

Summary
Background
The systemic immune system plays a role in inflammation and fibrogenesis associated with non‐alcoholic steatohepatitis (NASH) and has become a potential target for drug development. In particular, the gut immune system has been suggested as a means for generating signals that can target the systemic immune system.
Aim
To describe seven novel methods being developed for the treatment of NASH that target the gut immune system for alleviation of the systemic inflammatory response, including oral administration of fatty‐liver‐derived proteins, anti‐CD3 antibodies, tumour necrosis factor fusion protein, anti‐lipopolysaccharide antibodies, glucosylceramide, delayed‐release mercaptopurine, and soy‐derived extracts.
Methods
A search for these methods for oral immunotherapy for NASH was conducted.
Results
Oral administration of these compounds provides an opportunity for immune modulation without immune suppression, with the advantage of being independent of a single molecular/inflammatory pathway. These modes of oral immune therapy demonstrate superior safety profiles, such that the patient is not exposed to general immune suppression. Moreover, these approaches target the whole spectrum of the disease and may serve as adjuvants to other therapies, such that they provide a platform for treatment of concomitant disorders in patients with NASH, including diabetes and hyperlipidaemia. Most of the compounds reviewed are currently in phase II trials, and it is anticipated that the acquisition of more clinical data in the next few years will enable the use of this new class of drugs for the treatment of NASH.
Conclusion
Oral immunotherapy may provide a novel platform for the treatment of NASH.
Introduction: the difficulty associated with developing treatments for non‐alcoholic steatohepatitis
Globally, non‐alcoholic fatty liver disease (NAFLD) is currently the most common liver disease, affecting approximately one‐third of the Western world.1 The incidence and prevalence of this disease are rapidly rising to epidemic proportions around the globe. NAFLD comprises a spectrum of progressive liver diseases that include simple steatosis, non‐alcoholic steatohepatitis (NASH), fibrosis, and ultimately cirrhosis, end‐stage liver disease and primary liver cancer.2 NASH has been predicted to become the leading cause of liver transplantation in the USA by the year 2020.3 The incidence of hepatocellular carcinoma (HCC) is also rising; hence, HCC has become the primary cause of obesity‐related cancer death in middle‐aged men in the USA.1
During the process of drug development for NASH, numerous obstacles have been identified in recent years.4 While progress has been achieved in our understanding of the pathophysiology, diagnosis and natural history of NAFLD, no drugs have yet been approved for the treatment of NASH.4 Studies investigating the optimal therapy for NAFLD and NASH have not yet been able to develop a universal treatment protocol.5 Lack of a clear mechanism remains a major difficulty in drug development for NASH, which is caused by a complex interplay between host and environmental factors.3 Thus, the use of pharmacological agents as an adjunctive therapy to lifestyle modification is crucial, as weight loss is a difficult task to achieve and maintain.6 NAFLD is recognised as the hepatic component of the metabolic syndrome and is associated with liver‐related morbidity and mortality as well as an increased risk of cardiovascular disease, type 2 diabetes mellitus, hyperlipidaemia and abdominal obesity.7
Several pharmacological agents have been studied in an effort to improve insulin resistance and to improve the pro‐inflammatory mediators responsible for NASH progression.4 Examples of drugs that are effective only in the animal models of NASH but have failed in human trials are as follows: PDE4 inhibitors,8 caspase inhibitors,9 resveratrol,10 omega‐3 fatty acid preparations,11 anti‐tumour necrosis factor‐alpha (TNF‐α) and probiotics. In addition, most NASH patients use drugs to control type 2 diabetes, such as metformin, sulfonamides and insulin, although these drugs are ineffective for NASH.4, 12
The side effects of several compounds that have demonstrated efficacy in NASH trials are of major concern. Examples are pioglitazones, which were shown in the PIVENS trial to improve some histological features of NASH, and to achieve resolution of steatohepatitis.13 However, glitazones contribute to weight gain and to adipose tissue insulin resistance.14 Their long‐term use is associated with bone fractures in women, congestive heart failure and increased risk of bladder cancer.4, 15 Obeticholic acid (OCA) is a synthetic bile acid with picomolar agonistic activity towards the farnesoid X receptor (FXR), as shown in the FLINT trial to improve steatohepatitis and fibrosis.16 Side effects of pruritus and an increase in low‐density lipoprotein (LDL) cholesterol are of relevance for their chronic use.17 In the PIVENS trial, vitamin E was shown to improve steatosis, inflammation and ballooning, leading to the resolution of NASH in 36% of patients.13 However, long‐term vitamin E treatment can result in an increase in overall mortality,18 haemorrhagic stroke19 and prostate cancer in males older than 50 years.20
It was recently suggested that ‘an ideal drug choice for NASH should reduce hepatic inflammation and liver cell injury, be able to correct the underlying insulin resistance, and deliver anti‐fibrotic effects’.6 Drugs that target inflammation, even without having a direct anti‐fibrotic effect, may result in a subsequent reduction of fibrosis if sustained resolution of NASH is achieved. Alternatively, drugs that are exclusively anti‐fibrotic may not dampen inflammatory triggers for fibrogenesis.4
The immune system as a target for the treatment of NASH
Recent concepts in NASH development have proposed that multiple parallel hits are involved in disease initiation and progression. However, the ‘key switch’ between steatosis and NASH remains to be discovered.2 Genetic predisposition involving single nucleotide polymorphisms (SNPs) and mutations, with SNP distribution patterns along specific metabolic and cellular pathways, determines the susceptibility of a patient to NASH, while diet and lifestyle regulate the disease phenotype.4 It has been hypothesised that following these initiating events, inflammation and the associated fibrogenesis contribute to the perpetuation of disease. Another hypothesis states that the inflammatory processes are part of the intimating event in these patients. Consequently, chronic inflammation is a major contributor to NASH and thus represents a target for disease treatment.
Obesity is associated with chronic low‐grade inflammation, and inflammation is related to the disruption of metabolic homeostasis.21 The metabolic inflammatory state, termed ‘metaflammation’, is defined as low‐grade, chronic inflammation in response to excess nutrients and energy.22 The chronic inflammatory process involves the liver, adipose tissue, pancreas and muscle, leading to insulin resistance and metabolic dysfunction.23 Imbalances between pro‐ and anti‐inflammatory cytokines, alterations in insulin responses, β‐oxidation, lipid storage and transport, autophagy and nuclear receptor signalling all contribute to the progression from NAFLD to NASH, cirrhosis and cancer.
During the process of liver damage, tissue repair pathways are activated to restore tissue and metabolic homeostasis. Indeed, the balance between mechanisms of damage and repair defines the progression of the disease.24, 25 Liver inflammation can either be beneficial or detrimental.24 A mild inflammatory response exerts hepatoprotective effects and strengthens tissue restoration. However, an extreme inflammatory response induces hepatocyte damage and can generate irreversible liver damage, fibrosis and carcinogenesis. Liver injury is associated with the secretion of pro‐inflammatory factors by Kupffer cells, natural killer T (NKT) cells,26 hepatic stellate cells, sinusoidal endothelial cells, dendritic cells (DC), NK cells, monocytes and lymphocytes, which develop in response to injury or damage to the liver. These secreted factors include cytokines, chemokines, lipid messengers and reactive oxygen species that promote the apoptotic or necrotic demise of hepatocytes.24 A cycle of inflammation and cell death is generated by dying hepatocytes, releasing damage‐associated molecular patterns that bind to evolutionarily conserved pattern recognition receptors to activate cells of the innate immune system and stimulate the inflammatory process.24
Inflammatory genes are overexpressed in NASH. Increased expression of genes that regulate inflammation in patients with NAFLD and NASH have been noted in the subcutaneous adipose tissue (SAT), visceral adipose tissue (VAT) and phenotypes of adipose tissue macrophages in obese patients.27 A total of 111 genes associated with inflammation were differentially expressed between VAT and SAT, and the expression of these genes increased as the disease progressed from NAFLD to NASH. The levels of interleukin 8, chemokine (C–C motif) ligand 3 (CCL3) and TNF‐α are correlated with liver inflammation and NAFLD activity. Increased proportions of CD11c+CD206+ and CCR2+ macrophages in the VAT of patients with NASH result in increased levels of pro‐inflammatory cytokines and chemokines.27 Genetic manipulation of certain metabolic or stress response pathways, including one‐carbon metabolism, NF‐κB, PTEN and microRNAs, may contribute to the development of NASH and the regulation of carcinogenesis in NAFLD.1
Because the immune system plays a role in the pathogenesis of NASH, various treatments are being developed to directly or indirectly target the relevant inflammatory pathways.
Glitazones up‐regulate adiponectin, an insulin‐sensitising and anti‐steatogenic adipokine, and increase fatty acid β‐oxidation in liver and muscle.28, 29 PPARγ agonists also exert anti‐inflammatory effects on Kupffer cells, indicating a direct hepatoprotective effect.
FXR activation has a wide range of metabolic effects, such as improvement of glucose metabolism and peripheral insulin sensitivity,30 reduction of lipogenesis and enhancement of fatty acid β‐oxidation.31 FXR activation also has anti‐inflammatory actions,32, 33 and the histological features of NASH and fibrosis can be improved by treatment with OCA, an FXR agonist.34
Elafibiranor is a liver‐targeted insulin sensitiser, a dual PPARα/δ agonist that undergoes enterohepatic cycling. This drug improves hepatic and peripheral insulin sensitivity, dyslipidaemia and inflammatory markers.35 PPARδ activation is a potent metabolic regulator that induces hepatic fatty acid β‐oxidation, inhibits hepatic lipogenesis,36 reduces hepatic glucose production and improves hepatic inflammation.37, 38 Animal data have demonstrated the hepatoprotective effects of elafibranor due to reduced levels of steatosis, hepatic inflammation and pro‐inflammatory gene expression.39 A recent clinical trial showed that elafibranor resolved NASH without a worsening of fibrosis in the intention‐to‐treat analysis and in patients with moderate or severe NASH. However, the predefined end point was not met in the intention‐to‐treat population.40
Incretin mimetics, which are glucagon‐like peptide‐1 receptor (GLP‐1R) agonists, can be used to treat NASH.41 GLP, a peptide product of the L cells of the small intestine and proximal colon, stimulates insulin secretion from β cells and also inhibits glucagon secretion from α cells. The induction of PPARα and γ expression upon GLP‐1R agonist binding results in increased disposal towards fatty acids via β‐oxidation and lipid export in hepatocytes.42 GLP‐1R agonists improve hepatic insulin sensitivity.43 The beneficial effects of these drugs in humans with NASH were associated with reduced de novo lipogenesis, improved adipose tissue lipolysis and decreased inflammation.4, 44
Vitamin E and thiazolidinedione derivatives show anti‐inflammatory effects. Vitamin E also exerts protective effects against oxidative damage induced by free radicals, protecting against mitochondrial toxicity, blocking intrinsic apoptotic pathways and down‐regulating NF‐kB‐dependent inflammatory pathways.45, 46, 47 Vitamin E therapy, compared with placebo, was associated with a significantly higher rate of improvement in NASH (43% vs. 19%).13
Cenicriviroc (CVC), a dual‐CCR2/CCR5 antagonist, showed potent anti‐inflammatory and anti‐fibrotic activity in animal models,48 and its safety and pharmacokinetic results were recently published.49 This therapy is currently being tested in patients with NASH.
Several additional compounds that have been tested in NASH have direct and indirect anti‐inflammatory properties,50 that is, fatty acid–bile acid conjugates that serve as metabolic modulators, anti‐lysyl‐oxidase 2 monoclonal antibody, which has anti‐fibrotic effects, vitamin D, an immunomodulatory molecule and renin–angiotensin–aldosterone system blockers.4, 5
Massive weight loss as a result of bariatric surgery results in histological improvement and partial reversal of cirrhosis.51, 52 Bariatric surgery might also correct the pro‐inflammatory state associated with obesity,53, 54 reducing adipose tissue levels of pro‐inflammatory TNF‐α and IL‐6 and the expression of hepatic SOCS3, improving hepatic insulin resistance and inhibiting hepatic inflammation.55
Collectively, these data support the role of the immune system as a therapeutic target in NASH.
Oral immunotherapy: using the gut immune system as a target for generating signals that reach the systemic immune system to alleviate the inflammatory response
Gut–liver cross‐talk is implicated in the impairment of lipid and glucose homeostasis in steatogenesis and in the initiation of inflammation and fibrogenesis in NASH.56 Indeed, immune signals generated at the gut level and those generated by the gut microbiome affect the systemic immune system. Following the generation of these gut signals, activation of the innate immune system, which is accompanied by the activation of the adaptive system, can lead to steatosis, liver inflammation and fibrosis.57 These signals may exert a direct effect via absorption or a nondirect remote effect. Therefore, targeting the gut immune system represents a promising method for systemic immune modulation and for alleviating the liver inflammation in NASH.58 Figure 1 shows an outline of the potential role of the gut and the systemic immune systems in the pathogenesis of NASH, illustrating that both systems may serve as potential therapeutic targets.
Figure 1.
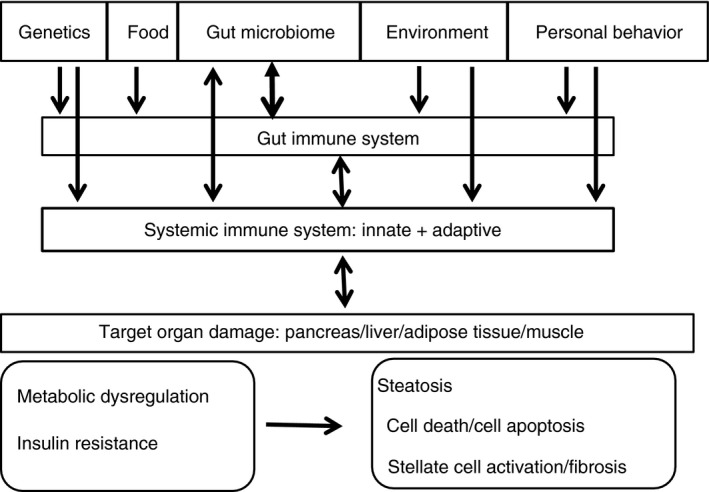
Pathogenesis of NASH: a schematic of the potential role of the gut immune system and the systemic immune system in the pathogenesis of NASH.
Dysbiosis and altered intestinal permeability play an important role in linking gut lumen antigenic/toxic substances and systemic conditions with liver inflammation in NAFLD and NASH.59, 60 A bidirectional interaction between the gut immune system and microbiota occurs, which interacts with food and bile. These interactions likely contribute to the progression from lean to obese states, steatosis, steatohepatitis and fibrosis.57 The gut microbiota induces the absorption and disposal of nutrients to the liver. Microbiome‐associated toll‐like receptor (TLR) ligands promote the production of hepatic pro‐inflammatory cytokines,56 and TLR2, TLR4, TLR5 and TLR9 are associated with the pathogenesis of NAFLD. Indeed, compositional changes in the gut microbiota are related to the development of NASH.61 An abundance of Firmicutes and Bacteroidetes may represent an underlying mechanism for the development of obesity and NAFLD. In particular, a decrease in Akkermansia muciniphila causes a thinner intestinal mucus layer and promotes gut permeability, which allows the leakage of bacterial components. Accordingly, an increase in A. muciniphila improves the metabolic parameters in subjects with obesity and NAFLD.61
The gut is composed of a physical barrier surface that prevents bacterial adhesion and regulates paracellular diffusion into the tissues and a functional barrier that differentiates pathogens from commensal microorganisms.62 The gut microbiota is part of the gut barrier, and it also competes with pathogens for energy resources, processes the molecules needed for mucosal integrity, and interacts with the immune components in the gut.62 The functional barrier comprises the gut immune system that regulates the generation of immune tolerance vs. immune responses to antigens.62 Several components of the gut barrier interact with the immune system. This bidirectional interaction between the gut immune system and the gut microbiome determines the signals that are delivered from the gut to the systemic immune system.63
The gut‐associated lymphoid tissue (GALT) is composed of both innate and adaptive immune cells. The GALT is responsible for antigen sampling in the lumen and for the generation of immune signals that are transported to the systemic immune system. The gut epithelial cells form a physical and immunological barrier, and the mucus layer that separates the intraluminal content from more internal layers containing anti‐microbial products and secretory IgA plays a role in the generation of these signals in the gut.62, 63 The GALT is constantly exposed to dietary and microbial‐derived foreign antigens. Proper responsiveness of all components of the mucosa is required for the maintenance of intestinal integrity and the ability to mount an effective immune response to potential pathogens.64
Dendritic cells are central for the initiation and differentiation of adaptive immune responses. In the intestinal mucosa, DCs are part of the GALT and are located diffusely throughout the intestinal lamina propria, Peyer's patches and smaller lymphoid aggregates, alongside intestinal‐draining lymph nodes including mesenteric lymph nodes (MLNs). MLNs serve as a site for the generation of immune signals between the gut and the systemic immune system. Lymphocytes influx into the gut‐draining MLNs, where they undergo antigen‐induced activation and priming to determine memory and effector phenotypes.65 During this process, these lymphocytes gain the capacity to migrate via the lymph and blood to the systemic organs.
Disruption of the gut barrier and accompanying effects on the immune system have been associated with many gastrointestinal diseases as well as extra‐intestinal disorders, such as the pathogenesis of NASH. Hence, the intestinal barrier can serve as a target for the treatment of these disorders. Drugs and compounds are currently being developed for the restoration of a normal intestinal barrier and immune function.62
Oral immunotherapy is based on the notion that targeting various components of the gut barrier can serve as a means to alter the immune signal that is transported from the gut to the systemic immune system, thereby affecting immune‐mediated damage in target organs.63 Oral tolerance is one of the mechanisms of oral immune therapy, such that exposure of the gut immune system to various antigens can alter the systemic immune signal.66, 67 This mechanism has been shown to be effective in animal models68, 69, 70, 71 and in several immune‐associated disorders in humans.72, 73, 74, 75 Figure 2 shows a schematic presentation of oral immunotherapy compounds that target the GALT and/or microbiome to generate an immune signal that affects the systemic immune system, specifically inducing regulatory T cells (Tregs) at the level of MLNs to alleviate the inflammatory response in the liver.76
Figure 2.
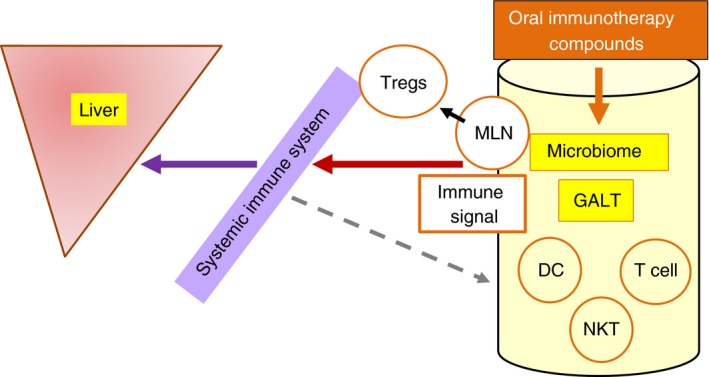
Oral immune therapy compounds target the gut‐associated lymphoid tissue (GALT) to generate immune signals that affect the systemic immune system and promote regulatory T cells (Tregs) at the level of the mesenteric lymph nodes (MLNs), thereby alleviating the inflammatory response in the liver. DC, dendritic cells; NKT, natural killer T cells.
Compounds being developed for the treatment of NASH by targeting the gut immune system
The inflammatory response is critical for initiating and sustaining the liver damage associated with NASH, making the immune system a target for treatment development.58 However, systemic immune suppression is nonspecific in numerous cases, and thus patients are exposed to a generalised immune suppression. Several methods have recently been investigated that target the gut immune system to generate an anti‐inflammatory signal that can redirect the systemic immune system. Herein, we review seven potential methods that have been demonstrated to be effective in pre‐clinical trials. Several of these drugs were previously shown to exert beneficial responses in phase I and II clinical trials in patients with NASH. Table 1 summarises some of the oral immune therapy compounds that are currently being developed for NASH and their status of development.
Table 1.
Compounds being developed for the treatment of NASH by targeting the gut immune system
| Compound | Description | Pre‐clinical data | Human triala |
|---|---|---|---|
| Liver‐extracted proteins | Fatty liver‐derived proteins | +77 | − |
| Anti‐CD3 | Anti‐CD3 antibodies | +76 | +119 |
| PRX106 | Anti TNF fusion protein | +124 | +a, 83 |
| Imm124E | Anti‐LPS antibodies | +109 | +123 |
| Glycosphingolipids | Glucosylceramide | +90, 91, 92 | +125 |
| DR‐MP | Delayed‐release mercaptopurine | − | +a, 72 |
| OS M1 | Soy‐derived extracts | +102 | − |
Tested for other indications.
Induction of oral tolerance towards fatty liver‐extracted proteins
To determine the effect of oral immune therapy towards liver‐extracted proteins on the metabolic syndrome, ob/ob mice and their lean littermates were orally administered liver extracts from wild‐type or ob/ob mice.77 The induction of immune regulation via the oral presentation of liver‐extracted proteins led to a significant reduction in hepatic fat content. Accordingly, a significant improvement in glucose intolerance was noted in treated mice. These changes were accompanied by alterations in NKT regulatory lymphocytes, with a significant elevation of anti‐inflammatory serum IL‐10 levels.77 Although this concept has not yet been tested in patients with NASH, a beneficial effect was observed in patients with inflammatory bowel disease (IBD) who received orally administered autologous colonic‐extracted proteins.72, 74 In a phase II randomised, double‐blind, placebo‐controlled trial, 58% of patients with moderate to severe Crohn's disease attained clinical remission compared with 29% in the placebo group.72, 74 Alterations in CD4+, CD8+ and NKT lymphocytes support the notion that oral immune therapy using non‐absorbable autologous proteins affects the systemic immune system. These data suggest that exposure of the gut immune system to disease‐associated antigens may serve as a means to down‐regulate the systemic inflammatory response.
PRX106: Oral administration of a non‐absorbable orally administered BY‐2 plant cell that expresses a recombinant anti‐TNF fusion protein
Parenteral administration of etanercept has been successfully used for the treatment of immune‐mediated disorders such as rheumatoid arthritis.78, 79, 80 PRX‐106 is a non‐absorbable, orally administered BY‐2 plant cell that expresses a recombinant anti‐TNF fusion protein consisting of the soluble form of the human TNF receptor (TNFR) fused to the Fc component of a human IgG1 antibody. This formulation has an amino acid sequence identical to that of etanercept. In vitro, this TNFR binds TNF‐α, inhibiting its binding to cellular TNFRs and hence blocking its downstream effects.81
In a high‐fat diet (HFD) animal model of NASH, C57B1/6 HFD mice were orally administered PRX‐106 daily for 22 weeks. A decrease in hepatic triglyceride content was observed in the treated mice, together with a decrease in serum triglycerides, aspartate aminotransferase (AST) levels, glucose levels and the homeostatic model assessment (HOMA) score.82 An altered distribution of CD4+CD25+FoxP3+ lymphocytes between the liver and spleen, an increase in the intrasplenic to intrahepatic CD4+CD25+FoxP3+ lymphocyte ratio and a decrease in the intrasplenic to intrahepatic CD8+CD25+FoxP3+ lymphocyte ratio were noted in treated mice. In addition, an increase in intrahepatic NKT cells was noted together with a decrease in the intrasplenic to intrahepatic NKT lymphocyte ratio. An assessment of the CD4–CD8 ratios revealed the sequestration of CD8+ lymphocytes in the liver.82
In a phase I/IIA clinical trial, three different doses (2, 8 or 16 mg/day) of PRX‐106 were orally administered for 5 consecutive days in 14 healthy volunteers.83 Oral administration PRX‐106 was safe and well tolerated. A pharmacokinetic study showed that PRX106 was not absorbed, and no effects on white blood cells or lymphocyte counts were noted. However, a dose‐dependent effect on systemic lymphocytes was observed. Oral administration of the three dosages was related to an increase in CD4+CD25+ cells and the CD8+CD25+ subset of suppressor T lymphocytes. A marked increased in CD4+CD25+FoxP3 Tregs was noted in the 8‐mg‐treated group. CD3+CD69+ NKT lymphocyte subsets also increased in response to treatment.83 A phase IIA clinical trial in patients with IBD is currently in progress (ClinicalTrials.gov Identifier: NCT02768974).
These data support the concept that oral administration of PRX‐106, a non‐absorbable recombinant anti‐TNF fusion protein, is safe and not associated with immune suppression yet induces favourable anti‐inflammatory immune modulation. Thus, PRX‐106 may provide an orally administered, safe and effective anti‐TNF‐α‐based immune therapy for the treatment of NASH.
Oral administration of beta‐glycosphingolipids as immune modulators
An altered lipidome, including alterations in glycosphingolipids, has been described as part of the ‘NASH signature’.84 Glucocerebroside (β‐glucosylceramide, GC) and lactosylceramide (LC) are intermediates in the metabolic pathways of glycosphingolipids.85 In vitro, GC inhibits NKT lymphocyte proliferation in the presence of DCs.86 In vivo, oral administration of GC alleviates liver damage in the concanavalin A (ConA) immune‐mediated hepatitis model86 and in models of immune‐mediated liver damage induced by graft‐versus‐host disease.87 These effects have been associated with an altered NKT lymphocyte distribution and a pro‐ to anti‐inflammatory cytokine shift.86, 88, 89
In the leptin‐deficient ob/ob model of diabetes and NASH, oral administration of GC decreased liver size and the hepatic fat content, which was associated with near normalisation of the glucose intolerance and a decrease in the serum triglyceride levels.90 These effects were associated with an altered NKT cell distribution and cytokine profile in the anti‐inflammatory direction.90
A synergistic beneficial effect was noted for the combination of GC and LC in animal models of diabetes and NASH.91, 92 In the treatment of Cohen diabetes‐sensitive rats, which is a lean model of non‐insulin‐resistant, nutritionally induced diabetes, a combination of GC and LC improved pancreatic and liver histology, reduced liver steatosis and improved glucose metabolism. These changes have been linked to an increase in intrahepatic trapping of CD8 T and NKT lymphocytes.92
Similarly, in the Psammomys obesus model of NASH and diabetes, beta‐glycosphingolipids ameliorate the hepatic injury, as shown by a decrease in liver enzymes, liver weight, hepatic fat and improved liver histology. Improved serum cholesterol and triglyceride levels have also been noted, and these effects are likely associated with decreased interferon‐gamma (IFN‐γ) serum levels.91 Preliminary studies in the CCL4 model of fibrosis further suggest that GC may exert anti‐fibrotic effects.93
In humans, oral administration of GC in patients with type 2 diabetes and NASH was tested in a randomised, double‐blind, placebo‐controlled trial.94 No treatment‐related adverse events were observed. In a per protocol analysis of the data, oral administration of GC decreased the hepatic fat content, as measured by magnetic resonance imaging (MRI) in the GC‐treated group compared with placebo. HbA1C levels were also decreased in patients treated with GC, which was associated with a milder decrease in serum high‐density lipoprotein levels. These beneficial effects were related to a decrease in CD4 and NKT cell subsets of lymphocytes.94 These data further support the concept that non‐absorbable glycosphingolipids exert an effect on the innate immune system of the gut that leads to systemic immune modulation and may thereby alleviate NASH and diabetes.
Oral administration of soy‐derived extracts
Soy‐derived molecules exert an adjuvant effect via the activation of innate immune cells in the gut, and they have been shown to be effective for the alleviation of immune‐associated disorders.95, 96, 97, 98 Indeed, soy products have been shown to decrease oxidative stress, pro‐inflammatory cytokine secretion and lipid peroxidation in the methionine–choline‐deficient (MCD) diet model of NASH.99 Epidemiological studies have confirmed that soy products may decrease the morbidity associated with NASH,100 lower serum lipids, with reduced lipid uptake by the liver, and improve insulin resistance.101
Soy extracts were orally administered to mice with ConA immune‐mediated hepatitis, and liver damage was alleviated based on a decrease in the serum levels of alanine aminotransferase (ALT) and AST.102 In the HFD model of NASH, oral administration of these extracts ameliorated liver injury, as indicated by a decrease in hepatic triglyceride levels, improved liver histology, decreased serum cholesterol and triglyceride levels, and improved insulin resistance.102 In the model of combined MCD and HFD, a decrease in hepatic triglycerides with improvement in blood glucose levels and liver histology were noted using soy extracts. These effects were linked to reduced serum TNF‐α levels and an altered Treg distribution.102 Based on these data and on the desirable safety profile of these extracts, they are currently being prepared for human studies in patients with NASH and diabetes.
DR6MP: oral administration of non‐absorbable delayed‐release mercaptopurine
The purine analogues azathioprine and mercaptopurine (MP) constitute the basis of long‐term maintenance therapy for several immune‐mediated disorders.103 However, their use is limited by their potential systemic side effects.104, 105, 106, 107 A novel formulation of non‐absorbable delayed‐release mercaptopurine (DR‐MP) was developed for oral immune therapy.108 Administration of a single dose of DR‐MP was shown to increase the number of systemic CD4+CD25+Foxp3+ Tregs. In a subsequent study, 70 patients with moderately active Crohn's disease were enrolled in a double‐blind, controlled phase II trial for 12 weeks.108 The time to maximal clinical response was 8 weeks for DR‐MP vs. 12 weeks for the control, purinethol, with a higher proportion of patients in the DR‐MP group achieving clinical remission and showing improvements in the IBD Questionnaire score. DR‐MP led to a decrease in CD62+ expression on T lymphocytes, indicating reduced cell adhesion to sites of inflammation. In addition, DR‐MP was safer than purinethol, with fewer adverse events. There was no evidence of drug‐induced leucopenia, and a lower proportion of hepatotoxicity was observed.108
These data suggest that oral administration of a non‐absorbable DR‐MP formulation is safe and biologically active in the gut and that it exerts a systemic anti‐inflammatory effect.
Imm124E: oral administration of non‐absorbable anti‐LPS antibodies with adjuvants
The gut microbiome and bacteria‐derived products are significant in the pathogenesis of NASH.60, 61 Imm124E is an IgG‐enriched fraction of enterotoxigenic Escherichia coli‐containing colostrum that consists of anti‐LPS antibody and several glycosphingolipid adjuvants. The induction of oral immune therapy using the non‐absorbable Imm124E formulation was shown to alter the systemic immune system to alleviate NASH in humans and animal models. In the ob/ob model of NASH and diabetes, oral administration of Imm124E decreased serum ALT levels and hepatic triglycerides and improved glucose intolerance.109 These effects were associated with a decrease in serum TNF‐α levels and an increase in CD4+CD25+ regulatory cells, CD4+CD25+Foxp3+ Tregs and NKT regulatory lymphocytes.
In the CCL4 fibrosis model, oral administration of Imm124E alleviated liver fibrosis, as determined by a significant progression of the Metavir liver fibrosis score and alpha smooth muscle actin levels.110 An improvement in liver injury was noted in treated mice based on a major decrease in liver enzymes and bilirubin levels, as well as amelioration of the decrease in body weight. Liver pathology was observed by staining with trichrome blue and Masson's red, which showed improvement in periportal necro‐inflammation, bridging and confluent necrosis, lobular necrosis, hepatocellular apoptosis and portal inflammation. These effects were associated with a decrease in hepatic F4/80+ macrophages, which are associated with liver injury and inflammation.110
In an open‐label trial conducted in humans, subjects with biopsy‐proven NASH and type 2 diabetes were orally administered Imm124E for 30 days.111 No treatment‐related adverse occurrences were observed, and no human anti‐bovine antibodies were detected, which suggested a lack of systemic drug absorption. Alleviation of insulin resistance, as determined by a decrease in fasting glucose levels, an increase in early insulin secretion during glucose administration and improvements in the glucose tolerance test and HbA1C levels, was noted. Treated patients showed a decrease in serum levels of triglycerides, total cholesterol, LDL cholesterol and liver enzymes.111 Increased serum levels of GLP‐1 and adiponectin, together with the promotion of CD25+ and CD4+CD25+Foxp3+ Tregs, were also noted. These data suggest that alterations of the gut microbiome together with the promotion of Tregs using oral immune therapy may alleviate NASH and its associated insulin resistance via a decrease in the systemic inflammatory response. A global large double‐blind, double‐dose, randomised, placebo‐controlled trial using Imm124E in patients with NASH is currently ongoing (Clinical Trial Gov. NCT02316717).
Anti CD3: oral administration of non‐absorbable anti‐CD3 antibodies as immune modulators
Intravenous administration of anti‐CD3 antibodies is effective in the transplantation setting and for the treatment of several immune‐mediated disorders via the induction of immune suppression.112 Studies have demonstrated a narrow therapeutic window for intravenous anti‐CD3‐based therapies, with lower doses being ineffective and higher pharmacologically active doses causing intolerable levels of adverse effects. In contrast, oral administration of anti‐CD3 promotes Tregs by affecting the gut immune system and MLNs.66 Orally delivered antibodies are not associated with generalised immune suppression, and they do not induce cytokine release syndromes.113
Orally administered anti‐CD3 was shown to alleviate experimental autoimmune encephalomyelitis in an animal model of multiple sclerosis, both prior to induction and at the peak of disease.114 This beneficial effect was associated with the induction of a unique type of Treg subset characterised by the expression of latency‐associated peptide (LAP) on their cell surface and functioning via a TGF‐β‐dependent mechanism.113, 115 A similar effect was shown in an animal model of type I diabetes.114 Oral and nasal administration of anti‐CD3 in mouse models of lupus was shown to suppress autoantibody production and prevent kidney damage.116, 117 In the leptin‐deficient model of NASH and diabetes, oral administration of anti‐CD3 with GC reduced pancreatic hyperplasia, hepatic fat accumulation and muscle inflammation. This treatment further alleviated diabetes and reduced liver enzyme, serum cholesterol and triglyceride levels.76 These effects were associated with the promotion of CD4+LAP+ Tregs and with an increase in the levels of TGF‐β and IL‐10, which were secreted from DCs and anti‐CD3‐activated peripheral blood lymphocytes (PBL).76
In an open‐label phase I clinical trial in healthy volunteers, oral anti‐CD3 was biologically active and well tolerated.118 Changes were not observed in CD3+ lymphocyte counts, and no human anti‐mouse antibodies were detected, implying non‐absorption of the antibodies. Suppression of the Th1/Th17 and IFN‐γ responses by increased numbers of CD4+CD25+ regulatory cells and CD8+CD25+ T cells was noted.118 A decrease in IFN‐γ and IL‐17 and an increase in TGF‐β secretion from anti‐CD3‐stimulated PBLs were observed.118 In a phase IIa clinical study of patients with NASH and type II diabetes, oral administration of several dosages of anti‐CD3 was biologically active, and no treatment‐related adverse events were reported. Decreases in plasma glucose and liver enzymes were noted in a dose‐dependent manner.119 The systemic promotion of Tregs was due to an increase in CD4+LAP+ and CD4+CD25+LAP+ cells, with a concomitant increase in serum TGF‐β levels. These data support a different mechanism of action for the oral route of anti‐CD3, which induces immune modulation without immune suppression, thereby alleviating the liver damage associated with NASH.
Advantages of using oral immune therapy for NASH
Oral immune therapy provides a way to overcome various obstacles that are currently encountered during the treatment of NASH. Table 2 summarises several unmet needs in the development of drugs for NASH, as well as the beneficial effects of using oral immune therapy‐based compounds for affected patients.
Table 2.
Several unmet needs in the development of drugs for NASH and beneficial effects of using oral immunotherapy‐based compounds
| Need | Advantages of oral immune therapy |
|---|---|
| Target the mechanism of disease | Takes advantage of the inherent ability of the gut immune system to deliver immune signals to control the systemic immune response |
| Target molecule or pathway | Not dependent on a specific molecular pathway |
| Promotes regulatory T cells | |
| Mode of administration | Oral administration |
| Safety profile | Desirable safety profile |
| Not associated with generalised immune suppression | |
| Compounds are non‐absorbable | |
| Spectrum of disease | Treatment of both the early and late stages of disease, including patients with inflammation without fibrosis |
| Can be used for prevention in patients with simple steatosis | |
| May be effective in patients with severe disease | |
| May be use for induction of remission as well as for maintenance | |
| Concomitant disorders | Can treat concomitant type 2 diabetes and hyperlipidaemia that affect the majority of patients with NASH |
| Long‐term use | Enables long‐term chronic use due to its desirable safety profile |
| Ease of administration | Easy to use |
| Dose | No absorption is required |
| A relatively low dose is sufficient to achieve a clinically meaningful effect | |
| Cost | Low cost |
| Adjuvant | Can serve as an adjuvant for other anti‐inflammatory and anti‐fibrotic drugs |
While the mechanism of NASH is complex, it is clear that two processes are involved in the pathogenesis of disease: inflammation and fibrogenesis. Although some of the drugs being developed for NASH are mainly anti‐fibrotics, others are commonly anti‐inflammatory. Oral immune therapy targets systemic inflammation, switching the systemic immune response from a pro‐ to an anti‐inflammatory state.7, 120, 121 In particular, oral therapy takes advantage of the inherent ability of the gut immune system to generate immune signals that are able to control the systemic immune response. This approach may have the advantage of affecting both the fibrotic and inflammatory mechanisms associated with liver disease. Some of the compounds described above have been suggested to possess possible anti‐fibrotic effects.93, 110 However, it remains to be determined whether the potent anti‐inflammatory effects described in animal models of NASH will translate into anti‐fibrotic effects of these compounds in humans.
Most of the drugs developed for NASH target a specific metabolic or inflammatory pathway. Some of the drugs under development target one molecule that is relevant to the disease process. However, it is clear that several pathways, in contrast to a single molecular pathway, underlie the pathogenesis of NASH. Oral immune therapy has the advantage of being ‘antigen‐ and molecular pathway‐independent’. Consequently, these compounds may provide broader ‘coverage’ of the inflammatory pathways relevant to NASH. Indeed, several oral immune therapy drugs have been shown to promote different subsets of Tregs that redirect the systemic immune system towards the anti‐inflammatory path, independent of a single molecular/inflammatory track.
Currently, numerous products developed for NASH are being tested in subpopulations of patients (e.g. patients with mild, moderate, or severe disease). Some of the drugs developed for NASH are also undergoing testing in cirrhotic patients. However, there is a need to develop therapeutics even for the early stages of NASH, especially in patients at risk of progression.122 Oral immune therapy compounds show potential for use in patients with all degrees of disease severity.
Drugs that have been tested for oral immune therapy show a relatively desirable safety profile; in particular, they are not associated with generalised immune suppression despite functioning as immune modulators. Most of these compounds are not absorbed and therefore exert an effect at the level of the gut immune system. Drugs under development for NASH are tested for relatively short periods of time, although it is anticipated that NASH will require long‐term treatment. The potential long‐term complications of several agents under development remain to be determined. However, oral immune therapy, due to its desirable safety profile, may be used for prevention (in patients with simple steatosis) and also in patients with end‐stage disease. It is likely that these oral compounds can be used to induce remission as well as long‐term maintenance.
The majority of patients with NASH suffer from concomitant disorders, including type 2 diabetes, hyperlipidaemia and atherosclerosis, all of which are part of the metabolic syndrome and are associated with chronic low levels of inflammation. As described above, some of the recently developed drugs for NASH may be associated with an exacerbation of these disorders. However, the data available for most oral immune therapeutic compounds suggest that they may be able to alleviate the concomitant disorders in NASH patients.
The process of oral administration and the lack of absorption make these compounds easy to use and well tolerated. Because no absorption is required and a relatively low dose is sufficient to achieve a clinically meaningful effect, these compounds are also expected to be relatively affordable for long‐term use.
Finally, it is expected that treatment for NASH, which is similar to that of hypertension, will require combination therapy consisting of several drugs that target different disease‐associated molecular pathways. Thus, oral immune therapy may serve as an adjuvant treatment for other anti‐inflammatory and anti‐fibrotic drugs. For example, pre‐clinical and clinical data already support an adjuvant effect in the gut for glycosphingolipids combined with anti‐CD3 or anti‐LPS antibodies.76, 118, 123
The future of using the gut for the treatment of NASH
Non‐alcoholic steatohepatitis is a complex disease, and novel treatments represent an unmet clinical need. Despite major efforts over the last decade, a final ‘HCV‐like’ therapy of ‘one pill for all’ is far from being realised. Targeting the gut immune system may provide a novel avenue for disease prevention, induction of remission, maintenance and development of adjuvant drugs for patients with NASH. Several of the compounds described above are already in phase IIB trials, and it is anticipated that in the next few years, considerable clinical data will facilitate the development of this new class of drugs for NASH.
Authorship
Guarantor of the article: Y. Ilan.
Author contribution: Y. Ilan has written the manuscript.
Acknowledgement
Declaration of personal interests: Ilan is a consultant for Immuron, Teva, Enzo Biochem, Protalix, Therapix, Nasvax and Natural Shield. As part of his consultation agreements YI owns options and shares in some of these companies.
Declaration of funding interests: This work was supported in part by a grant from The Roman‐Epstein Research Foundation (Y.I.).
The Handling Editor for this article was Professor Stephen Harrison, and this uncommissioned review was accepted for publication after full peer‐review.
References
- 1. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013; 10: 656–65. [DOI] [PubMed] [Google Scholar]
- 2. Noureddin M, Mato JM, Lu SC. Nonalcoholic fatty liver disease: update on pathogenesis, diagnosis, treatment and the role of S‐adenosylmethionine. Exp Biol Med 2015; 240: 809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis‐new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 2013; 10: 627–36. [DOI] [PubMed] [Google Scholar]
- 4. Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol 2015; 62(1 Suppl): S65–75. [DOI] [PubMed] [Google Scholar]
- 5. Milic S, Mikolasevic I, Krznaric‐Zrnic I, et al Nonalcoholic steatohepatitis: emerging targeted therapies to optimize treatment options. Drug Des Devel Ther 2015; 9: 4835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pearlman M, Loomba R. State of the art: treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol 2014; 30: 223–37. [DOI] [PubMed] [Google Scholar]
- 7. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013; 10: 330–44. [DOI] [PubMed] [Google Scholar]
- 8. Ratziu V, Bedossa P, Francque SM, et al Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2014; 12: 1724–30 e5. [DOI] [PubMed] [Google Scholar]
- 9. Ratziu V, Sheikh MY, Sanyal AJ, et al A phase 2, randomized, double‐blind, placebo‐controlled study of GS‐9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012; 55: 419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chachay VS, Macdonald GA, Martin JH, et al Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2014; 12: 2092–103 e1‐6. [DOI] [PubMed] [Google Scholar]
- 11. Sanyal AJ, Abdelmalek MF, Suzuki A, Cummings OW, Chojkier M, Group E‐AS . No significant effects of ethyl‐eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 2014; 147: 377–84 e1. [DOI] [PubMed] [Google Scholar]
- 12. Zelber‐Sagi S, Nitzan‐Kaluski D, Goldsmith R, et al Long term nutritional intake and the risk for non‐alcoholic fatty liver disease (NAFLD): a population based study. J Hepatol 2007; 47: 711–7. [DOI] [PubMed] [Google Scholar]
- 13. Sanyal AJ, Chalasani N, Kowdley KV, et al Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010; 362: 1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gastaldelli A, Harrison SA, Belfort‐Aguilar R, et al Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 2009; 50: 1087–93. [DOI] [PubMed] [Google Scholar]
- 15. Ferrara A, Lewis JD, Quesenberry CP Jr, et al Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care 2011; 34: 923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015; 385: 956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanyal AJ, Neuschwander‐Tetri BA, Tonascia J. End points must be clinically meaningful for drug development in nonalcoholic fatty liver disease. Gastroenterology 2016; 150: 11–3. [DOI] [PubMed] [Google Scholar]
- 18. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta‐analysis. JAMA 2007; 297: 842–57. [DOI] [PubMed] [Google Scholar]
- 19. Kurth T, Kase CS, Schurks M, Tzourio C, Buring JE. Migraine and risk of haemorrhagic stroke in women: prospective cohort study. BMJ 2010; 341: c3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein EA, Thompson IM Jr, Tangen CM, et al Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011; 306: 1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes 2005; 2008(32 Suppl. 7): S52–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011; 29: 415–45. [DOI] [PubMed] [Google Scholar]
- 23. Sztajnkrycer MJ, Bond GR. Chronic acetaminophen overdosing in children: risk assessment and management. Curr Opin Pediatr 2001; 13: 177–82. [DOI] [PubMed] [Google Scholar]
- 24. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013; 59: 583–94. [DOI] [PubMed] [Google Scholar]
- 25. Bohinc BN, Diehl AM. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis 2012; 16: 549–65. [DOI] [PubMed] [Google Scholar]
- 26. Kaur S, Venktaraman G, Jain M, Senapati S, Garg PK, Batra SK. Recent trends in antibody‐based oncologic imaging. Cancer Lett 2012; 315: 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Du Plessis J, van Pelt J, Korf H, et al Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology 2015; 149: 635–48 e14. [DOI] [PubMed] [Google Scholar]
- 28. Yu JG, Javorschi S, Hevener AL, et al The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 2002; 51: 2968–74. [DOI] [PubMed] [Google Scholar]
- 29. Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat‐derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 2003; 112: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 2006; 116: 1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watanabe M, Houten SM, Wang L, et al Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP‐1c. J Clin Invest 2004; 113: 1408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wagner M, Zollner G, Trauner M. Nuclear bile acid receptor farnesoid X receptor meets nuclear factor‐kappaB: new insights into hepatic inflammation. Hepatology 2008; 48: 1383–6. [DOI] [PubMed] [Google Scholar]
- 33. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008; 48: 1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pacana T, Sanyal AJ. Recent advances in understanding/management of non‐alcoholic steatohepatitis. F1000prime Rep 2015; 7: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cariou B, Zair Y, Staels B, Bruckert E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011; 34: 2008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin X, Xie X, Fan Y, et al Peroxisome proliferator‐activated receptor‐delta induces insulin‐induced gene‐1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008; 48: 432–41. [DOI] [PubMed] [Google Scholar]
- 37. Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest 2006; 116: 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bojic LA, Huff MW. Peroxisome proliferator‐activated receptor delta: a multifaceted metabolic player. Curr Opin Lipidol 2013; 24: 171–7. [DOI] [PubMed] [Google Scholar]
- 39. Staels B, Rubenstrunk A, Noel B, et al Hepatoprotective effects of the dual peroxisome proliferator‐activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013; 58: 1941–52. [DOI] [PubMed] [Google Scholar]
- 40. Ratziu V, Harrison S, Francque S, et al Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐alpha and ‐delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016; 150: 1147–59. [DOI] [PubMed] [Google Scholar]
- 41. Armstrong MJ, Houlihan DD, Rowe IA, et al Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta‐analysis of the LEAD program. Aliment Pharmacol Ther 2013; 37: 234–42. [DOI] [PubMed] [Google Scholar]
- 42. Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin‐4, a glucagon‐like protein‐1 (GLP‐1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006; 43: 173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gupta NA, Mells J, Dunham RM, et al Glucagon‐like peptide‐1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010; 51: 1584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Armstrong MJ, Hull D, Guo K, et al Glucagon‐like peptide 1 decreases lipotoxicity in non‐alcoholic steatohepatitis. J Hepatol 2015; 64: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soden JS, Devereaux MW, Haas JE, et al Subcutaneous vitamin E ameliorates liver injury in an in vivo model of steatocholestasis. Hepatology 2007; 46: 485–95. [DOI] [PubMed] [Google Scholar]
- 46. Morante M, Sandoval J, Gomez‐Cabrera MC, et al Vitamin E deficiency induces liver nuclear factor‐kappaB DNA‐binding activity and changes in related genes. Free Radic Res 2005; 39: 1127–38. [DOI] [PubMed] [Google Scholar]
- 47. Azzi A, Gysin R, Kempna P, et al Vitamin E mediates cell signaling and regulation of gene expression. Ann N Y Acad Sci 2004; 1031: 86–95. [DOI] [PubMed] [Google Scholar]
- 48. Friedman S, Sanyal A, Goodman Z, et al Efficacy and safety study of cenicriviroc for the treatment of non‐alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials 2016; 47: 356–65. [DOI] [PubMed] [Google Scholar]
- 49. Lefebvre E, Gottwald M, Lasseter K, et al Pharmacokinetics, safety, and CCR2/CCR5 antagonist activity of cenicriviroc in participants with mild or moderate hepatic impairment. Clin Transl Sci 2016; 9: 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ratziu V. Pharmacological agents for NASH. Nat Rev Gastroenterol Hepatol 2013; 10: 676–85. [DOI] [PubMed] [Google Scholar]
- 51. Kral JG, Thung SN, Biron S, et al Effects of surgical treatment of the metabolic syndrome on liver fibrosis and cirrhosis. Surgery 2004; 135: 48–58. [DOI] [PubMed] [Google Scholar]
- 52. Dixon JB, Bhathal PS, Hughes NR, O'Brien PE. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 2004; 39: 1647–54. [DOI] [PubMed] [Google Scholar]
- 53. Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444: 860–7. [DOI] [PubMed] [Google Scholar]
- 54. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 2008; 8: 923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moschen AR, Molnar C, Geiger S, et al Anti‐inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 2010; 59: 1259–64. [DOI] [PubMed] [Google Scholar]
- 56. Federico A, Dallio M, Godos J, Loguercio C, Salomone F. Targeting gut‐liver axis for the treatment of nonalcoholic steatohepatitis: translational and clinical evidence. Transl Res 2016; 167: 116–24. [DOI] [PubMed] [Google Scholar]
- 57. Mehal WZ. The Gordian knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol 2013; 10: 637–44. [DOI] [PubMed] [Google Scholar]
- 58. Ilan Y. Immune therapy for nonalcoholic steatohepatitis: are we there yet? J Clin Gastroenterol 2013; 47: 298–307. [DOI] [PubMed] [Google Scholar]
- 59. Scarpellini E, Lupo M, Iegri C, Gasbarrini A, de Santis A, Tack J. Intestinal permeability in non‐alcoholic fatty liver disease: the gut‐liver axis. Rev Recent Clin Trials 2014; 9: 141–7. [DOI] [PubMed] [Google Scholar]
- 60. Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 2012; 18: 2609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miura K, Ohnishi H. Role of gut microbiota and toll‐like receptors in nonalcoholic fatty liver disease. World J Gastroenterol 2014; 20: 7381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Viggiano D, Ianiro G, Vanella G, et al Gut barrier in health and disease: focus on childhood. Eur Rev Med Pharmacol Sci 2015; 19: 1077–85. [PubMed] [Google Scholar]
- 63. Ilan Y. Oral immune therapy: targeting the systemic immune system via the gut immune system for the treatment of inflammatory bowel disease. Clin Transl Immunol 2016; 5: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev 2014; 260: 86–101. [DOI] [PubMed] [Google Scholar]
- 65. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology 2015; 150: 340–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev 2011; 241: 241–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ilan Y. Oral tolerance: can we make it work? Hum Immunol 2009; 70: 768–76. [DOI] [PubMed] [Google Scholar]
- 68. Gotsman I, Beinart R, Alper R, Rabbani E, Engelhardt D, Ilan Y. Induction of oral tolerance towards hepatitis B envelope antigens in a murine model. Antiviral Res 2000; 48: 17–26. [DOI] [PubMed] [Google Scholar]
- 69. Ilan Y, Weksler‐Zangen S, Ben‐Horin S, et al Treatment of experimental colitis by oral tolerance induction: a central role for suppressor lymphocytes. Am J Gastroenterol 2000; 95: 966–73. [DOI] [PubMed] [Google Scholar]
- 70. Kolker O, Klein A, Alper R, et al Early expression of interferon gamma following oral antigen administration is associated with peripheral tolerance induction. Microbes Infect 2003; 5: 807–13. [DOI] [PubMed] [Google Scholar]
- 71. Trop S, Samsonov D, Gotsman I, Alper R, Diment J, Ilan Y. Liver‐associated lymphocytes expressing NK1.1 are essential for oral immune tolerance induction in a murine model. Hepatology 1999; 29: 746–55. [DOI] [PubMed] [Google Scholar]
- 72. Israeli E, Zigmond E, Lalazar G, et al Oral mixture of autologous colon‐extracted proteins for the Crohn's disease: a double‐blind trial. World J Gastroenterol 2015; 21: 5685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Israeli E, Ilan Y. Oral administration of Alequel, a mixture of autologous colon‐extracted proteins for the treatment of Crohn's disease. Therap Adv Gastroenterol 2010; 3: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Margalit M, Israeli E, Shibolet O, et al A double‐blind clinical trial for treatment of Crohn's disease by oral administration of Alequel, a mixture of autologous colon‐extracted proteins: a patient‐tailored approach. Am J Gastroenterol 2006; 101: 561–8. [DOI] [PubMed] [Google Scholar]
- 75. Israeli E, Safadi R, Melhem A, et al Induction of oral immune regulation towards liver‐extracted proteins for treatment of chronic HBV and HCV hepatitis: results of a phase I clinical trial. Liver Int 2004; 24: 295–307. [DOI] [PubMed] [Google Scholar]
- 76. Ilan Y, Maron R, Tukpah AM, et al Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci U S A 2010; 107: 9765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Elinav E, Pappo O, Sklair‐Levy M, et al Amelioration of non‐alcoholic steatohepatitis and glucose intolerance in ob/ob mice by oral immune regulation towards liver‐extracted proteins is associated with elevated intrahepatic NKT lymphocytes and serum IL‐10 levels. J Pathol 2006; 208: 74–81. [DOI] [PubMed] [Google Scholar]
- 78. Scott LJ. Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs 2014; 74: 1379–410. [DOI] [PubMed] [Google Scholar]
- 79. Olivieri I, D'Angelo S, Palazzi C, Padula A. Advances in the management of psoriatic arthritis. Nat Rev Rheumatol 2014; 10: 531–42. [DOI] [PubMed] [Google Scholar]
- 80. Armuzzi A, Lionetti P, Blandizzi C, et al Anti‐TNF agents as therapeutic choice in immune‐mediated inflammatory diseases: focus on adalimumab. Int J Immunopathol Pharmacol 2014; 27(1 Suppl.): 11–32. [DOI] [PubMed] [Google Scholar]
- 81. Shaaltiel Y BYaA, Shabbat Y, Zolotarov L, et al A novel method for anti‐TNF based‐oral immunotherapy: oral administration of a plant cell‐expressed recombinant anti‐TNF fusion protein for treating of Crohn's disease. Gastroenterology 2014; 146: S 901 (A 2029). [Google Scholar]
- 82. Shaaltiel Y, Ya'acov AB, Gingis‐Velitski S, Almon E, Aviezer D, Ilan Y. A novel method for anti‐TNF based‐oral immunotherapy: oral administration of a plant cell‐expressed recombinant anti‐TNF fusion protein for treating of fatty liver disease. Hepatology 2014; 60: 663A. [Google Scholar]
- 83. Almon E, Khoury T, Drori A, et al Oral administration of a recombinant anti‐TNF fusion protein is biologically active in the gut promoting regulatory T cells: results of a phase I clinical trial using a novel oral anti‐TNF alpha‐based therapy. Gastroenterology 2016; 150: S804 (A1881). [DOI] [PubMed] [Google Scholar]
- 84. Anjani K, Lhomme M, Sokolovska N, et al Circulating phospholipid profiling identifies portal contribution to NASH signature in obesity. J Hepatol 2015; 62: 905–12. [DOI] [PubMed] [Google Scholar]
- 85. Radin NS, Inokuchi J. Glucosphingolipids as sites of action in the chemotherapy of cancer. Biochem Pharmacol 1988; 37: 2879–86. [DOI] [PubMed] [Google Scholar]
- 86. Margalit M, Ghazala SA, Alper R, et al Glucocerebroside treatment ameliorates ConA hepatitis by inhibition of NKT lymphocytes. Am J Physiol Gastrointest Liver Physiol 2005; 289: G917–25. [DOI] [PubMed] [Google Scholar]
- 87. Ilan Y, Ohana M, Pappo O, et al Alleviation of acute and chronic graft‐versus‐host disease in a murine model is associated with glucocerebroside‐enhanced natural killer T lymphocyte plasticity. Transplantation 2007; 83: 458–67. [DOI] [PubMed] [Google Scholar]
- 88. Zigmond E, Preston S, Pappo O, et al Beta‐glucosylceramide: a novel method for enhancement of natural killer T lymphoycte plasticity in murine models of immune‐mediated disorders. Gut 2007; 56: 82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ilan Y, Elstein D, Zimran A. Glucocerebroside: an evolutionary advantage for patients with Gaucher disease and a new immunomodulatory agent. Immunol Cell Biol 2009; 87: 514–24. [DOI] [PubMed] [Google Scholar]
- 90. Margalit M, Shalev Z, Pappo O, et al Glucocerebroside ameliorates the metabolic syndrome in OB/OB mice. J Pharmacol Exp Ther 2006; 319: 105–10. [DOI] [PubMed] [Google Scholar]
- 91. Zigmond E, Tayer‐Shifman O, Lalazar G, et al Beta‐glycosphingolipids ameliorated non‐alcoholic steatohepatitis in the Psammomys obesus model. J Inflamm Res 2014; 7: 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zigmond E, Zangen SW, Pappo O, et al Beta‐glycosphingolipids improve glucose intolerance and hepatic steatosis of the Cohen diabetic rat. Am J Physiol Endocrinol Metab 2009; 296: E72–8. [DOI] [PubMed] [Google Scholar]
- 93. Margalit M, Safadi R, Zigmond E, et al Amelioration of hepatic fibrosis via beta‐glucoslyceramide mediated immune modulation is associated with altered CD8 and NKT lymphoycte distribution. Hepatology 2005; 42: 1046A. [DOI] [PubMed] [Google Scholar]
- 94. Lalazar GZE, Zangen SW, Pappo O, et al Treatment of insulin resistance and non‐alcoholic steatohepatitis by administration of beta glucosylceramide controlled trial. Hepatology 2009; 50(Suppl 4): 200A. [Google Scholar]
- 95. Friedman M, Brandon DL. Nutritional and health benefits of soy proteins. J Agric Food Chem 2001; 49: 1069–86. [DOI] [PubMed] [Google Scholar]
- 96. Erdman JW Jr. Control of serum lipids with soy protein. N Engl J Med 1995; 333: 313–5. [DOI] [PubMed] [Google Scholar]
- 97. Hermansen K, Sondergaard M, Hoie L, Carstensen M, Brock B. Beneficial effects of a soy‐based dietary supplement on lipid levels and cardiovascular risk markers in type 2 diabetic subjects. Diabetes Care 2001; 24: 228–33. [DOI] [PubMed] [Google Scholar]
- 98. Zhang HM, Chen SW, Zhang LS, Feng XF. The effects of soy isoflavone on insulin sensitivity and adipocytokines in insulin resistant rats administered with high‐fat diet. Nat Prod Res 2008; 22: 1637–49. [DOI] [PubMed] [Google Scholar]
- 99. Ustundag B, Bahcecioglu IH, Sahin K, et al Protective effect of soy isoflavones and activity levels of plasma paraoxonase and arylesterase in the experimental nonalcoholic steatohepatitis model. Dig Dis Sci 2007; 52: 2006–14. [DOI] [PubMed] [Google Scholar]
- 100. Nomura H, Kashiwagi S, Hayashi J, Kajiyama W, Tani S, Goto M. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn J Med 1988; 27: 142–9. [DOI] [PubMed] [Google Scholar]
- 101. Yang HY, Tzeng YH, Chai CY, et al Soy protein retards the progression of non‐alcoholic steatohepatitis via improvement of insulin resistance and steatosis. Nutrition 2011; 27: 943–8. [DOI] [PubMed] [Google Scholar]
- 102. Khoury T, Ben Ya'acov A, Shabat Y, Zolotarovya L, Snir R, Ilan Y. Altered distribution of regulatory lymphocytes by oral administration of soy‐extracts exerts a hepatoprotective effect alleviating immune mediated liver injury, non‐alcoholic steatohepatitis and insulin resistance. World J Gastroenterol 2015; 21: 7443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Waters OR, Lawrance IC. Understanding the use of immunosuppressive agents in the clinical management of IBD. Curr Drug Targets 2011; 12: 1364–71. [DOI] [PubMed] [Google Scholar]
- 104. Gilissen LP, Wong DR, Engels LG, et al Therapeutic drug monitoring of thiopurine metabolites in adult thiopurine tolerant IBD patients on maintenance therapy. J Crohns Colitis 2012; 6: 698–707. [DOI] [PubMed] [Google Scholar]
- 105. Hanauer SB, Korelitz BI, Rutgeerts P, et al Postoperative maintenance of Crohn's disease remission with 6‐mercaptopurine, mesalamine, or placebo: a 2‐year trial. Gastroenterology 2004; 127: 723–9. [DOI] [PubMed] [Google Scholar]
- 106. Mate‐Jimenez J, Hermida C, Cantero‐Perona J, Moreno‐Otero R. 6‐mercaptopurine or methotrexate added to prednisone induces and maintains remission in steroid‐dependent inflammatory bowel disease. Eur J Gastroenterol Hepatol 2000; 12: 1227–33. [DOI] [PubMed] [Google Scholar]
- 107. Markowitz J, Grancher K, Kohn N, Lesser M, Daum F. A multicenter trial of 6‐mercaptopurine and prednisone in children with newly diagnosed Crohn's disease. Gastroenterology 2000; 119: 895–902. [DOI] [PubMed] [Google Scholar]
- 108. Israeli E, Goldin E, Fishman S, et al Oral administration of non‐absorbable delayed release 6‐mercaptopurine is locally active in the gut, exerts a systemic immune effect and alleviates Crohn's disease with low rate of side effects: results of double blind Phase II clinical trial. Clin Exp Immunol 2015; 181: 362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Adar T, Ben Ya'acov A, Lalazar G, et al Oral administration of immunoglobulin G‐enhanced colostrum alleviates insulin resistance and liver injury and is associated with alterations in natural killer T cells. Clin Exp Immunol 2012; 167: 252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Abd Alrahem M, Zolotarov L, Shabat Y, Khatib A, Mizrahi M. Alleviation of liver damage and hepatic fibrosis by oral administration of Imm 124 colostrums in Carbon tetrachloride (CCl4) model is mediated by decrease of hepatic F4/80 macrophages activation. Hepatology 2013; 58(Suppl.): 588A. [Google Scholar]
- 111. Mizrahi MLG, Shabat Y, Adar T, Ben Ya'acov A, Ilan Y. Alleviation of insulin resistance and liver damage by oral administration of etec colostrums is mediated by increased GLP‐1, adiponectin serum levels and tregs: results of a phase I/II clinical trial in NASH. Hepatology 2010; 52: 163A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chatenoud L. CD3‐specific antibody‐induced active tolerance: from bench to bedside. Nat Rev Immunol 2003; 3: 123–32. [DOI] [PubMed] [Google Scholar]
- 113. Ochi H, Abraham M, Ishikawa H, et al New immunosuppressive approaches: oral administration of CD3‐specific antibody to treat autoimmunity. J Neurol Sci 2008; 274: 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of autoimmune diabetes by oral administration of anti‐CD3 monoclonal antibody. Diabetes 2007; 56: 2103–9. [DOI] [PubMed] [Google Scholar]
- 115. Ochi H, Abraham M, Ishikawa H, et al Oral CD3‐specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25‐ LAP+ T cells. Nat Med 2006; 12: 627–35. [DOI] [PubMed] [Google Scholar]
- 116. Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti‐CD3: inducible CD4+CD25‐LAP+ regulatory T cells control the expansion of IL‐17+ follicular helper T cells. Lupus 2009; 18: 586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wu HY, Quintana FJ, Weiner HL. Nasal anti‐CD3 antibody ameliorates lupus by inducing an IL‐10‐secreting CD4+ CD25‐ LAP+ regulatory T cell and is associated with down‐regulation of IL‐17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J Immunol 2008; 181: 6038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ilan Y, Zigmond E, Lalazar G, et al Oral administration of OKT3 monoclonal antibody to human subjects induces a dose‐dependent immunologic effect in T cells and dendritic cells. J Clin Immunol 2010; 30: 167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lalazar G, Mizrahi M, Turgeman I, et al Oral Administration of OKT3 MAb to patients with NASH, promotes regulatory T‐cell induction, and alleviates insulin resistance: results of a phase IIa blinded placebo‐controlled trial. J Clin Immunol 2015; 35: 399–407. [DOI] [PubMed] [Google Scholar]
- 120. Eckert C, Klein N, Kornek M, Lukacs‐Kornek V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front Immunol 2015; 6: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol 2015; 62: 720–33. [DOI] [PubMed] [Google Scholar]
- 122. Sanyal AJ, Friedman SL, McCullough AJ, et al Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases‐U.S. Food and Drug Administration Joint Workshop. Hepatology 2015; 61: 1392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mizrahi M, Shabat Y, Ben Ya'acov A, et al Alleviation of insulin resistance and liver damage by oral administration of Imm124‐E is mediated by increased Tregs and associated with increased serum GLP‐1 and adiponectin: results of a phase I/II clinical trial in NASH. J Inflamm Res 2012; 5: 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Shaaltiel Y, Ya'acov AB, SGingis‐Velitski S, Almon E, Aviezer D, Ilan Y. A novel method for anti‐TNF based‐oral immunotherapy: oral administration of a plant cell‐expressed recombinant anti‐TNF fusion protein for treating of fatty liver disease. Hepatology 2014; 60: 663. [Google Scholar]
- 125. Zigmond E, Pappo O, Zangen S, et al Treatment of non‐alcoholic steatohepatitis by B‐glucosylceramide: a phase I/II clinical study. Hepatology 2006; 44: 180A. [Google Scholar]