

Gupta et al. use single-channel electrophysiology to investigate the gating mechanism of acetylcholine receptor ion channels. They propose that channel opening starts at the M2–M3 linker and ligand-binding sites and proceeds through four brief intermediate conformations before ending with the collapse of a gate bubble.
Abstract
Nicotinic acetylcholine receptors are allosteric proteins that generate membrane currents by isomerizing (“gating”) between resting and active conformations under the influence of neurotransmitters. Here, to explore the mechanisms that link the transmitter-binding sites (TBSs) with the distant gate, we use mutant cycle analyses to measure coupling between residue pairs, phi value analyses to sequence domain rearrangements, and current simulations to reproduce a microsecond shut component (“flip”) apparent in single-channel recordings. Significant interactions between amino acids separated by >15 Å are rare; an exception is between the αM2–M3 linkers and the TBSs that are ∼30 Å apart. Linker residues also make significant, local interactions within and between subunits. Phi value analyses indicate that without agonists, the linker is the first region in the protein to reach the gating transition state. Together, the phi pattern and flip component suggest that a complete, resting↔active allosteric transition involves passage through four brief intermediate states, with brief shut events arising from sojourns in all or a subset. We derive energy landscapes for gating with and without agonists, and propose a structure-based model in which resting→active starts with spontaneous rearrangements of the M2–M3 linkers and TBSs. These conformational changes stabilize a twisted extracellular domain to promote transmembrane helix tilting, gate dilation, and the formation of a “bubble” that collapses to initiate ion conduction. The energy landscapes suggest that twisting is the most energetically unfavorable step in the resting→active conformational change and that the rate-limiting step in the reverse process is bubble formation.
Introduction
Neuromuscular acetylcholine (ACh) receptors (AChRs) have two transmitter-binding sites (TBSs) in the extracellular domain (ECD) and a gate in the transmembrane domain (TMD). AChRs activate constitutively, but agonists bind with a higher affinity to the active-O(pen) versus resting-C(losed) conformation to increase the probability of being open (PO) over the basal level (Monod et al., 1965; Karlin, 1967; Jackson, 1989; Auerbach, 2012). The complete C↔O allosteric transition (“gating”) is global, so rearrangements at the TBSs (that undergird affinity) and the gate (that undergird conductance) are linked.
AChRs have a molecular mass of ∼300 kD and share a common architecture with other pentameric ligand-gated ion channels (pLGICs; Fig. 1 A, left; Miyazawa et al., 1999; Sine, 2012; Taly et al., 2014; Cecchini and Changeux, 2015; Jaiteh et al., 2016). The ECD is mostly β-barrel (β1–10, in each subunit) with each TBS located midlevel at a subunit interface. The TMD is mostly α-helix (M1–4) with a hydrophobic gate located near the equator and formed by M2 from all five subunits. X-ray structures of other pLGICs show that in C→O there is a rotation between these two domains, a compaction of the ECD, a rearrangement of TMD helices that includes a radial tilt of M2 and straightening of M1, and dilation of the gate (Hibbs and Gouaux, 2011; Sauguet et al., 2013, 2014; Althoff et al., 2014). In AChRs, neurotransmitter affinity is determined mainly by a group of aromatic residues in loops at the TBS.
Figure 1.

Structures. (A, left) Torpedo AChR (PDB accession code 2BG9): α subunit, tan; γ subunit, white. The TBS is ∼50 Å from the gate. (right) AChR α subunit diliganded gating phi values mapped onto Caenorhabditis elegans GluCl (PDB accession code 3RIF); colors assigned by statistical criteria (Fig. 6 A; Purohit et al., 2013). Amino acids at the TBS and in αM2M3 have phi ∼1 (purple) and are separated by a domain of phi ∼0.8 residues. For clarity, some amino acids were removed at the ECD–TMD interface and M3. (B, top) Ligand-binding site of the Lymnaea stagnalis ACh-binding protein (PDB accession code 3WIP; AChR numbers). In adult AChRs, affinity is mainly determined by αY190, αY198, and αW149 (green), but in fetal AChRs, αY93 and γW55 also contribute (yellow). (bottom) ECD–TMD interface of human α4β2 AChRs (PDB accession code 5KXI). (inset) In GluCl, the M2 helix is displaced upward relative to M1 in O versus C (PDB accession codes 3RIF and 4TNV). (C) M2M3 sequence alignments. *, αP265 in mouse muscle AChRs. Highlighted residues are loop.
Actions at the ECD–TMD interface are important in the gating isomerization, where loops connect β-strands, the ECD–TMD, and the M2–M3 helices (M2M3; Fig. 1 B, bottom; Bouzat, 2012; Bertozzi et al., 2016; Morales-Perez et al., 2016). In mouse muscle AChRs, mutations of αM2M3 residues influence substantially the gating equilibrium constant and PO (Grosman et al., 2000a; Bafna et al., 2008). Unnatural amino acid substitutions of a conserved M2M3 proline in 5HT3A receptors led to the proposal that a cis-trans isomerization here opens the pore (Lummis et al., 2005), and simulations of other pLGICs support the idea that this proline is a “handle” that pulls open M2 (Zhu and Hummer, 2010).
There have been several suggestions regarding the mechanism of TBS–gate communication. In one, the TBS and the β1–β2 loop (loop2) at the base of the ECD are linked mechanically (by a rigid-body motion), so that agonist occupancy perturbs the β2 strand, then loop2, M2M3, the top of M2 (“pin-into-socket”) and the gate (Miyazawa et al., 2003; Kash et al., 2004; Lee et al., 2008). In another, agonist occupancy perturbs loop C and the β10 strand, then an interfacial salt bridge near the base of β10, loop2, M2, and the gate (Lee and Sine, 2005). A third proposal derives from phi values, which provide a snapshot of the relative free energy of an amino acid at the gating transition state (TS) on a scale from 1 (O-like) to 0 (C-like; see Materials and methods). In AChRs, diliganded gating α subunit phi values decrease in steps between the TBS and the gate (Fig. 1 A, right), which led to the proposal that in C→O the protein passes through a series of brief intermediate states, as a conformational cascade (Grosman et al., 2000b; Auerbach, 2005). A similar phi pattern has been observed in CFTR (Sorum et al., 2015). Structures of GLIC having an O-like ECD and a C-like TMD (Prevost et al., 2012; Sauguet et al., 2014) and an approximate microsecond shut component in high-resolution electrophysiology recordings (“flip” or “primed”; Lape et al., 2008; Mukhtasimova et al., 2016) are more-direct evidence for an intermediate state or states in pLGIC gating.
Basal activity of WT receptors is nearly zero and neurotransmitter application causes rapid and nearly complete activation, so it is understandable that in all of the above proposals the opening process starts with a perturbation at the TBS that propagates to the gate. However, some observations challenge this orthodoxy. WT AChRs expressed in muscle cells open and close without agonists (Jackson, 1984, 1986). The unliganded PO of WT AChRs is small, but many mutations throughout the protein can increase this value substantially (as much as an agonist), apparently by making the same energy changes and with approximately the same phi values as when agonists are present (Purohit and Auerbach, 2009; Jadey et al., 2011). Furthermore, most mutations of TBS residues have little or no effect on the unliganded gating equilibrium constant, so this region is not special when it does not contain an agonist. Removal of loop C (β9–β10) or a key, complementary tryptophan on β2 does not affect unliganded gating (Purohit and Auerbach, 2013; Nayak and Auerbach, 2015). Finally, the top-down gradient in phi is not perfectly uniform. With agonists, some residues in αM2M3 have phi values as high as in the TBS (Bafna et al., 2008), and without agonists, gating phi values of some TBS residues become smaller and similar to those in the rest of the ECD (Purohit and Auerbach, 2010). These results question the assumptions that the AChR opening process starts at the TBS and that long-range, TBS–gate communication occurs via a discrete, mechanical pathway.
To investigate the AChR gating process, we used double mutant cycle analysis (MCA) of single-channel currents to measure interaction energies between αM2M3 with the TBS and proximal loops and linear free energy relationships (phi values) to sequence amino acid gating rearrangements. The phi pattern was used to compute energy landscapes of the gating TS ensemble (TSE), and simulations were used to reproduce the approximate microsecond flip/primed shut component. We propose a modified version of the conformational cascade model, in which C→O starts at αM2M3 and the TBS, to promote a twist of the ECD, a tilt of the TMD helices, a dilation of the gate, and the formation of a gate “bubble” that collapses to initiate ion conduction.
The paper is organized as follows. In the Materials and methods, in addition to describing experimental protocols, we provide summaries of the cyclic activation mechanism and phi value analysis. After a brief consideration of structures, we describe remote and local interactions with residues in the αM2M3 linker. We then show phi analyses that indicate αM2M3 is the first region of the protein to reach the TSE in unliganded gating and use simulations to show that flip reflects sojourns in the TSE mainly associated with gate bubble formation. We describe methods to compute a TSE energy profile from phi and combine phi and flip measurements to explore energy landscapes and structure-based models for AChR gating with and without agonists. Animations based on the models are shown at different time scales in Video 1.
Materials and methods
Mutagenesis, expression, and electrophysiology
Mutations were made to mouse muscle AChR cDNAs using the QuikChange site-directed mutagenesis kit (Agilent Technologies) and were verified by nucleotide sequencing. HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) fetal bovine serum and 1% (vol/vol) penicillin-streptomycin, pH 7.4. The cell cultures were transfected with a mixture of cDNAs encoding WT or mutant subunits by calcium phosphate precipitation, by incubating for ∼20 h with 3.5–5.0 µg DNA per 35-mm culture dish. The AChR subunit cDNAs were added at a ratio of 2:1:1:1 (α/β/δ/ε or γ); cDNA for GFP (0.3 µg/µl) was also added to the mixture to facilitate the selection of AChR-expressing cells. The media was changed, and electrophysiology recordings commenced within 16–18 h.
Single-channel currents were recorded in the cell-attached patch configuration at 23°C. Pipettes were pulled from borosilicate capillaries and coated with Sylgard (Dow Corning). For the coupling measurements, an agonist concentration at least five times higher than the resting equilibrium dissociation constant was added to the pipette (≥1 mM ACh or ≥20 mM choline). To reduce channel block by the agonist, the pipette potential was −100 mV, which corresponds to a membrane potential (Vm) of ∼70 mV. The bath and pipette solutions were Dulbecco’s phosphate-buffered saline containing (mM) 137 NaCl, 0.9 CaCl2, 2.7 KCl, 1.5 KH2PO4, 0.5 MgCl2, and 8.1 Na2HPO4, pH 7.4.
Data acquisition and kinetic analyses were performed by using QUB software (Nicolai and Sachs, 2013). Currents were low-pass filtered at 20 kHz and digitized at a sampling frequency of 50 kHz. At high agonist concentrations, openings occurred in clusters that represent C↔O gating activity of individual AChRs with the long, silent intervals between clusters reflecting periods when all of the AChRs in the patch were desensitized. Clusters were selected by eye, and the intra-cluster currents were idealized into noise-free intervals after digitally low-pass filtering at 12 kHz and by using the segmental k-means algorithm. Rate constants were estimated from the idealized interval durations by using a maximum log-likelihood algorithm and a two state, C↔O model after imposing an approximate dead time correction of 25 µs. When necessary, an additional shut state was added to the model and connected to O to eliminate from the rate constant estimation gaps associated with sojourns in an approximate millisecond, apparently desensitized state (Salamone et al., 1999; Elenes et al., 2006). Without the inclusion of this state, the opening rate and gating equilibrium constants would be underestimated. The diliganded gating equilibrium constant was calculated as the ratio of the gating rate constants. Our time resolution was such that the approximate microsecond shut intermediate component (flip/primed) was not detected but rather was incorporated into C↔O TSE, represented by the arrow.
Methods for analyzing modal activity are described in detail elsewhere (Vij et al., 2015). In brief, clusters were selected by eye, and PO was calculated from intra-cluster interval durations. In these experiments, which did not involve rate constant estimation, idealization was after low-pass filtering at 5 kHz and by using a half-amplitude criterion. A k-means clustering algorithm was used to determine the number of PO populations and to segregate the clusters. An adult WT background was used for αP265A and αP265A+δW57A (100 µM ACh, Vm = −100 mV). For the loop C mutants, coupling was measured for each mode using [ACh] = 100 mM (to saturate the TBS) and Vm = 70 mV (to reduce channel block). To prolong the open duration and facilitate rate constant estimation, a background mutation was added to these constructs, βT456F for αY190F or αC418Y for αY198A. Without the αP265A mutation, there were no modes with either the backgrounds or the loop C mutations on these backgrounds.
Activation cycle
AChRs activate by a closed allosteric cycle (no external energy) that involves C↔O gating with and without bound agonists and agonist binding to both C and O conformations (Fig. S1). In adult mouse AChRs, the two binding sites are equivalent and independent for ACh and choline (Nayak et al., 2014), so
| (1) |
E2 is the diliganded gating equilibrium constant, E0 is the unliganded gating equilibrium constant, and Kd/Jd is the ratio of equilibrium dissociation constants to C versus O (the coupling constant). In WT adult mouse AChRs at −100 mV (23°C), E0 is 7.4 × 10−7, KdACh = 170 µM, E2ACh = 25, and the coupling constant is ∼5,800 (Auerbach, 2010). The log of an equilibrium constant is proportional to the free energy difference between the end states. Taking the log of Eq. 1,
| (2) |
where ΔG2 is the diliganded O minus C energy difference, ΔG0 is the unliganded O minus C energy difference, and ΔΔGcc is the log of the coupling constant and is the per-site difference in agonist binding free energy (bound minus free) to O versus C. The presence of intermediate states between C and O does not affect these energy differences. In WT adult mouse AChRs, at −100 mV, ΔG0 is 8.3 kcal/mol and ΔΔGcc is −5.1 kcal/mol for ACh and −3.3 kcal/mol for choline. For the TBS residues we studied, the effect of a mutation on ΔΔGcc was much larger than on ΔG0, so the pairwise interaction values involving these amino acids are mainly with regard to ΔΔGcc.
Protein engineering
In about half of the experiments, a background mutation was added to adjust E0 so that the emergent interval durations with agonists were in a readily measurable range (0.1–10 ms; Jadey et al., 2011). The background mutations only influenced E0 (ΔG0) and had no effect on the coupling constant. ACh and choline provide different energies from the low to high affinity change (ΔΔGcc values), which when added to the engineered ΔG0 produced diliganded gating equilibrium constants (ΔG2 values) that placed PO into the optimal range for analysis (Eq. 2). By correcting the apparent rate constants for the effects of depolarization and the background mutation or mutations, gating equilibrium and rate constants were normalized to a standard condition (WT, −100 mV, 23°C). This approach eliminates many of the technical difficulties associated with idealization and modeling of events close to the time resolution of the patch clamp and extends greatly the range over which gating constants can be estimated. The backgrounds are shown in Tables S1 and S2.
Coupling measurements
The main method we used to estimate the interaction between side chains is MCA. A pair of residues was selected and mutated. The fold change in the diliganded gating equilibrium constant (E2mut/E2WT) was measured separately for each mutant. Multiplying the natural logarithm of the fold change by −RT (where R is the gas constant and T is the absolute temperature; RT = 0.59 at 23°C) gives the effect of the mutation as a free energy change (kcal/mol). All MCA experiments were with fully liganded AChRs.
Next, the fold change in E2 was measured for the mutation pair. A deviation of the sum of the individual energy changes from that of the pair indicates an interaction ("coupling") between the two mutants. The coupling values are expressed as pair minus sum. For αP265 coupling, only the predominant, highest PO mode was used, except for with αY198A and αY190F, for which two modes were measured. The coupling values were all from mouse endplate AChRs and either are new (n = 79) or were published previously (n = 98; Cadugan and Auerbach, 2010; Purohit and Auerbach, 2010; Jadey et al., 2011; Jha et al., 2012; Gupta et al., 2013).
We estimate that we are able to measure an approximately twofold change in E2 (∼0.4 kcal/mol). Assuming independent errors, by the law of propagation of errors (Taylor, 1997), the combined error for a mutation pair is √(0.42 + 0.42), or ∼0.57 kcal/mol. Another way to estimate the error in the coupling energy estimate is from the SD of experimental values for residues separated by >15 Å (excluding αM2M3), which was 0.56 kcal/mol. The agreement indicates that the errors in the equilibrium constant estimates are independent. Below, we set the threshold for an interaction being significant at ±0.6 kcal/mol.
An independent method of estimating interactions between the agonist and a mutation is by comparing mutational fold changes in E2 and E0 (Eq. 1). The two TBSs are approximately equivalent for the agonists we used, ACh and choline (Jha and Auerbach, 2010; Nayak et al., 2014), so √(E2/E0) is equal to the coupling constant. A deviation in ΔΔGcc ≥0.6 kcal/mol compared with the WT value indicates an interaction with the TBS.
Phi analysis
A mutation that changes the gating equilibrium constant causes the relative free energy (structure) of O versus C to be different than in the WT. This happens because the substituted side chain is more (less) stable in the local environment compared with the WT. So far, we have not found an AChR mutation away from the TBS that changes significantly the ΔΔGcc of any agonist or that has an effect on ΔG2 that is agonist dependent. This suggests that in AChRs, the agonist and separated mutations have mainly independent and short-range effects. Consequently, gating energy (structure) changes can be localized to the mutated amino acid and put into sequence by using phi value analysis, as follows.
Phi is the slope of a log-log plot of the full C→O (opening) rate constant versus the full C↔O (gating) equilibrium constant for a series of mutations of one residue. This slope gives the relative energetic character of that amino acid at the gating TS on a scale from 1 (O-like) to 0 (C-like). The opening rate constant is proportional to the free energy difference between the gating TS and C (ΔGTS − ΔGC), and the gating equilibrium constant is proportional to the free energy difference between O and C (ΔGO − ΔGC). Phi is the ratio of these free energy differences. Phi = 1 (the two energy differences in the previous sentence are equal) indicates that ΔGTS and ΔGO changed to the same extent with all mutations of that amino acid, and phi = 0 indicates that ΔGTS and ΔGC changed to the same extent. Energy is related to structure, so a residue with a high phi is structurally more O-like at the TS than one with a low phi. Phi gives the relative position in the forward isomerization at which a side chain changes energy (structure) from C to O (see supplemental text). Methods for determining and phi values and population means (the color assignments in Fig. 1 A, right) are described in detail elsewhere (Purohit et al., 2013).
Unliganded gating phi values of αM2M3 amino acids were estimated after adding background mutations that increased constitutive activity (Table S2). The level of activity of αP265 mutants was extremely low, and many patches were required before observing single-channel clusters, perhaps because of low expression.
AChR diliganded gating phi values are modal (mean values of ∼1, 0.8, 0.6, 0.3, and 0) and follow a longitudinal spatial gradient (Purohit et al., 2013). There are several interpretations regarding the physical meaning of a fractional phi value. (1) In the “multiple intermediates” view, each phi population represents a short-lived gating intermediate state (see supplemental text; Auerbach, 2005). The five AChR phi populations thus reflect a sequence of transitions between six states: C, four short-lived intermediates (which together comprise the TSE), and O. Higher phi residues change energy (structure) earlier in this reaction chain. (2) In the “multiple paths” view, there are two gating reaction trajectories connecting C and O, with TSs at phi = 1 and 0. A fractional phi value thus reflects the relative probability of taking either pathway (Ternström et al., 1999; Oliveberg, 2001). Phi still gives the position in the reaction where the mutated amino acid reaches the TS, but possibly with no metastable intermediate states between C and O. (3) In the “multiple perturbation” view, an amino acid can experience an energy perturbation at more than one point in a single reaction trajectory (Jha et al., 2009). For instance, a TBS residue might be bumped in the low→high affinity change and again when the ECD compacts, or an M2M3 linker residue might be perturbed at the onset of opening and again when the TMD helices rearrange. The emergent phi of an amino acid is thus a weighted mean of the energy perturbations at multiple positions in the reaction trajectory. In the case of a reaction with one metastable intermediate state separated from the end states by barriers at phi = 1 and 0, equal energy perturbations in both transitions would yield phi ∼0.5. These three interpretations are not mutually exclusive.
Of these three interpretations, only the multiple intermediate view predicts intermediate states and modal phi values that are organized spatially. The multiple path and multiple perturbation views (that only have barriers at phi = 1 and 0) both predict a continuum of fractional phi values and say nothing regarding spatial organization. We therefore use the multiple intermediate interpretation, with four metastable TSE states (T1–4) separated by five microscopic energy barriers (the arrows). The relative energies of the barriers in the TSE were calculated from the diliganded gating phi values as described in the supplemental text and elsewhere (Zhou et al., 2005).
(Scheme 1).

According to this interpretation, phi values reveal the relative heights of the TSE energy barriers in the reaction chain but nothing regarding the energy wells. To estimate these, we incorporated knowledge regarding flip/primed, assuming that this signal arises from sojourns in one or more of the TSE states (T1–4, in brackets in Scheme 1). We simulated single-channel currents using QUB and Scheme 1 assuming all of the TSE states are nonconducting and using different values of the unconstrained rate constants while maintaining correspondence with the phi values. The objective was to match the apparent lifetime of flip/primed. We used parameters that mimic a high-resolution patch clamp experiment (sampling frequency of 1 MHz and low-pass filtering at 30 kHz) with idealization by using a half-amplitude criterion. After simulation using Scheme 1, the idealized interval durations were fitted using QUB using Scheme 2 after invoking a dead time of 8 µs, where sojourns in C′ correspond to those in the T states of Scheme 1 and the flip/primed shut component. In experiments with human adult AChRs, the lifetime of C′ is ∼9 µs, with left and right exit rate constants of ∼82 and 19 ms−1 (Mukhtasimova et al., 2016).
(Scheme 2).
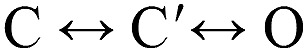
In the Scheme 1 simulations, the rate constants were changed to adjust the depths of the TSE wells while keeping the relative barrier heights (set by the phi values) constant. For each simulation, we calculated an energy landscape from the rate constants (k) from the relationship ΔG‡ = −0.59ln(k/A*) (in kcal/mol), using A* = 2 × 106 s−1 (Jones et al., 1992; Chakrapani and Auerbach, 2005). Changing A* alters the absolute, but not relative, heights of the barriers and therefore does not affect phi values. As described in the supplemental text, we also calculated a committor (‡), which is the point in the trajectory where there is a 50% probability of rapidly entering C or O (Bolhuis et al., 2002; Zhu and Hummer, 2012), and a transmission coefficient, defined as the inverse of the mean number of times the system crosses ‡ before reaching the other end state.
Structure analyses
Distances were measured using GluCl as the structural template (PDB accession code 3RIF) using VMD (Humphrey et al., 1996). In the MCA analyses, separations were calculated as distances between α carbons (αC). The secondary structural assignment, loop versus helix, was by the measurement of the backbone dihedral angles and hydrogen bonds using the DSSP program (Kabsch and Sander, 1983; Touw et al., 2015). The conformational energy of the M2M3 backbone was calculated as the combination of bonded energy terms (bond, angle, Urey-Bradley, dihedral, and improper energy) using the CHARMM27 force field with CMAP corrections (MacKerell et al., 1998; Brooks et al., 2009). The O and C structures were GluCl 3RIF and 4TNV (residues 265–276; Hibbs and Gouaux, 2011; Althoff et al., 2014), and GLIC 4HFI and 4NPQ (244–255; Sauguet et al., 2013, 2014).
Online supplemental material
The supplemental text describes the method for computing the TSE energy landscape from phi values. Fig. S1 shows the allosteric activation cycle, and Fig. S2 shows energy landscapes for simulations with stabilized intermediate states. Video 1 describes the model and shows cartoon structures, energy landscapes, and single-channel currents for simulations of gating at different time scales. Tables S1 and S2 report the background mutations used to make the measurements. Tables S3 and S4 give the simulation parameters and results.
Results
Definitions
Classically, the global C↔O allosteric isomerization is called "gating," with "opening" as the forward process and "closing" as the backward process. The detection of a shut gating intermediate (C↔C′↔O; Scheme 2) has suggested a change in this nomenclature, with the first step called "flipping" (or "priming") and the second called "gating." Below, we present evidence that there are four intermediate shut states between C and O and name each of the five connecting steps according to a correlated structural change. In this paper, we will continue the classical use of "gating" to refer to the full transition between the stable end states of the reaction. For example, here "closing" is O→C′→C (where C′ represents all intermediate shut states) and is not necessarily the same as a step decrease in single-channel current that could reflect O↔C′↔O.
Structures
Each TBS is at an α subunit interface, ∼30 Å from the ECD–TMD interface and ∼50 Å from the gate (Fig. 1). The principal side (α subunit, in heteromeric AChRs) of each TBS is formed by loops A (β4–5), B (β7–8), and C (β9–10). At the two adult sites (αδ and αε), three aromatic amino acids provide most of the ACh binding energy, αY190 (loop C) > αW149 (loop B) ≈ αY198 (loop C), but at the fetal αγ site, the energetically coupled pair αY93 (loop A) and γW55 also contribute (Nayak and Auerbach, 2013). Mutations of two TBS glycines and two complementary side prolines also influence resting affinity (Sine et al., 1995; Ohno et al., 1996; Purohit and Auerbach, 2011; Gupta et al., 2013; Jadey et al., 2013; Nayak et al., 2016).
Fig. 1 A (right) shows a map of the relative free energy (structure) of residues in the α subunit at the diliganded gating TS (Purohit et al., 2013). The highest, phi ∼1 residues (purple) are at the TBS and in αM2M3 (αI260, αP265, and αS268). The ECD is mostly phi ∼0.8 (blue), and the TMD is mostly phi ∼0.6 (green), with a discrete border near a proline-induced π-helix in M1 (Purohit et al., 2015). The lowest, phi ∼0.3 residues (red) are at the gate and lipid-facing residues in M3 (not depicted). At the TS, the TBS and some residues in αM2M3 are O-like, the gate is C-like, and the ECD and TMD are intermediate. The phi map indicates that in the C→O conformational change, the α subunit TBS+αM2M3, ECD, transmembrane helices, and gate reach the TS in that order.
In mouse AChRs, the high-phi and large-effect amino acids in the TMD align with the αM2M3 region of α4β2 AChRs (Fig. 1, B and C; Morales-Perez et al., 2016). In the related pLGICs GluCl and GLIC, in O versus C structures, the M2M3 linker is displaced upward relative to M1 (Fig. 1 B, bottom inset). In both of these pLGICs, the calculated backbone conformational energy in C is 4 and 2 kcal/mol less stable than in O. We also calculated the nonbonded interactions of these residues with their local environment, and for both receptors, the C configuration was 5.2 kcal/mol less stable. These calculations did not consider side chain energies but suggest that the M2M3 backbone may be locally more stable in O versus C.
αM2M3–TBS coupling
Interaction free energies were estimated by using MCA as the free energy difference between the effects of a mutation pair on the gating equilibrium constant minus the sum for the two mutations separately (Fig. 2 A). Below, we use “coupling” specifically to refer to the MCA interaction energy, “significant” to characterize an absolute interaction 0.6–1.2 kcal/mol, and “strong” for one that is >1.2 kcal/mol (see Materials and methods).
Figure 2.

αM2M3–TBS coupling. (A) Examples of MCA. Single-channel currents are C↔O clusters (see Fig. 3 B); open is down. Arrows, gating free energy change caused by the mutation or mutations. αS268 is in αM2M3, and αW149/αY93 are at the TBS; αY93 shows no interaction (pair = sum), but αW149 is coupled significantly (sum > pair by 0.9 kcal/mol). (B) Map of αM2M3–TBS coupling. Connecting lines mark interacting amino acids: thick-dashed, strong (>1.2 kcal/mol); and thin-solid, significant (0.6–1.2 kcal/mol). TBS structure is AChBP (PDB accession code 3WIP), and M2M3 structure is α4β2 AChR (PDB accession code 5KXI); spheres are αC (TBS: green, aromatic; pink, proline; blue, glycine). (C) MCA coupling values. Dashed lines are at ±0.6 kcal/mol. Main, TBS aromatics; inset, TBS prolines and glycines. TBS mutations were to Ala except where indicated; the M2M3 mutations are shown except for *, Pro and **, Gly. All pairs are adult-type AChRs except for αY190F and αY198A (f, for fetal; 1 and 2 indicate modes; see Fig. 3 B).
We used MCA of diliganded AChRs to estimate coupling between mutations of four αM2M3 residues (to A or G) and five TBS aromatic residues (Fig. 2, B and C). αS268A interacts strongly with αY190F and significantly with αW149A, two mutations that have large (∼2 kcal/mol) effects on ACh affinity in adult AChRs. αP265A showed strong interactions with all five aromatics. A glycine mutation of αL263 interacts significantly with the TBS aromatics. Overall, 74% of the αM2M3–aromatic pairs were coupled significantly, with all but one producing a smaller effect than predicted from independence.
Fig. 2 C (inset) shows coupling values for these αM2M3 amino acids and prolines or glycines at the TBS. 11 of the 14 residue pairs showed a significant degree of absolute coupling. αP265A was coupled significantly with αG147, εP121, and γP112, a residue in loop E that influences affinity. There was a strong interaction between αS268P and αY190P. For the glycine and aromatic TBS substitutions, the coupling values pertain to mutations in two α subunits, whereas for the proline mutants only one non–α subunit was substituted. Hence, the mean absolute coupling with the lone P121 (0.8 kcal/mol; n = 7) may indicate a stronger per-residue interaction than for the double glycines (0.9 kcal/mol; n = 7) and aromatics (1.1 kcal/mol; n = 22).
All of the coupling energies are summarized as a function of separation in Fig. 3 A. αM2M3 and the TBS interact more strongly than expected given their ∼30-Å separation. Also, many closely apposed residues do not interact significantly. Of the four αM2M3 residues we examined, αP265 and αL263 were the most interactive. At the TBS, strong interactions were divided approximately equally between the five aromatics.
Figure 3.

Coupling summary and modal activity. (A) Interaction energy versus separation. αC distance is that between α-carbons of the mutations using GluCl as the template structure. Circles, MCA coupling free energy (open, αM2M3–TBS); squares, TMD–TBS interaction energies estimated from gating constants (see Materials and methods). Lines mark ±0.6 kcal/mol. (inset) Histograms of coupling energies for separations >15 Å (mean ± SD [n]): αM2M3–TBS, −0.68 ± 1.02 (47); all others, 0.09 ± 0.56 (87). (B) αP265A induces kinetic modes. Clusters reflect C↔O gating (O is down) and long silent periods are D(esensitized). WT clusters are homogeneous (PO = 0.88 ± 0.02; mean ± SD), αP265A are modal (0.02 ± 0.01, 0.51 ± 0.07, and 0.94 ± 0.03), and αP265A+δW57A are homogeneous (0.34 ± 0.03). δW57A eliminates modes, but αY190F and αY198A do not.
Modes
In WT AChRs, kinetic properties of clusters are essentially homogeneous, but loop C mutations (in particular at αP197) generate discrete kinetic modes arising from differences in affinity at the αδ TBS (Vij et al., 2015). Fig. 3 B shows that modes are also apparent with the αP265A mutation in M2M3. As was the case with αP197 mutations, the αP265A modes disappear when δW57A is added, suggesting that these, too, arise from affinity differences at αδ. However, the αP265A and αP197A modes are not identical. αP265A modes persist with the addition of either αY190F or αY198A, whereas with αP197, the addition of αY190F but not αY198A reduces modal activity. Also, the addition of δW57A to αP265 causes a large reduction in PO but has a smaller effect with αP197A. Regardless, the modal activity of αP265A independently supports the existence of long-range interactions between αM2M3 and the TBS. The mechanism for this connection is not known, but given the significant MCA coupling between αP265A and both εP121 and γP112, this could possibly involve long-distance backbone interactions between αM2M3 and the β-sheet of the complementary subunit at the TBS (Nayak et al., 2016).
αM2M3 and the domain interface
Fig. 4 A shows coupling values for alanine mutations in αM2M3 (except αA270G) and nearby regions at the ECD–TMD interface. Regarding αloop2, strongly coupled pairs were Q48–I260/V261 (in the M2 helix) and E45–P265. Q48 and E262 do not interact, and neither of the phi ∼1 M2M3 residues (P265 and S268) interact with V46. Only a few αloop7 pairs were measured, with the only significant coupling being between V132–S268. Regarding εloop9, previous experiments showed there is strong coupling (−1.7 kcal/mol) between εG183I–αP265G (Jha et al., 2012). The new measurements show significant coupling between εG183 and T267–S268, but not with S266. Regarding εpreM1, εR219 interacts strongly with both S266 and T267, but εL221 was not coupled.
Figure 4.

Short-range coupling. (A) Short-range αM2M3 coupling at the ECD–TMD interface (PDB accession code 5KXI; mouse α subunit muscle AChR numbering). Lines: red, strong; white, significant. αP265 interacts strongly with αloop2 (Q48 and E45), αM2 (I260), and εloop9 (G183). (bottom) MCA values; blue bars are significant (≥0.6 kcal/mol). (B) Interactions across the fetal αγ TBS interface (PDB accession code 3WIP).
We also examined four alanine mutant pairs within αM2M3. αI260–αP265 are coupled by −0.6 kcal/mol, but the interactions between αI260–αS268, αS268–αK276, and αP272–αK276 were insignificant.
Overall, 10 of 29 local pairs showed strong or significant MCA coupling. For the 10 residue pairs that were coupled significantly, the absolute interaction free energy was 1.5 ± 0.2 kcal/mol. There were strong intra-subunit interactions with loop2 and cross-subunit interactions with εloop9 and εpreM1. αP265 was the only M2M3 amino acid coupled to three different structural elements (αM2, αloop2, and εloop9).
Fig. 4 B shows cross-subunit coupling at the diliganded fetal (αγ) TBS. Previously, it was found that in adult-type AChRs, εP121A interacts strongly with αG147A (2.6 kcal/mol) when ACh is present (Gupta et al., 2013). At the fetal site, γP121A interacts strongly with the three aromatic amino acids that provide most of the ACh binding energy from the α subunit (αY190 and αY198 in loop C and αW149 in loop B) and significantly with αG153. γP121A did not interact with its neighbor γP120A, αG147A, or the pair of residues that provide substantial binding energy at the fetal site and are themselves coupled (αY93 and γW55).
Phi and flip
The MCA results indicate that some long-range interactions between αM2M3 and the TBS are nearly as strong as short-range ones at the domain and TBS interfaces. In the next set of experiments, we compared phi values at αM2M3 and the TBS, with versus without agonists.
Fig. 5 A shows unliganded gating rate-equilibrium linear free energy relationships (REFERs; slope is phi) for four αM2M3 residues. Without agonists, these amino acids have a mean phi = 0.89 ± 0.05 (mean ± SEM), which is the same as with agonists (0.90 ± 0.03; Fig. 5 B). However, TBS amino acid phi values without agonists (0.79 ± 0.03) are smaller than with agonists (0.95 ± 0.02; Purohit and Auerbach, 2010). The agonist, which we think of as a removable TBS side chain, has a phi value of 0.93 (Grosman et al., 2000b). These results indicate that at the unliganded gating TS, αM2M3 is more O-like than the TBS.
Figure 5.

Phi analysis of the TBS and αM2M3. (A) Unliganded gating REFERs for αM2M3 amino acids. The slope (phi) is given in each plot. (B) Comparison of phi values with versus without agonists (dashed lines, mean). For TBS residues, phi is higher with versus without agonists, but for αM2M3 residues, it is the same.
We attempted to measure diliganded gating phi values for residues in the β10 strand of the α subunit. However, the effects of 14 mutations on the gating equilibrium constant were all small (<0.5 kcal/mol), and phi values could not be estimated (Purohit et al., 2013). The mutations were αT202A/C, αY203A/C, αH204A/C/Y, αF205A/C/T, and αV206A/C/I/W.
In AChR diliganded gating, amino acid phi values are distributed as five Gaussians with means of ∼0.95, 0.79, 0.58, 0.33, and ∼0.06 (Fig. 6 A). The phi populations also show a clear spatial organization, with the first four mapping to the α subunit TBS+M2M3, ECD, TMD, and gate, respectively (Fig. 1 A), and the last mapping mainly to gate residues in non–α subunits. (Lipid-facing residues in αM3 also have a phi ∼0.3.) We used Scheme 1 to model the intermediate states of the gating TSE, with the ratios of the exit rate constants from each of these (r values) calculated from phi (supplemental text) so that 6 of the 10 rate constants were constrained. When the overall C↔O equilibrium constant is 1, Scheme 1 becomes Scheme 3. The rate constants are ms−1, and the bracketed states (T1–4) comprise the intermediates of the TSE. Each arrow in Scheme 3 is associated with a microscopic barrier crossing representing a structural change that can be localized spatially according to the phi map.
Figure 6.

Combining phi and flip. (A) Phi values in diliganded AChR gating (Purohit et al., 2013). There are five populations, the first four of which correspond in decreasing order to purple, blue, green, and red in Fig. 1 A. (B) Simulations based on a model with four intermediate states (center; k = 300 ms−1; see supplemental text). (middle) The TSE are states T1–4 (all shut); each state is associated with a cartoon structure is in Fig. 7. (top left) Currents and perfect idealization. (top right) Energy landscape corresponding to the model (calibration, 1 kcal/mol; filled circle/‡, committor). (bottom) Histograms; sojourns in the TSE generate five shut components (gray lines), four of which are short-lived (time constants given); open sojourns are described by a single exponential. In standard patch recordings (∼25-µs resolution), the time constant of the open (long-shut) component is the inverse of the C↔O closing (opening) rate constant. (C) Fitting the simulations of panel B by C↔C′↔O after low-pass filtering to 30 kHz. There is only a single, brief component (flip/primed) apparent in the shut interval duration histogram. (D) Simulations using different values for k. Left y axis is the slowest shut interval TSE component (filled circles) calculated from the model, and right y axis (open squares) is the fitted backward/forward exit rate constant ratio from C′ using Scheme 2 (panel C). (E) Energy landscapes. (middle) The model in panel B with k = 300 ms−1; (top) the first two steps of unliganded opening are steeply uphill; (bottom) opening with two bound ACh molecules starts rapidly. In all conditions, the second step of the opening process (ECD twisting) is relatively the most difficult energetically, and the closing process is rate limited by gate bubble formation.
(Scheme 3).
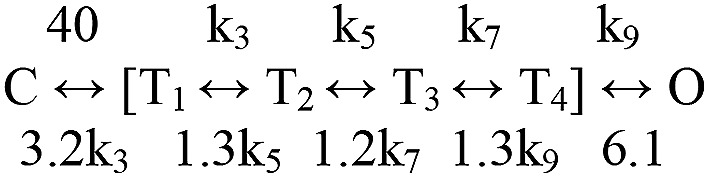
We hypothesize that the αM2M3 and TBS rearrangements are synchronized because of coupling, so that the first step (C↔T1) incorporates gating movements in both regions. The subsequent transitions in Scheme 3 are rearrangements of the ECD (T1↔T2), the TMD (T2↔T3), the gate (T3↔T4), and water entry into the gate region (T4↔O). An energy landscape corresponding to Scheme 3 is shown in Fig. 6 B (right). In the opening process, the relative TSE barrier heights (calculated from the phi values) show an increase associated with ECD movement, followed by a nearly flat region. The committor (‡; the position in the TSE that leads rapidly to C or O with equal probabilities) is near T3 (Fig. 6 B). Because of the upward tilt of the TSE barriers in opening, when C→T1 is slow relative to exits from the TSE, a mean of ∼20 exits from C must be attempted before O is achieved (supplemental text). Because of the downward tilt of the TSE in the reverse direction, closing is more efficient with only approximately three exit attempts from O required for a complete transit to C.
Next, we adjusted the unconstrained rate constants of Scheme 3 while maintaining consistency with the phi distribution. The goal was to set the TSE well depths so that the lifetime in T1–T4 was consistent with flip/primed. In experiments, this duration was ∼9 µs and arose from left and right exit rate constants from C′ (Scheme 2) of 82 and 19 ms−1 (Mukhtasimova et al., 2016). Fig. 6 B (left) shows example simulated single-channel currents and, below, corresponding open and shut interval duration histograms.
First, we used a single value k for all of the unconstrained rate constants (in Scheme 3: k3, k5, k7, and k9 all equal to 300 ms−1). This kinetic scheme predicts a broad distribution of four brief-shut components. Fig. 6 C shows the results of filtering and fitting by Scheme 2. In facsimile high-resolution patch-clamp recordings, the four components merge to become the tail of a single exponential with a lifetime of τflip ∼6.6 µs. Fig. 6 B shows the corresponding energy landscape and that ‡ is right of center.
Fig. 6 D (and Table S3) shows the simulated TSE and flip time constants and the ratio of the C′ exit rate constants estimated by using Scheme 2 as a function of k. Values of k <200 ms−1 generate τflip values that are too long-lived to be consistent with the experimental results.
In the next experiments, we used a large value of k (500 ms−1) to make most intermediate-state sojourns extremely brief and made just one or two of the TSE wells deeper (Fig. S2 and Table S4). These schemes approximate a single intermediate shut state while maintaining consistency with the phi pattern. It was possible to reproduce an ∼7-µs shut interval by lowering any one of the wells by 0.5–1 kcal/mol. Stabilizing T1 and T2 together, each by 0.5 kcal/mol, also produced a flip-like event. Apparently, there are many possible combinations of k values and energy wells that can generate an approximate microsecond intermediate shut event, but we did not attempt to simulate these exhaustively.
In the simulations, the lifetime of each intermediate state can be calculated as 1/(kleft + kright). Using Scheme 3 and with a uniform k = 300 ms−1, these lifetimes are 0.8, 1.4, 1.5, and 1.4 µs for T1–T4, respectively. With a uniform k = 500 ms−1, these are 0.5, 0.9, 0.9, and 0.9 µs. The lifetimes of the TSE microstates in landscapes having 1 or 2 wells stabilized are shown in Fig. S2.
Discussion
Coupling
The coupling values (Fig. 3 A) show that (a) except for αM2M3–TBS pairs, most significant interactions are apparent with αC separations of less than ∼15 Å, and (b) many residues that are this close (or closer) to each other do not interact significantly. Evidently, for many amino acids, the C versus O free energy change is not influenced by those of other residues. In the AChR allosteric transition, side chains appear simply to “resettle” to influence the gating equilibrium constant and PO by whatever changes happen to their local (within ∼15 Å) environment.
αM2M3–TBS coupling and the modal activity of αP265A indicate that these separated regions interact long range. The results show only a fuzzy “cloud” of communication with the TBS rather than a specific set of interacting amino acids. We suspect that the strengths of the αM2M3–TBS interactions are sufficient to account for their common phi values when agonists are present because they are on par with short-range coupling magnitudes. We hypothesize that long-distance interactions synchronize the gating rearrangements and energy changes at αM2M3 and the diliganded TBS.
With agonists, and for the TBS mutations we investigated, the main energy perturbation was with regard to the coupling constant (ΔΔGcc; Eq. 2). Although we did not measure M2M3–TBS communication in unliganded gating, evidence suggests that it is minimal. Mutations that induce a high constitutive PO also enforce a high affinity (Purohit and Auerbach, 2013), so the unliganded TBS appears to undergo a low→high affinity rearrangement without agonists, with an associated energy change that is part of ΔG0. Without agonists, aromatic TBS residues have phi values that are similar to the rest of the ECD (Fig. 5 B). This indicates that their gating energy changes occur not with the affinity rearrangement (phi ∼1) but rather with ECD rotation/compaction (phi ∼0.8). Also, phi values of αM2M3 and TBS residues are similar in liganded but different in unliganded gating. We speculate that without agonists, M2M3 and the TBS do not communicate.
The coupling mechanism has been suggested to involve a mechanical link between the TBS and M2M3, such as the displacement of a β strand or a domino-like cascade of side chain/domain bumping. The structural elements of a rigid-body movement (or a cascade) should all reach the TS together and therefore have the same phi. However, with agonists, the TBS and αM2M3 are separated by a sea of lower-phi amino acids (Fig. 1 A), and with or without agonists, phi values of residues in αloop2 (base of β2), the salt bridge (base of β10), and αloop7 (base of β7) are characteristic of the rest of the ECD rather than of the TBS and αM2M3. The current phi map does not support a mechanical link, but phi values for many ECD residues have not yet been determined (including in β9 and β10), and a high-phi, rigid-body connection remains a possibility.
Proteins are not static objects—bonds fluctuate, side chains adopt rotamers, and domain linkages are flexible. One way to move energy (between M2M3 and the TBS) and leapfrog an intervening region (the ECD β-barrel) is by correlated changes in backbone dynamics (Taly et al., 2005; Bu and Callaway, 2011; Changeux, 2014; Garret et al., 2014). Long-range communication, including allostery, can be mediated by changes in protein “breathing” motions rather than by changes in mean atomic positions (Popovych et al., 2006; Tsai et al., 2008; Zheng and Thirumalai, 2009; Ma et al., 2011). The structural changes in gating at the AChR ECD–TMD domain interface are not known, but it is conceivable that the upward displacement of the M2M3 linker apparent in GluCl and GLIC could be sufficient to influence the dynamics of both M2M3 and TBS loops, to link these separated regions energetically without perturbing the intervening ECD. For example, a loop↔helix transition is correlated with change in structure and function at a distant site in kinesin (Kikkawa et al., 2001), myosin (Skolnick, 1987; Harrington et al., 1988), GPCRs (Kjeldgaard et al., 1996; Sprang, 1997), and AcrR (Manjasetty et al., 2016). Certainly, M2M3–TBS coupling in pLGICs could be by both a rigid-body link and backbone dynamics, as these mechanisms are not mutually exclusive.
The short-range interactions between αM2M3 and other loops at the domain interface are complex (Fig. 4 A). Side chains in the linker interact with those both in the same subunit (loop2 and loop7) and the adjacent subunit (εpreM1 and εloop9). Consistent with the pin-into-socket hypothesis, the N-terminal αM2 positions αI260 and αV261 interact with αQ48 in loop2. Interestingly, amino acids at the top of M2 (αE262 and αL263) also interact long range with the TBS. Within αM2M3, the only significant interaction was between αI260 and αP265, both of which are phi ∼1 amino acids.
Overall, it appears that αP265 plays a central role in orchestrating gating rearrangements at the domain interface. This proline is highly conserved (Jaiteh et al., 2016), has a phi ∼1 with and without agonists, induces affinity modes, is coupled to most TBS amino acids, and interacts with the ECD and TMD of the same and the adjacent subunit. These results are consistent with the previous suggestion that a change in the M2M3 proline omega dihedral angle initiates the global opening transition, although perhaps by a partial rather than a complete cis-trans isomerization. However, this proline was our focus from the outset, and more coupling measurements elsewhere at the interface are needed to confirm whether or not it is uniquely important.
The strong coupling between αQ48A and the top of M2 supports the notion that gating movements of loop2 and M2 are linked. The αQ48A mutation has a “catalytic” effect on gating (Chakrapani et al., 2004), which suggests that the loop2–M2 interaction mainly influences the energy barrier for ECD rotation/compaction rather than the stability of the active-state configuration. It is possible that in the opening process, an extension of the M2 helix (Fig. 1 C) facilitates ECD twisting by a direct interaction with loop2.
Although we did not find many αM2M3 residues coupled with αloop7, we note that only a few pairs were examined and not with the absolutely conserved proline here because all mutations of αP136 in AChRs eliminate expression or function.
γP121 on the complementary side of the TBS interacts significantly and long range with two high-phi αM2M3 residues, αS268 and αP265 (Fig. 2). At the fetal, liganded TBS, this proline also interacts locally and strongly with residues in loops B and C (Fig. 4 B). Hence, part of the energy connection between αP265 and the TBS may involve γP121. It has not escaped our notice that many of the long-distance interactions involve prolines, for instance γP112 (loopE), αP197 (loopC), γP121 (β5′), and αP265 (M2M3) and that αP221 (M1) forms the discrete ECD–TMD phi border (Purohit et al., 2015). It is possible that in AChRs these (and other) prolines are nodes in a network for long-distance communication via the backbone.
TSE
Neither phi nor flip provide complete descriptions of the intermediate states that separate C from O, but together, they begin to illuminate the gating TSE. Phi suggests the number of intermediates, the sequence and locations of the rearrangements, and the relative energy barriers associated with each, but it does not provide temporal information. Flip/primed shows intermediate states directly along with lifetime information, but it is at the extreme edge of the patch clamp’s capabilities and therefore requires two assumptions, that the brief shut intervals all have exactly zero current (but see Cymes and Grosman [2012]) and arise from a single exponential component (a single intermediate state) even if all of the detected events are longer than that time constant. The results suggest that the modal phi distribution and the flip/primed component are complementary and lead us to propose that the TSE of AChR gating is a single reaction trajectory with four intermediate states that each have a lifetime of ∼1 µs.
We are at present unable to derive a unique energy landscape for the AChR gating TSE. Flip/primed could reflect sojourns in the ensemble of all four TSE intermediates, a combination of a few, or perhaps just one, as proposed. It is easy to imagine a far more complex TSE landscape. For instance, the ECD may not rotate and compact synchronously, or the TMD helices might rearrange in a specific sequence. Revealing these microscopic features is now beyond the resolution of phi and flip analyses.
Model
The results indicate that (a) many mutational energy changes have only short-range effects (Fig. 3 A), (b) phi values are modal (Fig. 6 A), and (c) with each phi population mapping to a discrete structural domain (Fig. 1 A). Also, a reaction chain with four brief intermediate states can account for both (d) the phi distribution (supplemental text) and (e) the flip/primed shut component (Fig. 6). Together, these observations suggest the following model for AChR gating (Fig. 7 and Video 1). (Again, C and O represent stable structures and gating refers to the full C↔O isomerization.) We use AChR α subunit diliganded phi values to locate, sequence, and define an energy landscape for the gating rearrangements, flip/primed to provide temporal information about the intermediates, MCA values to describe interactions as being local versus nonlocal, and other pLGICs to associate the on–off configurations with intermediate structures.
Figure 7.

Structure-based model of AChR gating. Eight components undergo local off↔on transitions (black↔red): five are protein (left labels) plus the agonist (black/red filled circle), ions (green filled circle), and water (blue). Only the end state C (all components off) and O (all on) structures are stable. The gate region can be a hydrophobic cluster (gray square), a bubble (red open circle), or water. The four, short-lived intermediate states of the TSE (T1–4) have a mixture of on/off components. The five steps in the opening process are M2M3–TBS click-and-hold, ECD twist, TMD tilt, gate dilate, and gate wetting (bubble collapse). The energy landscapes (Fig. 6 E and Fig. S2) calculated from phi and flip suggest that ECD twisting is the most energetically unfavorable step in the opening process. Channel closing is the reverse of opening and starts with (and is rate limited by) the spontaneous reformation of a gate bubble. Animations of this scheme at different time scales are shown in Video 1.
The model has five moving protein parts that switch between a locally off or locally on configuration: M2M3, TBS, ECD, TMD, and gate. In resting-C, all component structures are off, and in active-O, all are on. Between these two structures, there is a mixture. For clarity, each reversible on↔off local structural transition is named as follows: “click” (upward displacement of M2M3), “hold” (rearrangements at the TBS that underpin the affinity change), “twist” (rotation and compaction of the ECD), “tilt” (rearrangements of the TMD helices including straightening of M1 and radial bending of M2), and “dilate” (unpacking of the hydrophobic gate to form a bubble). The lipid, too, is perturbed in the dilate step (Purohit et al., 2013). The nonprotein components of the model are the agonist, ions, and water. “Wetting” of the gate region (collapse of the gate bubble) initiates fusion of the extra- and intracellular water compartments and ion conduction. We assume that M2M3–TBS coupling synchronizes click-and-hold so there are only four short-lived TSE intermediates (preopen states), all assumed to be nonconducting, which in some combination give rise to flip/primed. The energy barriers separating the TSE intermediates are small, so there is a thermal, random walk across the barrier region. As parts of a Brownian machine, the protein components are a linear actuator (M2M3), a latch (TBS) with a pin (agonist), a screw cap (ECD), a valve stem (TMD), and a plug (gate/bubble).
In the first step of the diliganded opening process, M2M3 and the TBS move (phi ∼0.95). These regions are coupled, so the agonist sites switch to the high-affinity conformation (hold) at approximately the same point in the reaction as the upward displacement of M2M3 (click). The second step is ECD twisting (rotation/compaction; phi ∼0.8). The relative heights of the exit barriers from T1 indicate that this twisting is more difficult than reversing click-and-hold, so the receptor returns rapidly (<1 µs) to C a mean of ∼3.2 times before the ECD twist is achieved (Fig. 6 B). It is possible that twist and hold stabilize each other by virtue of favorable and local interactions at the TBS. Continuing, the ECD twist, when it eventually does happen, perturbs the domain interface to favor TMD tilting (phi ∼0.6) by short-range interactions that involve ECD β strands, interfacial loops, and TMD helices. Tilting causes gate dilation (phi ∼0.3), perhaps by a direct mechanical linkage with the αM2–13′ amino acid that, based on its relatively high phi ∼0.5, has been suggested to hold together the hydrophobic cluster (Purohit et al., 2013). We speculate that unpacking the gate creates a bubble (Sauguet et al., 2016) that in the final step of opening (phi ∼0) collapses and fills with water to start ion flow (Roth et al., 2008; Zhu and Hummer, 2010). The closing process is the reverse of these steps and begins with a spontaneous separation of the water into intra- and extracellular compartments, without the need for agonist dissociation (Calimet et al., 2013). In the model, gating appears as a staggering and reversible conformational cascade (Video 1).
The simulations suggest that the approximate microsecond shut intervals observed in high-resolution electrophysiology recordings correspond to sojourns in the TSE, entered from O. In the model, these events mainly reflect bubble reformation. We cannot distinguish whether flip is a pause in the reaction chain in just one TSE micro-well (Scheme 2) or whether this component reflects sojourns in an ensemble of intermediate states (Scheme 3). A time resolution of <1 µs might distinguish these possibilities and determine whether the brief shut component is a single exponential or the tail of several closely spaced exponentials.
Gating with versus without agonists
With two bound ACh molecules, the apparent opening and closing rate constants (C↔O, full transits across the TSE) in WT mouse adult AChRs are ∼50 and 2.5 ms−1, to yield shut- and open-channel lifetimes of 20 and 400 µs (−100 mV, 23°C, ∼25-µs time resolution). Using Scheme 3 with 300 ms−1 for the unconstrained rate constants to match phi and flip, and assuming 1,000 ms−1 for the C→T1 rate constant (close to the maximum; Chakrapani and Auerbach, 2005), the extended kinetic model with all states fully liganded (gating with 2 ACh) is shown in Scheme 4. The rate constants are in ms−1. In Scheme 4, the lifetime of diliganded C is only 1 µs, whereas the experimental shut duration associated with the C→O transition is 20 µs. This discrepancy is because, on average, approximately three exits from C and sojourns in the TSE (lifetime ∼6 µs; Fig. 6 B) are required to achieve O (Eq. S7 in the supplemental text). The experimental lifetime of diliganded O is ∼150 µs, which in the model is the time required for the gate bubble to reform. This also happens, on average, approximately three times per full passage back to C, to give an apparent open lifetime of ∼500 µs. There is no single rate-limiting step in opening with ACh, but ECD twisting appears to be the most difficult. In contrast, in the model, bubble formation is rate limiting for channel closing. Bubble reformation requires ∼150 µs, which is at least 100 times longer than the time required for bubble collapse. Apparently, the bubble is unstable compared with contiguous water.
(Scheme 4).
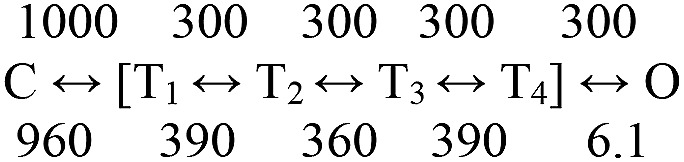
The apparent opening and closing rate constants for unliganded gating of WT AChRs are ∼10−3s−1/104 s−1 (Purohit and Auerbach, 2009; Jha and Auerbach, 2010). There is no phi map or flip measurement for unliganded gating, so we cannot calculate this TSE. However, we know that in a REFER the unliganded data point falls on the same line as that for agonists (Fig. 2 in Auerbach [2010]) and that many residues in the TMD have similar phi values with and without agonists. These results indicate that the unliganded TSE profile is similar to that for diliganded gating, except for “early” barriers close to C.
We speculate that unliganded WT AChRs have a small opening rate constant because with only water at the binding sites, the energy barriers for TBS hold and ECD twist both are large. Scheme 5 divides these energy penalties equally and reproduces approximately the apparent unliganded gating rate constants (gating without agonists). The increase in the T1↔T2 barrier (Fig. 6 E) without a concomitant one for C↔T1 predicts a different phi pattern without ligands. However, the calculated phi values of Scheme 5 are 0.99, 0.83, 0.61, 0.35, and 0.0 (supplemental text), which are indistinguishable from the measured diliganded ones (Fig. 6 A). We hypothesize that the unliganded opening rate constant is tiny because both hold and twist are difficult, so that ∼18,000 exit attempts from C are required before O is finally reached.
(Scheme 5).
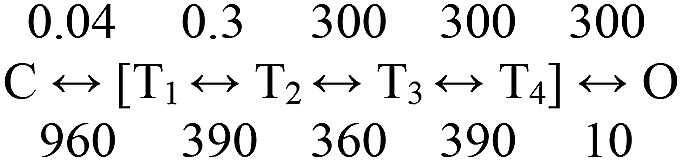
Both with and without agonist, the apparent gating rate constants measured in a standard patch clamp recording do not give accurate estimates of the exit rates from C and O. Either ∼3 (with ACh at both TBS), ∼20 (with choline), or ∼18,000 (with water) exit attempts from C are required to reach O, to make the apparent opening rate constant underestimate the exit rate from C by these factors. The model predicts that during silent periods in electrophysiology experiments, AChRs are dynamic structurally and undergo partial gating rearrangements without generating a current signal (Video 1).
The kinetic and structural models for diliganded AChR gating are consistent with local energy changes, phi values, locally closed GLIC structures, flip/primed, a central role for the M2M3 proline, and suggestions that a gate bubble is a metastable barrier to ion flow. They are also consistent with mechanical interactions at the domain interface that have been suggested to be involved in opening, including those involving the β2 and β10 strands. However, in our model, these strains are mainly local and are initiated by ECD twisting rather than being linked directly to actions at the TBS.
Supplementary Material
Acknowledgments
We thank Marlene Shero, Mary Merrit, and Mary Teeling for technical assistance, Tapan Kumar Nayak for helpful discussions, and Aashutosh Vihani for data shown in Fig. 3 A.
This work is supported by National Institutes of Health grants NS-023513 and NS-064969.
The authors declare no competing financial interests.
Author contributions: S. Gupta, S. Chakraborty, and R. Vij performed research and analyzed data; S. Chakraborty derived the equations in the supplemental text; A. Auerbach designed experiments, simulated currents, and wrote the paper.
Sharona E. Gordon served as editor.
Footnotes
Abbreviations used:
- ACh
- acetylcholine
- AChR
- ACh receptor
- ECD
- extracellular domain
- MCA
- mutant cycle analysis
- pLGIC
- pentameric ligand-gated ion channel
- REFER
- rate-equilibrium linear free energy relationship
- TBS
- transmitter-binding site
- TMD
- transmembrane domain
- TS
- transition state
- TSE
- TS ensemble
References
- Althoff T., Hibbs R.E., Banerjee S., and Gouaux E.. 2014. X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature. 512:333–337. 10.1038/nature13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. 2005. Gating of acetylcholine receptor channels: Brownian motion across a broad transition state. Proc. Natl. Acad. Sci. USA. 102:1408–1412. 10.1073/pnas.0406787102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. 2010. The gating isomerization of neuromuscular acetylcholine receptors. J. Physiol. 588:573–586. 10.1113/jphysiol.2009.182774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. 2012. Thinking in cycles: MWC is a good model for acetylcholine receptor-channels. J. Physiol. 590:93–98. 10.1113/jphysiol.2011.214684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafna P.A., Purohit P.G., and Auerbach A.. 2008. Gating at the mouth of the acetylcholine receptor channel: energetic consequences of mutations in the alphaM2-cap. PLoS One. 3:e2515 10.1371/journal.pone.0002515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi C., Zimmermann I., Engeler S., Hilf R.J., and Dutzler R.. 2016. Signal transduction at the domain interface of prokaryotic pentameric ligand-gated ion channels. PLoS Biol. 14:e1002393 (published erratum appears in PLoS Biol. 2016. 14:e1002390) 10.1371/journal.pbio.1002393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis P.G., Chandler D., Dellago C., and Geissler P.L.. 2002. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 53:291–318. 10.1146/annurev.physchem.53.082301.113146 [DOI] [PubMed] [Google Scholar]
- Bouzat C. 2012. New insights into the structural bases of activation of Cys-loop receptors. J. Physiol. Paris. 106:23–33. 10.1016/j.jphysparis.2011.09.012 [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Brooks C.L. III, Mackerell A.D. Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., et al. 2009. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30:1545–1614. 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu Z., and Callaway D.J.. 2011. Proteins move! Protein dynamics and long-range allostery in cell signaling. Adv. Protein Chem. Struct. Biol. 83:163–221. 10.1016/B978-0-12-381262-9.00005-7 [DOI] [PubMed] [Google Scholar]
- Cadugan D.J., and Auerbach A.. 2010. Linking the acetylcholine receptor-channel agonist-binding sites with the gate. Biophys. J. 99:798–807. 10.1016/j.bpj.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calimet N., Simoes M., Changeux J.P., Karplus M., Taly A., and Cecchini M.. 2013. A gating mechanism of pentameric ligand-gated ion channels. Proc. Natl. Acad. Sci. USA. 110:E3987–E3996. 10.1073/pnas.1313785110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini M., and Changeux J.P.. 2015. The nicotinic acetylcholine receptor and its prokaryotic homologues: Structure, conformational transitions & allosteric modulation. Neuropharmacology. 96:137–149. 10.1016/j.neuropharm.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Chakrapani S., and Auerbach A.. 2005. A speed limit for conformational change of an allosteric membrane protein. Proc. Natl. Acad. Sci. USA. 102:87–92. 10.1073/pnas.0406777102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S., Bailey T.D., and Auerbach A.. 2004. Gating dynamics of the acetylcholine receptor extracellular domain. J. Gen. Physiol. 123:341–356. 10.1085/jgp.200309004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux J.P. 2014. Protein dynamics and the allosteric transitions of pentameric receptor channels. Biophys. Rev. 6:311–321. 10.1007/s12551-014-0149-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymes G.D., and Grosman C.. 2012. The unanticipated complexity of the selectivity-filter glutamates of nicotinic receptors. Nat. Chem. Biol. 8:975–981. 10.1038/nchembio.1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenes S., Ni Y., Cymes G.D., and Grosman C.. 2006. Desensitization contributes to the synaptic response of gain-of-function mutants of the muscle nicotinic receptor. J. Gen. Physiol. 128:615–627. 10.1085/jgp.200609570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garret M., Boué-Grabot E., and Taly A.. 2014. Long distance effect on ligand-gated ion channels extracellular domain may affect interactions with the intracellular machinery. Commun. Integr. Biol. 7:e27984 10.4161/cib.27984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C., Salamone F.N., Sine S.M., and Auerbach A.. 2000a The extracellular linker of muscle acetylcholine receptor channels is a gating control element. J. Gen. Physiol. 116:327–340. (published erratum appears in J. Gen. Physiol. 2000. 116:327) 10.1085/jgp.116.3.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C., Zhou M., and Auerbach A.. 2000b Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 403:773–776. 10.1038/35001586 [DOI] [PubMed] [Google Scholar]
- Gupta S., Purohit P., and Auerbach A.. 2013. Function of interfacial prolines at the transmitter-binding sites of the neuromuscular acetylcholine receptor. J. Biol. Chem. 288:12667–12679. 10.1074/jbc.M112.443911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington W.F., Ueno H., and Davis J.S.. 1988. Helix-coil melting in rigor and activated cross-bridges of skeletal muscle. Adv. Exp. Med. Biol. 226:307–318. [PubMed] [Google Scholar]
- Hibbs R.E., and Gouaux E.. 2011. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 474:54–60. 10.1038/nature10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., and Schulten K.. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Jackson M.B. 1984. Spontaneous openings of the acetylcholine receptor channel. Proc. Natl. Acad. Sci. USA. 81:3901–3904. 10.1073/pnas.81.12.3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.B. 1986. Kinetics of unliganded acetylcholine receptor channel gating. Biophys. J. 49:663–672. 10.1016/S0006-3495(86)83693-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.B. 1989. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 86:2199–2203. 10.1073/pnas.86.7.2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey S.V., Purohit P., Bruhova I., Gregg T.M., and Auerbach A.. 2011. Design and control of acetylcholine receptor conformational change. Proc. Natl. Acad. Sci. USA. 108:4328–4333. 10.1073/pnas.1016617108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey S., Purohit P., and Auerbach A.. 2013. Action of nicotine and analogs on acetylcholine receptors having mutations of transmitter-binding site residue αG153. J. Gen. Physiol. 141:95–104. 10.1085/jgp.201210896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiteh M., Taly A., and Hénin J.. 2016. Evolution of pentameric ligand-gated ion channels: Pro-loop receptors. PLoS One. 11:e0151934 10.1371/journal.pone.0151934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A., and Auerbach A.. 2010. Acetylcholine receptor channels activated by a single agonist molecule. Biophys. J. 98:1840–1846. 10.1016/j.bpj.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A., Purohit P., and Auerbach A.. 2009. Energy and structure of the M2 helix in acetylcholine receptor-channel gating. Biophys. J. 96:4075–4084. 10.1016/j.bpj.2009.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A., Gupta S., Zucker S.N., and Auerbach A.. 2012. The energetic consequences of loop 9 gating motions in acetylcholine receptor-channels. J. Physiol. 590:119–129. 10.1113/jphysiol.2011.213892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.M., Ansari A., Henry E.R., Christoph G.W., Hofrichter J., and Eaton W.A.. 1992. Speed of intersubunit communication in proteins. Biochemistry. 31:6692–6702. 10.1021/bi00144a008 [DOI] [PubMed] [Google Scholar]
- Kabsch W., and Sander C.. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- Karlin A. 1967. On the application of “a plausible model” of allosteric proteins to the receptor for acetylcholine. J. Theor. Biol. 16:306–320. 10.1016/0022-5193(67)90011-2 [DOI] [PubMed] [Google Scholar]
- Kash T.L., Trudell J.R., and Harrison N.L.. 2004. Structural elements involved in activation of the γ-aminobutyric acid type A (GABAA) receptor. Biochem. Soc. Trans. 32:540–546. 10.1042/bst0320540 [DOI] [PubMed] [Google Scholar]
- Kikkawa M., Sablin E.P., Okada Y., Yajima H., Fletterick R.J., and Hirokawa N.. 2001. Switch-based mechanism of kinesin motors. Nature. 411:439–445. 10.1038/35078000 [DOI] [PubMed] [Google Scholar]
- Kjeldgaard M., Nyborg J., and Clark B.F.. 1996. The GTP binding motif: variations on a theme. FASEB J. 10:1347–1368. [PubMed] [Google Scholar]
- Lape R., Colquhoun D., and Sivilotti L.G.. 2008. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 454:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.Y., and Sine S.M.. 2005. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 438:243–247. 10.1038/nature04156 [DOI] [PubMed] [Google Scholar]
- Lee W.Y., Free C.R., and Sine S.M.. 2008. Nicotinic receptor interloop proline anchors β1-β2 and Cys loops in coupling agonist binding to channel gating. J. Gen. Physiol. 132:265–278. 10.1085/jgp.200810014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lummis S.C., Beene D.L., Lee L.W., Lester H.A., Broadhurst R.W., and Dougherty D.A.. 2005. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 438:248–252. 10.1038/nature04130 [DOI] [PubMed] [Google Scholar]
- Ma B., Tsai C.J., Haliloğlu T., and Nussinov R.. 2011. Dynamic allostery: linkers are not merely flexible. Structure. 19:907–917. 10.1016/j.str.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- Manjasetty B.A., Halavaty A.S., Luan C.H., Osipiuk J., Mulligan R., Kwon K., Anderson W.F., and Joachimiak A.. 2016. Loop-to-helix transition in the structure of multidrug regulator AcrR at the entrance of the drug-binding cavity. J. Struct. Biol. 194:18–28. 10.1016/j.jsb.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A., Fujiyoshi Y., Stowell M., and Unwin N.. 1999. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J. Mol. Biol. 288:765–786. 10.1006/jmbi.1999.2721 [DOI] [PubMed] [Google Scholar]
- Miyazawa A., Fujiyoshi Y., and Unwin N.. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 423:949–955. 10.1038/nature01748 [DOI] [PubMed] [Google Scholar]
- Monod J., Wyman J., and Changeux J.P.. 1965. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12:88–118. 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- Morales-Perez C.L., Noviello C.M., and Hibbs R.E.. 2016. X-ray structure of the human α4β2 nicotinic receptor. Nature. 538:411–415. 10.1038/nature19785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N., daCosta C.J., and Sine S.M.. 2016. Improved resolution of single channel dwell times reveals mechanisms of binding, priming, and gating in muscle AChR. J. Gen. Physiol. 148:43–63. 10.1085/jgp.201611584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T.K., and Auerbach A.. 2013. Asymmetric transmitter binding sites of fetal muscle acetylcholine receptors shape their synaptic response. Proc. Natl. Acad. Sci. USA. 110:13654–13659. 10.1073/pnas.1308247110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T.K., and Auerbach A.. 2015. Differences in agonist energy at the neurotransmitter binding sites in the neuromuscular acetylcholine receptors. Biophys. J. 108:429a–430a. 10.1016/j.bpj.2014.11.2348 [DOI] [Google Scholar]
- Nayak T.K., Bruhova I., Chakraborty S., Gupta S., Zheng W., and Auerbach A.. 2014. Functional differences between neurotransmitter binding sites of muscle acetylcholine receptors. Proc. Natl. Acad. Sci. USA. 111:17660–17665. 10.1073/pnas.1414378111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T.K., Chakraborty S., Zheng W., and Auerbach A.. 2016. Structural correlates of affinity in fetal versus adult endplate nicotinic receptors. Nat. Commun. 7:11352 10.1038/ncomms11352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C., and Sachs F.. 2013. Solving ion channel kinetics with the QuB software. Biophys. Rev. Lett. 08:191–211. 10.1142/S1793048013300053 [DOI] [Google Scholar]
- Ohno K., Wang H.L., Milone M., Bren N., Brengman J.M., Nakano S., Quiram P., Pruitt J.N., Sine S.M., and Engel A.G.. 1996. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 17:157–170. 10.1016/S0896-6273(00)80289-5 [DOI] [PubMed] [Google Scholar]
- Oliveberg M. 2001. Characterisation of the transition states for protein folding: towards a new level of mechanistic detail in protein engineering analysis. Curr. Opin. Struct. Biol. 11:94–100. 10.1016/S0959-440X(00)00171-8 [DOI] [PubMed] [Google Scholar]
- Popovych N., Sun S., Ebright R.H., and Kalodimos C.G.. 2006. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13:831–838. 10.1038/nsmb1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevost M.S., Sauguet L., Nury H., Van Renterghem C., Huon C., Poitevin F., Baaden M., Delarue M., and Corringer P.J.. 2012. A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat. Struct. Mol. Biol. 19:642–649. 10.1038/nsmb.2307 [DOI] [PubMed] [Google Scholar]
- Purohit P., and Auerbach A.. 2009. Unliganded gating of acetylcholine receptor channels. Proc. Natl. Acad. Sci. USA. 106:115–120. 10.1073/pnas.0809272106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., and Auerbach A.. 2010. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J. Gen. Physiol. 135:321–331. 10.1085/jgp.200910384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., and Auerbach A.. 2011. Glycine hinges with opposing actions at the acetylcholine receptor-channel transmitter binding site. Mol. Pharmacol. 79:351–359. 10.1124/mol.110.068767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., and Auerbach A.. 2013. Loop C and the mechanism of acetylcholine receptor-channel gating. J. Gen. Physiol. 141:467–478. 10.1085/jgp.201210946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., Gupta S., Jadey S., and Auerbach A.. 2013. Functional anatomy of an allosteric protein. Nat. Commun. 4:2984 10.1038/ncomms3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., Chakraborty S., and Auerbach A.. 2015. Function of the M1 π-helix in endplate receptor activation and desensitization. J. Physiol. 593:2851–2866. 10.1113/JP270223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth R., Gillespie D., Nonner W., and Eisenberg R.E.. 2008. Bubbles, gating, and anesthetics in ion channels. Biophys. J. 94:4282–4298. 10.1529/biophysj.107.120493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone F.N., Zhou M., and Auerbach A.. 1999. A re-examination of adult mouse nicotinic acetylcholine receptor channel activation kinetics. J. Physiol. 516:315–330. 10.1111/j.1469-7793.1999.0315v.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L., Poitevin F., Murail S., Van Renterghem C., Moraga-Cid G., Malherbe L., Thompson A.W., Koehl P., Corringer P.J., Baaden M., and Delarue M.. 2013. Structural basis for ion permeation mechanism in pentameric ligand-gated ion channels. EMBO J. 32:728–741. 10.1038/emboj.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L., Shahsavar A., Poitevin F., Huon C., Menny A., Nemecz À., Haouz A., Changeux J.P., Corringer P.J., and Delarue M.. 2014. Crystal structures of a pentameric ligand-gated ion channel provide a mechanism for activation. Proc. Natl. Acad. Sci. USA. 111:966–971. 10.1073/pnas.1314997111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L., Fourati Z., Prangé T., Delarue M., and Colloc’h N.. 2016. Structural basis for xenon inhibition in a cationic pentameric ligand-gated ion channel. PLoS One. 11:e0149795 10.1371/journal.pone.0149795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine S.M. 2012. End-plate acetylcholine receptor: structure, mechanism, pharmacology, and disease. Physiol. Rev. 92:1189–1234. 10.1152/physrev.00015.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine S.M., Ohno K., Bouzat C., Auerbach A., Milone M., Pruitt J.N., and Engel A.G.. 1995. Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 15:229–239. 10.1016/0896-6273(95)90080-2 [DOI] [PubMed] [Google Scholar]
- Skolnick J. 1987. Possible role of helix-coil transitions in the microscopic mechanism of muscle contraction. Biophys. J. 51:227–243. 10.1016/S0006-3495(87)83328-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorum B., Czégé D., and Csanády L.. 2015. Timing of CFTR pore opening and structure of its transition state. Cell. 163:724–733. 10.1016/j.cell.2015.09.052 [DOI] [PubMed] [Google Scholar]
- Sprang S.R. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66:639–678. 10.1146/annurev.biochem.66.1.639 [DOI] [PubMed] [Google Scholar]
- Taly A., Delarue M., Grutter T., Nilges M., Le Novère N., Corringer P.J., and Changeux J.P.. 2005. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys. J. 88:3954–3965. 10.1529/biophysj.104.050229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taly A., Hénin J., Changeux J.P., and Cecchini M.. 2014. Allosteric regulation of pentameric ligand-gated ion channels: an emerging mechanistic perspective. Channels (Austin). 8:350–360. 10.4161/chan.29444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.R. 1997. An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements. Second edition University Science Books, New York. 327 pp. [Google Scholar]
- Ternström T., Mayor U., Akke M., and Oliveberg M.. 1999. From snapshot to movie: phi analysis of protein folding transition states taken one step further. Proc. Natl. Acad. Sci. USA. 96:14854–14859. 10.1073/pnas.96.26.14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touw W.G., Baakman C., Black J., te Beek T.A., Krieger E., Joosten R.P., and Vriend G.. 2015. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 43:D364–D368. 10.1093/nar/gku1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.J., del Sol A., and Nussinov R.. 2008. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 378:1–11. 10.1016/j.jmb.2008.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vij R., Purohit P., and Auerbach A.. 2015. Modal affinities of endplate acetylcholine receptors caused by loop C mutations. J. Gen. Physiol. 146:375–386. 10.1085/jgp.201511503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., and Thirumalai D.. 2009. Coupling between normal modes drives protein conformational dynamics: illustrations using allosteric transitions in myosin II. Biophys. J. 96:2128–2137. 10.1016/j.bpj.2008.12.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Pearson J.E., and Auerbach A.. 2005. Phi-value analysis of a linear, sequential reaction mechanism: theory and application to ion channel gating. Biophys. J. 89:3680–3685. 10.1529/biophysj.105.067215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., and Hummer G.. 2010. Pore opening and closing of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. USA. 107:19814–19819. 10.1073/pnas.1009313107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., and Hummer G.. 2012. Theory and simulation of ion conduction in the pentameric GLIC channel. J. Chem. Theory Comput. 8:3759–3768. 10.1021/ct2009279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.