagy proteins may affect both autophagy-dependent and -independent processes that have myriad effects on disease outcomes. Furthermore, any attempts to stimulate xenophagy may be futile if the pathogen efficiently blocks or avoids a downstream step in the pathway (Figures 1 and 2). Importantly, if a pathogen blocks a downstream step of autophagy– such as autophagolysosome formation– then induction of autophagy may not be effective and could result in a buildup of intermediates (undegraded autophagosomes) that may be detrimental to the cell. Therefore, a mechanistic understanding of such evasion mechanisms is critical to the design of successful therapeutic strategies.
Box 5. Clinician’s Corner.
-
-
Autophagy is an intracellular process that plays diverse roles in the host, including affecting cell adaptation, starvation, as well as eliminating intracellular organisms (a process called xenophagy).
-
-
There is great interest in targeting autophagy for the treatment of non-communicable diseases as well as infectious diseases.
-
-
Several currently FDA-approved agents induce autophagy and may increase xenophagic targeting of bacteria. However, these drugs also impact other pathways in the cell, leading to broad-spectrum effects that may negate the benefits. Furthermore, it is currently unknown whether the clinical benefits of any of these are due to autophagy or alternative effects of these drugs.
-
-
Ongoing efforts focus on developing tissue-specific modulators of autophagy, including gene therapy strategies.
-
-
Most, if not all, intracellular bacterial pathogens encode specific inhibitors of autophagy and, therefore a better understanding of bacterial evasion mechanisms is warranted for the development of potential specific therapeutic approaches targeting evasion.
-
-
Autophagy has been shown to play a role in various common bacterial infection comorbidities including HIV-1 infection, cancer, and diabetes. It will be critical to determine the impact of manipulating autophagy on these comorbidities when perusing options for clinical intervention.
The first strategy being pursued to target autophagy in the treatment of bacterial infections is the repurposing of pharmacologic agents that are approved by the FDA and present pleiotropic effects, including the induction of autophagy[102]. Notably, it is unknown whether any of these agents exert their clinical benefits through autophagy. The most widely studied example is rapamycin, an agent used to prevent transplant rejection. The use of rapamycin in cell cultures has been shown to decrease the survival of various intracellular bacterial pathogens including GBS[41], P. aeruginosa[103], Salmonella[44], B. pseudomallei[104], and M. tuberculosis[105]. However, the differences in bacterial burden tend to be less than 2.5 fold[41,44,103,104]. In addition, a recent study found that in macrophages coinfected with HIV and M. tuberculosis, stimulation of autophagy with rapamycin actually led to increased M. tuberculosis survival[106]. Thus, it will be critical to determine the impact of comorbidities such as HIV when perusing options for clinical intervention. Rapamycin has been tested in mouse models of intranasal P. aeruginosa infection, resulting in a twofold reduction in bacterial burdens in the lungs compared to control mice, but there were no differences in survival between rapamycin and control-treated mice[107]. Therefore, it is unclear if this would lead to better outcomes following infection. In addition, rapamycin diminished the bactericidal activity of neutrophils and increased the bacterial burden in P. aeruginosa-infected corneas in a mouse model of P. aeruginosa keratitis[108]. This may be related to the immunosuppressive effects of rapamycin, which could also cause reactivation and progression of some infections, outweighing any benefit of activating autophagy.
AR-12, identified as a suppressor of tumor cell viability, is another agent that modulates several pathways within mammalian cells including autophagy, and has been tested for effects on bacterial infections in mouse models. Administration of AR-12 to mice during S. Typhimurium infection resulted in lower bacterial burdens in the liver and spleen in some, but not all, mice, and a significant but incremental improvement in survival[109]. More impressive results were found when AR-12 was combined with aminoglycosides, and AR-12 improved the efficacy of this class of antibiotics in terms of prolonging survival of S. Typhimurium-infected mice[110]. Other FDA approved pharmacologic agents that induce autophagy, in addition to their other effects, have been shown to improve control of M. tuberculosis in mouse models, and include Gefitinib[111], an epidermal growth factor receptor (EGFR) inhibitor used to treat cancer, and statins[112]. However, the effects of these treatments on bacterial burden have been very modest in cultured macrophages and in mice, and it is unclear whether the effects on bacterial burden are due to autophagy. Furthermore, because M. tuberculosis blocks Rab7-mediated fusion with lysosomes[18], a process that is shared between autophagy, phagocytosis, and LAP, it is unknown whether stimulating upstream steps of these pathways (i.e., inducing autophagosome formation) will be able to increase killing of M. tuberculosis to a level that is clinically significant (without including a direct bacterial inhibitor).
The likely complications resulting from the pleiotropic effects of these pharmacologic agents have raised interest in more specific ways to modulate autophagy with limited side effects. One such approach is lentivirus- or adenovirus-based gene therapy for tissue-specific expression of autophagy genes. Positive results for gene therapy targeting autophagy have been obtained in animal models for a number of non-communicable diseases[99], but it is unknown whether these approaches will prove to be effective for the treatment of infectious diseases. Another approach being pursued is the use of cell-penetrating autophagy-inducing peptides. Tat-beclin 1 peptide is the best-developed agent in this class, and is believed to stimulate autophagy by interfering with a negative regulator of autophagy (GAPR-1)[113]. Tat-beclin 1 peptide treatment reduces the replication of an L. monocytogenes ΔactA strain during infection of macrophages, although the ΔactA mutant is unable to shield itself from autophagic recognition[113]. Development of therapeutics that specifically interfere with the molecules used by bacteria to restrict autophagy may be an effective approach, and could avoid problems associated with targeting autophagy at a systemic level. Furthermore, this approach may be effective in combination with autophagy-inducing therapeutics to provide a more potent, shorter course of treatment [102].
Concluding Remarks
The rise in incidence of antibiotic-resistant bacterial infections and the dearth of antibiotics in the pipeline has raised interest in applying host-directed therapies to the treatment of bacterial infections. In particular, there are efforts currently underway to develop modulators of autophagy as a therapeutic strategy. Much of the focus in these areas constitutes identifying agents that induce xenophagy to target intracellular bacterial pathogens for degradation. Although there has recently been an upsurge in these investigations, many open questions remain that will need to be addressed to be successful in this approach (See Outstanding Questions). A major obstacle is that most intracellular bacterial pathogens have evolved to avoid or exploit targeting by xenophagy. Thus far, xenophagy has not proved effective in controlling bacterial pathogens, at least in cell culture experiments and inbred mouse models. It remains to be seen whether stimulation of xenophagy will overcome these defenses to a clinically relevant degree, or whether simultaneously targeting bacterial defense mechanisms will be necessary. It is also necessary to establish whether cell culture experiments represent an accurate model for testing the efficacy of targeting autophagy, particularly since phenotypes observed in cell culture have thus far been modest and do not always correlate well with results from in vivo studies. Alternative therapeutic strategies could focus on roles for autophagy proteins outside of xenophagy that may have a significant impact on the outcomes of infection and investigating the synergy between autophagy-modulators and antibiotics may be fruitful. These areas, similar to targeting xenophagy, still require a significant amount of research to fully understand their clinical potential, but may hold promise for future therapeutic strategies.
Outstanding Questions.
-
-
Can we overcome bacterial defense mechanisms by activating autophagy using host-directed therapies?
-
-
Are there other pathways involving autophagy-associated proteins that would be more effective targets in host-directed therapies than xenophagy?
-
-
What is the mechanistic basis for the autophagy-independent role of ATG5 in controlling Mycobacterium tuberculosis infection, and can this be exploited in therapeutic interventions?
-
-
Can we engineer specific modulators of pathways involving autophagy-associated proteins to avoid off-target effects during system-wide autophagy manipulation?
-
-
Can differences in pharmacologically-induced autophagy observed in cell cultures correlate with in vivo efficacy?
Supplementary Material
Trends.
Xenophagy is a type of selective macroautophagy that targets intracellular pathogens for lysosomal degradation.
Intracellular bacterial pathogens have evolved mechanisms of evading xenophagy, inimizing its effectiveness as an innate immune response to these infections.
Mycobacterium tuberculosis is an example of a pathogen that is not effectively targeted by xenophagy in myeloid cells. In fact, no autophagy-dependent pathway is required in myeloid cells to control M. tuberculosis infection in mouse models. However, ATG5 functions independently of autophagy in myeloid cells to limit neutrophil-mediated inflammation, suggesting that autophagy-related proteins can function in multiple ways to control infection.
Some intracellular bacteria exploit autophagy to promote pathogenesis, requiring autophagic flux for survival and replication.
Autophagy-associated proteins play diverse roles in inflammation and immunity and can exert both positive and negative effects for the host. Many of these effects result from autophagy-independent functions and can vary depending on the cell type, host species, and interactions with particular pathogens.
There is interest in developing novel therapeutic strategies aimed at targeting autophagy to treat bacterial infection; however, this will require a better understanding of the roles of autophagy-associated proteins during infection and how bacteria evade these defenses.
Acknowledgments
C.L.S. is supported by a Beckman Young Investigator Award from the Arnold and Mabel Beckman Foundation and the NIH NIAID, award AI111696. J.M.K. is supported by a National Science Foundation Graduate Research Fellowship DGE-1143954 and the NIGMS Cell and Molecular Biology Training Grant GM007067.
Glossary
- 3-Methyladenine(3-MA)
prevents autophagosome formation by interfering with class III phosphatidylinositol3-kinases (PI3Ks)
- Apoptosis
a highly regulated and controlled process of programmed cell death. Generally results in very little inflammation
- AR-12
an antitumor celecoxib derivative that is used in clinical trials as an anticancer agent. Has many effects, including stimulation of autophagy
- Autophagic cell death
a form of cell death dependent on autophagy genes, and is associated with morphological features of increased autophagic vacuoles. Distinct from autosis and apoptosis
- Autophagic flux
a measure of autophagic degradation activity, that is, the rate of substrate sequestration and degradation by autophagy. Distinct from the number of autophagosomes in the cell, which can be caused by increased autophagosome formation or decreased autophagosome degradation
- Autosis
a mode of cell death dependent on autophagy genes. Mediated by the Na+, K+-ATPase pump and is distinct from autophagic cell death, apoptosis, and necrosis
- B cell
adaptive immune cells which respond to infection in an antigen-specific manner, and are important in the production of antibodies
- Bafilomycin A1
prevents maturation of autophagic vacuoles by inhibiting vacuolar H+ ATPase (V-ATPase), and decreases fusion between autophagosomes and lysosomes
- Chloroquine
prevents endosomal acidification and increases lysosomal pH, leading to inhibition of both fusion of autophagosome with lysosome and lysosomal protein degradation. Used to inhibit autophagosomal degradation
- Damage-associated molecular patterns (DAMPs)
host biomolecules that are released from cells upon damage that can initiate and perpetuate a noninfectious inflammatory response
- Dendritic cell (DC)
innate immune cells which are important for antigen presentation
- Efferocytosis
the process by which apoptotic cells are removed by phagocytosis (most often by macrophages)
- Entosis
a process by which a living cell is internalized by another living cell, often observed in epithelial cells after detachment from the extracellular matrix
- Focal adhesion kinase (FAK)
a non-receptor tyrosine kinase that is recruited to the Salmonella-containing vacuole and promotes intracellular survival in macrophages. Leads to inhibition of autophagy through effects on signaling through the Akt–mTOR axis
- LC3-associatedphagocytosis (LAP)
a type of phagocytosis which induces components of the autophagy machinery (includingLC3) to associate with the phagosome, promoting its fusion to lysosomes
- LysM+ cells
cells which express lysozyme M, that is, myeloid-derived cells such as macrophages, monocytes, neutrophils, and myeloid-derived dendritic cells. Typically referenced in the context of LysM–Cre, which results in conditional deletion in LysM+ cells
- Macroautophagy
a process in which cellular contents are sequestered by double-membraned structures called autophagosomes and degraded by lysosomes. Specific subtypes of macroautophagy are named based on the substrate targeted for degradation such as ribophagy (ribosomes), mitophagy (mitochondria), and pathogen (xenophagy)
- Macrophage
innate immune cells are important for phagocytosis of pathogens and cell debris
- Mammalian target of rapamycin (mTOR)
a serine/threonine protein kinase that regulates cell growth, cell proliferation, cell motility, cell survival, protein synthesis, autophagy, transcription
- Mannose-capped lipoaribomannan (ManLAM)
a glycolipid primarily produced by pathogenic mycobacterial species, and represents an important immunomodulatory virulence factor for Mycobacterium tuberculosis
- Mitophagy
a type of selective macroautophagy that specifically targets mitophagy to lysosomes
- Necrosis
a form of cell injury which results in the premature death of cells by autolysis. Causes inflammation and is typically detrimental to the host
- Pathogen-associated molecular patterns (PAMPs)
molecules associated with groups of pathogens that are recognized by the innate immune system and can modulate inflammation
- Phosphatidylinositol3-kinases (PI3Ks)
a family of enzymes involved in initiation of autophagy, as well as diverse cellular functions including cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking
- Rapamycin
inhibitor of the Ser/Thr protein kinase mTOR, which regulates cell growth and metabolism in response to environmental cues. Often used to induce autophagy, as inhibition of mTOR mimics cellular starvation by blocking signals required for cell growth and proliferation
- Retinoic acid-induciblegene1 (RIG-I)
a pattern recognition receptor that recognizes double-stranded RNA
- Stimulator of interferon genes (STING)
functions to increase Type I interferon production through its action as a direct cytosolic DNA sensor and an adaptor protein in Type I interferon
- T cells
adaptive immune cells which respond to infection in an antigen-specific manner. Can be cytotoxic (cause lysis of target cells) or support immune functions through the production of cytokines
- Toll-like receptor (TLR)
a class of proteins that play a key role in the innate immune system by recognizing structurally conserved molecules derived from microbes
- Wortmannin
an inhibitor of autophagy that functions through inhibition ofphosphatidylinositol3-kinase
- Xenophagy
a type of selective macroautophagy that specifically targets intracellular pathogens to lysosomes
References
- 1.Zumla A, et al. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis. 2016;16:e47–63. doi: 10.1016/S1473-3099(16)00078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, et al. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 3.Rikihisa Y. Glycogen autophagosomes in polymorphonuclear leukocytes induced by rickettsiae. Anat Rec. 1984;208:319–27. doi: 10.1002/ar.1092080302. [DOI] [PubMed] [Google Scholar]
- 4.Dutta RK, et al. IL-6 inhibits IFN-γ induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol. 2012;44:942–54. doi: 10.1016/j.biocel.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 5.Juárez E, et al. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol. 2012;42:880–9. doi: 10.1002/eji.201142105. [DOI] [PubMed] [Google Scholar]
- 6.Watson RO, et al. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–15. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seto S, et al. Autophagy Adaptor Protein p62/SQSTM1 and Autophagy-Related Gene Atg5 Mediate Autophagosome Formation in Response to Mycobacterium tuberculosis Infection in Dendritic Cells. PLoS One. 2013;8:e86017. doi: 10.1371/journal.pone.0086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakowski ET, et al. Ubiquilin 1 Promotes IFN-γ-Induced Xenophagy of Mycobacterium tuberculosis. PLOS Pathog. 2015;11:e1005076. doi: 10.1371/journal.ppat.1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayaswal S, et al. Identification of host-dependent survival factors for intracellular Mycobacterium tuberculosis through an siRNA screen. PLoS Pathog. 2010;6:e1000839. doi: 10.1371/journal.ppat.1000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JJ, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–68. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 2013;9:e1003697. doi: 10.1371/journal.ppat.1003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castillo EF, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109:E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–62. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimmey JM, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–9. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manzanillo PS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–6. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mira MT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. 2004;427:636–40. doi: 10.1038/nature02326. [DOI] [PubMed] [Google Scholar]
- 17.Ali S, et al. PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol. 2006;144:425–31. doi: 10.1111/j.1365-2249.2006.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandra P, et al. Mycobacterium tuberculosis Inhibits RAB7 Recruitment to Selectively Modulate Autophagy Flux in Macrophages. Sci Rep. 2015;5:16320. doi: 10.1038/srep16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergne I, et al. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med. 2003;198:653–9. doi: 10.1084/jem.20030527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shui W, et al. Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J Proteome Res. 2011;10:339–48. doi: 10.1021/pr100688h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seto S, et al. Coronin-1a inhibits autophagosome formation around Mycobacterium tuberculosis-containing phagosomes and assists mycobacterial survival in macrophages. Cell Microbiol. 2012;14:710–27. doi: 10.1111/j.1462-5822.2012.01754.x. [DOI] [PubMed] [Google Scholar]
- 22.Shin DM, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010;6:e1001230. doi: 10.1371/journal.ppat.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim KH, et al. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A. 2012;109:7729–34. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Case EDR, et al. The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 2014;16:862–77. doi: 10.1111/cmi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabadi SM, et al. Antioxidant Defenses of Francisella tularensis Modulate Macrophage Function and Production of Proinflammatory Cytokines. J Biol Chem. 2016;291:5009–21. doi: 10.1074/jbc.M115.681478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele S, et al. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 2013;9:e1003562. doi: 10.1371/journal.ppat.1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Checroun C, et al. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–83. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–40. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 29.Dortet L, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011;7:e1002168. doi: 10.1371/journal.ppat.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tattoli I, et al. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013;32:3066–78. doi: 10.1038/emboj.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell G, et al. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun. 2015;83:2175–84. doi: 10.1128/IAI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogawa M, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–31. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 33.Baxt LA, Goldberg MB. Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS One. 2014;9:e94653. doi: 10.1371/journal.pone.0094653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayath CA, et al. Escape of intracellular Shigella from autophagy requires binding to cholesterol through the type III effector, IcsB. Microbes Infect. 2010;12:956–66. doi: 10.1016/j.micinf.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Campbell-Valois FX, et al. Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. MBio. 2015;6:e02567–14. doi: 10.1128/mBio.02567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko Y, et al. Active escape of Orientia tsutsugamushi from cellular autophagy. Infect Immun. 2013;81:552–9. doi: 10.1128/IAI.00861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi JH, et al. Orientia tsutsugamushi subverts dendritic cell functions by escaping from autophagy and impairing their migration. PLoS Negl Trop Dis. 2013;7:e1981. doi: 10.1371/journal.pntd.0001981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Cruze T, et al. Role for the Burkholderia pseudomallei type three secretion system cluster 1 bpscN gene in virulence. Infect Immun. 2011;79:3659–64. doi: 10.1128/IAI.01351-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdulrahman BA, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–70. doi: 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q, et al. Burkholderia pseudomallei survival in lung epithelial cells benefits from miRNA-mediated suppression of ATG10. Autophagy. 2015;11:1293–307. doi: 10.1080/15548627.2015.1058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cutting AS, et al. The role of autophagy during group B Streptococcus infection of blood-brain barrier endothelium. J Biol Chem. 2014;289:35711–23. doi: 10.1074/jbc.M114.588657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang L, et al. Superoxide dismutase of Streptococcus suis serotype 2 plays a role in anti-autophagic response by scavenging reactive oxygen species in infected macrophages. Vet Microbiol. 2015;176:328–36. doi: 10.1016/j.vetmic.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Kreibich S, et al. Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe. 2015;18:527–37. doi: 10.1016/j.chom.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 44.Tattoli I, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–75. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 45.Owen KA, et al. Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog. 2014;10:e1004159. doi: 10.1371/journal.ppat.1004159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Birmingham CL, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 47.Zheng YT, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–16. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 48.Wu S, et al. Inhibition of macrophage autophagy induced by Salmonella enterica serovar typhi plasmid. Front Biosci (Landmark Ed. 2014;19:490–503. doi: 10.2741/4220. [DOI] [PubMed] [Google Scholar]
- 49.Yasir M, et al. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun. 2011;79:4019–28. doi: 10.1128/IAI.05308-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al-Younes HM, et al. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy. 2011;7:814–28. doi: 10.4161/auto.7.8.15597. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen HTT, et al. Crohn’s disease-associated adherent invasive escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. 2014;146:508–519. doi: 10.1053/j.gastro.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 52.Chargui A, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One. 2012;7:e51727. doi: 10.1371/journal.pone.0051727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choy A, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–6. doi: 10.1126/science.1227026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rolando M, et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc Natl Acad Sci U S A. 2016;113:1901–6. doi: 10.1073/pnas.1522067113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fedrigo GV, et al. Serratia marcescens is able to survive and proliferate in autophagic-like vacuoles inside non-phagocytic cells. PLoS One. 2011;6:e24054. doi: 10.1371/journal.pone.0024054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pujol C, et al. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun. 2009;77:2251–61. doi: 10.1128/IAI.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moreau K, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. 2010;12:1108–23. doi: 10.1111/j.1462-5822.2010.01456.x. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 59.O’Neill AM, et al. Cytosolic Replication of Group A Streptococcus in Human Macrophages. MBio. 2016;7:e00020–16. doi: 10.1128/mBio.00020-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Seaghdha M, Wessels MR. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 2013;9:e1003394. doi: 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu SL, et al. Insufficient Acidification of Autophagosomes Facilitates Group A Streptococcus Survival and Growth in Endothelial Cells. MBio. 2015;6:e01435–15. doi: 10.1128/mBio.01435-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barnett TC, et al. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe. 2013;14:675–82. doi: 10.1016/j.chom.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niu H, et al. Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell Microbiol. 2008;10:593–605. doi: 10.1111/j.1462-5822.2007.01068.x. [DOI] [PubMed] [Google Scholar]
- 64.Niu H, et al. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci U S A. 2012;109:20800–7. doi: 10.1073/pnas.1218674109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vázquez CL, Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010;17:421–38. doi: 10.1038/cdd.2009.129. [DOI] [PubMed] [Google Scholar]
- 66.Winchell CG, et al. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun. 2014;82:2229–38. doi: 10.1128/IAI.01236-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berón W, et al. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–21. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newton HJ, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014;10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez E, et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A. 2016;113:E3260–9. doi: 10.1073/pnas.1522811113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El-Awady AR, et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog. 2015;10:e1004647. doi: 10.1371/journal.ppat.1004647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gutierrez MG, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005;7:981–93. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 72.Dorn BR, et al. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bélanger M, et al. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–70. doi: 10.4161/auto.2828. [DOI] [PubMed] [Google Scholar]
- 74.Schnaith A, et al. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. 2007;282:2695–706. doi: 10.1074/jbc.M609784200. [DOI] [PubMed] [Google Scholar]
- 75.Liu PF, et al. IsaB Inhibits Autophagic Flux to Promote Host Transmission of Methicillin-Resistant Staphylococcus aureus. J Invest Dermatol. 2015;135:2714–22. doi: 10.1038/jid.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamer I, et al. Replication of Brucella abortus and Brucella melitensis in fibroblasts does not require Atg5-dependent macroautophagy. BMC Microbiol. 2014;14:223. doi: 10.1186/s12866-014-0223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Starr T, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo F, et al. Autophagy favors Brucella melitensis survival in infected macrophages. Cell Mol Biol Lett. 2012;17:249–57. doi: 10.2478/s11658-012-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Symington JW, et al. ATG16L1 deficiency in macrophages drives clearance of uropathogenic E. coli in an IL-1β-dependent manner. Mucosal Immunol. 2015;8:1388–99. doi: 10.1038/mi.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang YH, et al. Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One. 2010;5:e10844. doi: 10.1371/journal.pone.0010844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang YH, et al. The autophagic induction in Helicobacter pylori-infected macrophage. Exp Biol Med (Maywood) 2009;234:171–80. doi: 10.3181/0808-RM-252. [DOI] [PubMed] [Google Scholar]
- 82.Tang B, et al. Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori. Autophagy. 2012;8:1045–57. doi: 10.4161/auto.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li P, et al. Streptococcus pneumoniae induces autophagy through the inhibition of the PI3K-I/Akt/mTOR pathway and ROS hypergeneration in A549 cells. PLoS One. 2015;10:1–15. doi: 10.1371/journal.pone.0122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deng Q, et al. Pseudomonas aeruginosa Triggers Macrophage Autophagy To Escape Intracellular Killing by Activation of the NLRP3 Inflammasome. Infect Immun. 2016;84:56–66. doi: 10.1128/IAI.00945-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Su C, et al. Helminth infection impairs autophagy-mediated killing of bacterial enteropathogens by macrophages. J Immunol. 2012;189:1459–66. doi: 10.4049/jimmunol.1200484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shibutani ST, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16:1014–1024. doi: 10.1038/ni.3273. [DOI] [PubMed] [Google Scholar]
- 87.Levine B, et al. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deretic V, et al. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma Y, et al. Autophagy and cellular immune responses. Immunity. 2013;39:211–227. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 90.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–69. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benjamin JL, et al. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013;13:723–34. doi: 10.1016/j.chom.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conway KL, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145:1347–57. doi: 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Inoue J, et al. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch Biochem Biophys. 2012;521:95–101. doi: 10.1016/j.abb.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 95.Maurer K, et al. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe. 2015;17:429–40. doi: 10.1016/j.chom.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuang E, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012;8:e1003007. doi: 10.1371/journal.pgen.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang C, et al. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci U S A. 2012;109:11008–13. doi: 10.1073/pnas.1203952109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marchiando AM, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. 2013;14:216–24. doi: 10.1016/j.chom.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Levine B, et al. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romano PS, et al. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life. 2012;64:387–96. doi: 10.1002/iub.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong X, Levine B. Autophagy and viruses: adversaries or allies? J Innate Immun. 2013;5:480–93. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rubinsztein DC, et al. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–30. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yuan K, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125:507–15. doi: 10.1242/jcs.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cullinane M, et al. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4:744–53. doi: 10.4161/auto.6246. [DOI] [PubMed] [Google Scholar]
- 105.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 106.Andersson AM, et al. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci Rep. 2016;6:28171. doi: 10.1038/srep28171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Junkins RD, et al. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One. 2013;8:e72263. doi: 10.1371/journal.pone.0072263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang K, et al. miR-155 suppresses bacterial clearance in Pseudomonas aeruginosa-induced keratitis by targeting Rheb. J Infect Dis. 2014;210:89–98. doi: 10.1093/infdis/jiu002. [DOI] [PubMed] [Google Scholar]
- 109.Chiu HC, et al. Eradication of intracellular Salmonella enterica serovar Typhimurium with a small-molecule, host cell-directed agent. Antimicrob Agents Chemother. 2009;53:5236–44. doi: 10.1128/AAC.00555-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lo JH, et al. Sensitization of intracellular Salmonella enterica serovar Typhimurium to aminoglycosides in vitro and in vivo by a host-targeted antimicrobial agent. Antimicrob Agents Chemother. 2014;58:7375–82. doi: 10.1128/AAC.03778-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stanley SA, et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014;10:e1003946. doi: 10.1371/journal.ppat.1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parihar SP, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209:754–63. doi: 10.1093/infdis/jit550. [DOI] [PubMed] [Google Scholar]
- 113.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–6. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lamb CA, et al. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 115.Kuballa P, et al. Autophagy and the Immune System. Annu Rev Immunol. 2012;30:611–646. doi: 10.1146/annurev-immunol-020711-074948. [DOI] [PubMed] [Google Scholar]
- 116.Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 117.Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424–425. doi: 10.4161/auto.22931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 119.Delgado MA, et al. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12:101–14. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stolz A, et al. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 122.Matsumoto G, et al. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–42. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- 123.Pilli M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–34. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bauckman KA, et al. Selective autophagy: xenophagy. Methods. 2015;75:120–7. doi: 10.1016/j.ymeth.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mizushima N, et al. Methods in Mammalian Autophagy Research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bonilla DL, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39:537–547. doi: 10.1016/j.immuni.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez J, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Deretic V, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. doi: 10.1172/JCI73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boxx GM, Cheng G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe. 2016;19:760–769. doi: 10.1016/j.chom.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sahoo M, et al. Role of the Inflammasome, IL-1b, and IL-18 in Bacterial Infections. Sci World J. 2011;11:2037–2050. doi: 10.1100/2011/212680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Harris J, et al. Autophagy controls IL-1b secretion by targeting Pro-IL-1b for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dupont N, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–11. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang M, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy- mediated secretion. Elife. 2015;4:1–23. doi: 10.7554/eLife.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367–76. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tait SWG, et al. Die another way–non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127:2135–44. doi: 10.1242/jcs.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yousefi S, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 139.Bestebroer J, et al. Hidden behind autophagy: The unconventional roles of ATG proteins. Traffic. 2013;14:1029–1041. doi: 10.1111/tra.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186:7110–9. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martin CJ, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. 2012;12:289–300. doi: 10.1016/j.chom.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.