

Abstract
N1,N6-ethenoadenine, ε-A, nucleos(t)ides have been previously applied as fluorescent probes in numerous biochemical systems. However, these ε-A analogues lack the H-bonding capability of adenine. To improve the fluorescence characteristics while preserving the H-bonding pattern required for molecular recognition, we designed a novel probe: N2,N3-etheno-adenosine, (N2,N3-ε-A). Here, we describe four novel syntheses of the target ε-nucleoside and related analogues. These methods are short, facile, and provide the product regiospecifically. In addition, we report the absorption and emission spectra of N2,N3-ε-A and the dependence of the spectral features on the pH and polarity of the medium. Specifically, maximum emission of N2,N3-ε-A in water is observed at 420 nm (ϕ=0.03, excitation at 290 nm). The biochemical relevance of the new probe was evaluated with respect to the P2Y1 receptor and NTPDases 1 and 2. N2,N3-ε-ATP was found to be almost equipotent with ATP at the P2Y1 receptor and was hydrolyzed by NTPDases 1 and 2at about 80% of the rate of ATP. Furthermore, protein binding does not seem to shift the fluorescence of N2,N3-ε-ATP. Based on the fluorescence and full recognition by ATP-binding proteins, we propose N2,N3-ε-ATP and related nucleo(s)tides as unique probes for the investigation of adenine nucleo(s)tide-binding proteins as well as for other biochemical applications.
Keywords: fluorescent probes, NTPDase, nucleosides, nucleotides, P2Y1 receptor
Introduction
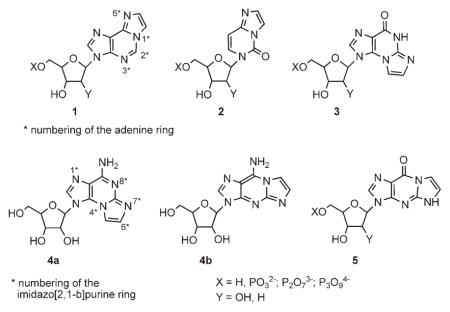
Adenine has poor fluorescence properties.[1] However, bridging the adenine N1,N6-positions with an etheno moiety, 1, improves the fluorescence of the parent adenine system (e.g., for 1: λmax=415 nm; ϕ=0.56).[2, 3] Over the past three decades, N1,N6-etheno-adenosine, 1 (ε-A), N3,N4-etheno-cytidine, 2 (ε-C), N2,N3-etheno-guanosine, 3 (ε-G), and the corresponding nucleotide analogues have been extensively studied as fluorescent nucleos(t)ide probes. All these probes bear an etheno bridge that causes minimal perturbation to the natural system.[4, 5]
N1,N6-Etheno-adenine nucleotides are commonly applied as fluorescent probes for various biochemical studies, such as the structure and function of nucleic acids,[2, 4, 6–8] protein visualization,[7] enzymatic studies,[2, 6, 7,9–11] investigation of nucleotide-binding sites,[2, 12–14] conformational analysis of nucleotides,[10] and pharmacology of nucleosides/nucleotides.[5, 15]
Although useful probes, N1,N6-ε-A nucleo(s)tides cannot be applied to biochemical systems requiring H-bonding-based molecular recognition. Specifically, the engagement of the N1 and N6 nitrogens of ε-A in an imidazole ring results in the loss of the adenine natural H-bonding capability. Consequently, the molecular recognition of these probes by target proteins or nucleic acids is reduced or lost.
So far, no fluorescent analogue of adenosine or the corresponding nucleotides with free N1,N6-positions has been investigated.[16] To address this challenge, we decided to extend the natural adenine chromophore at the C2,N3-positions to form analogue 4a. Specifically, we considered substitution of an amino group at the adenine C2-position, as substituents that donate electrons to the π system enhance the absorption of light and increase fluorescence. A good example is 2-aminopurine, which is a fluorescent purine analogue with a quantum yield of 0.6 in water and an emission maximum at 370 nm.[17] Next, we planned the incorporation of the adenine C2-amino group into an imidazole ring fused to the adenine ring, to form 9-amino-3-β-D-ribofuranosylimidazo[2,1-b]purine (4a), denoted here N2,N3-etheno-adenosine or N2,N3-ε-A.
Here, we describe four novel syntheses of the target ε-nucleoside, 4a and related analogues. These methods are short (3- to 4-step syntheses), facile, and provide the product regiospecifically in a reasonable yield. We also report on the spectral properties of the novel analogue, including absorption and emission spectra, and the dependence of the spectral features on the pH and polarity of the medium. Finally, we report on the biochemical relevance of the new fluorescent nucleotide, N2,N3-ε-ATP, and related analogues with respect to several ATP-binding proteins: the P2Y1 receptor and NTPDases 1 and 2.
Results and Discussion
Synthesis
The extension of natural nucleobases to form fluorophores 1–3 is achieved by treating the nucleobases with α-haloaldehydes at pH 4.5.[3, 18] For instance, the fluorescent angular isomer, N2,N3-ε-G (3; emission maximum in water at 410 nm)[19] is obtained from O6-benzyl-guanosine and bromoacetaldehyde at pH 4.5.[20] However, at an elevated pH, a nonfluorescent[19, 21] linear isomer, N1,N2-ε-G(5) is obtained from guanosine.[21, 22]
Since we targeted the novel angular heterocyclic system N2,N3-ε-A (4a) rather than the related angular N1,N6-ε-A (1) or linear N1,N2-ε-A (4b) isomers, we had to direct the regiochemistry of the ring closure to N2,N3- rather than to the N1,N6- or N1,N2-positions. For this purpose, we selected 6-Cl-, 6-NHAr-, or 6-SMe-purine riboside analogues as starting materials that would direct the reaction with bromoacetaldehyde, at pH 4.5, to the purine N2,N3-positions. With these starting materials, we developed several efficient syntheses of the target compound 4a and its analogues.
Thus, in the first synthesis, 6-chloro-2-aminopurine riboside 6 was treated with freshly prepared bromoacetaldehyde at pH 4.5 and 38°C for 30 h (Scheme 1), and N2,N3-ε-guanosine (3) was obtained in 86% yield. Initially, N2,N3-ε-6-chloropurine riboside was formed and then hydrolyzed to the corresponding guanosine derivative 3. Apparently, formation of the imidazole ring is faster than the hydrolysis of the chloro substituent, as guanosine is not reactive at all with α-haloaldehyde at pH 4.5.[19]
Scheme 1.

Synthesis of N2,N3-ε-A from 2-amino-6-chloro-purine riboside. Reagents and conditions: a) BrCH2CHO, EtOH/buffer (pH 4.5), 38°C, 30 h (86%); b) Ac2O/Py, RT, 48 h (63%); c) POCl3, N,N-dimethylaniline, CH3CN, 70°C, 1 h (45%); d) 2 M NH3/EtOH, 100°C, 24 h (43 %).
The replacement of the C6 carbonyl oxygen in 3 by NH2 was achieved in three steps: 1) The ribose hydroxyls were peracetylated to provide product 7; 2) This product was then chlorinated with phosphoryl chloride in the presence of N,N-dimethylaniline to provide 8; 3) Finally, 8 was treated with ethanolic NH3 at 100 °C to provide product 4a.
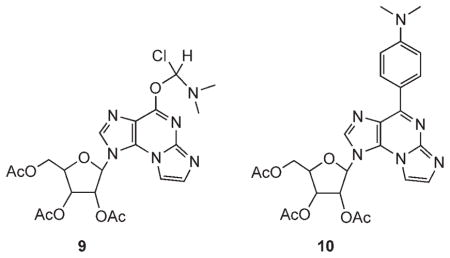
The moderate yield of the chlorination step prompted us to explore more drastic conditions for this conversion.[23] Thus, 7, in neat SOCl2 solution and one drop of DMF, was heated at 100°C for 3 h. Product 9 was obtained almost quantitatively, resulting from the reaction of 7 with a Vilsmeier reagent formed in situ from thionyl chloride and DMF.[24] Likewise, treatment of 7 in neat phosphoryl chloride solution in the presence of N,N-dimethylaniline and tetraethylammonium chloride at 120°C for 3 h resulted in the quantitative formation of product 10, due to the attack of N,N-dimethylaniline on the initially formed chloro product 8.
To avoid the loss of the chloro substituent at C6, due to hydrolysis (conversion of 6 to 3), we attempted the N2,N3-ring closure reaction under anhydrous conditions. Instead of using the aqueous bromoacetaldehyde solution, we attempted in situ generation of the aldehyde under anhydrous conditions in dry acetonitrile from bromoacetaldehyde diethyl acetal, BF3/etherate, and NaI under reflux.[25] Peracetylated 2-amino-6-chloropurine riboside 12 was then added to the solution at 38 °C; this gave rise to a complex mixture of products.
Therefore, instead of working under anhydrous conditions, we repeated the reaction of 6 with bromoacetaldehyde in CHCl3/buffer (pH 4.5) solution (Scheme 2). To increase the solubility of 6 in chloroform, we used 2′,3′,5′-triacetylated-2-amino-6-chloropurine riboside 12, obtained by chlorination of peracetylated guanosine 11 or acetylation of 6. Thus, 12 in CHCl3 was treated with bromoacetaldehyde and tetrabutylammonium bromide buffer (pH 4.5) at 38 °C for 48 h. The desired product, 8, was obtained in a 38% yield as the major product. In addition, etheno-guanosine analogue 7 was obtained in an 8% yield. This minor product was converted back to 8 in neat POCl3 in the presence of N,N-dimethylaniline at 120 °C for 20 min. Heating product 8 in ethanolic ammonia replaced C6-chloro by amine, and removed the acetate protective groups to furnish 4a in 43% yield.
Scheme 2.

Two-phase synthesis of N2,N3-ε-A from 2-amino-6-chloropurine riboside. Reagents and conditions: a) Ac2O/Py, RT, 48 h (68 %); a′) POCl3, N,N-dimethylaniline, CH3CN, 70°C, 1 h (49 %); b) BrCH2CHO, CHCl3/EtOH/buffer (pH 4.5), 38°C, 48 h (8: 38%, 7: 8%); c) 2 M NH3/EtOH, 100 °C, 24 h (43 %).
To improve the overall yield of 4a, we attempted alternative pathways. The first approach involved the early substitution of the C6-Cl by an aryl amine to yield 13, followed by treatment with bromoacetaldehyde to form etheno analogue 14, and finally, attempted removal of the N6-amine protecting group to produce 4a (Scheme 3).
Scheme 3.

Attempted synthesis of 4a from 2-amino-6-arylamino-purine riboside. Reagents and conditions: a) 2,4-di(O-methyl)benzylamine, EtOH, 60°C, 36 h (91%); b) BrCH2CHO, CHCl3, buffer (pH 4.5), 38 °C, 30 h (73 %); c) DDQ, CHCl3, H2O, RT, 12 h.
We selected 2,4-(dimethoxy)benzyl as an amine protective group due to the relatively mild conditions needed for its removal compared to those for the N-benzyl group. Our preliminary attempts to remove a benzyl protective group from N6-exocyclic amine in 14a included hydrogenation at ~5 atm over Pd/C with or without HOAc,[26, 27] and oxidation of the benzyl carbon by RuO2/NaIO4 [28] or NH4S2O8.[29] However, these attempts were unsuccessful as 14a remained unchanged under the hydrogenation conditions, and oxidation with persulfate caused complete cleavage of the etheno bridge. Alternatively, 12 was treated with 2,4-(dimethoxy)benzylamine in EtOH to provide 13b in 91% yield. Product 13b was in turn treated with bromoacetaldehyde, as described above, to provide, regiospecifically, the angular etheno product 14b in a 73% yield. However, the attempted removal of the aryl protective group of 14b by oxidation with 2,3-dichloro-5,6-(dicyano)-benzoquinone (DDQ)[30] in CHCl3/H2O caused complete cleavage of both the glycosidic bond and the etheno bridge in 14b, whereas the 2,4-(dimethoxy)benzylamino group remained intact.
Nonetheless, nucleotide analogues of 14a, b, and c (the last was prepared from 12 and dibenzylamine) were synthesized (Scheme 6, below) to evaluate their molecular recognition by our target proteins.
Scheme 6.

Preparation of nucleotides 18a–f. Reagents and conditions: a) POCl3, PO(OMe)3, proton sponge; b) (Bu3NH+)2H2P2O72−; c) 0.2 M TEAB.
The best synthetic approach, for both directing the ring cyclization to N2,N3-positions and facilitating the eventual substitution of C6 by an amine, involved purine riboside as the starting material (compound 16; Scheme 4). Compound 16 was prepared by treating 6 with NaSMe in DMSO.[31] Alternatively, 16 was obtained in a 84% yield by methylation of thioguanosine 15.[32] Bromoacetaldehyde solution was added to 16 in EtOH buffer (pH 4.5) at 60 °C for 8 h to provide the etheno product 17a in a 64% yield and absolute regiospecificity. Finally, product 17a was treated with ethanolic ammonia at 100°C for 14 h to provide product 4a in a 68% yield.
Scheme 4.

Synthesis of 4a from 2-amino-6-(thiomethyl)purine riboside. Reagents and conditions: a) NaSMe, DMSO, 60°C, 40 h (26%); b) NaOH(aq), MeI (84%); c) BrCH2CHO, EtOH/buffer (pH 4.5), 60°C, 8 h (64%); d) 2 M NH3/EtOH, 100°C, 14 h (68%); e) BrCH2CHO, EtOH/buffer (pH 6.4), 38°C, 30 h (47 %).
Regiochemistry of the ring-cyclization reaction
To assign the regiochemistry of the ring-closure product as either 4a or 4b, we recorded the 1H,1H NOESY spectra of intermediate ε-products 7, 14a (Figure 1), and 17a. A clear cross peak between the etheno H5 and the ribose H1′ proved the angular geometry of these products. A cross peak between H2 and H2′ indicated the anti geometry of the products.
Figure 1.

Assignment of the regiochemistry of an N2,N3-ε-A product. A) 1H,1H-NOESY spectrum of product 14a; B) NOE interactions in product 14a.
Mechanism and regiochemistry control of the cyclization reaction
In all the synthetic routes we attempted, we observed regiospecific N2,N3-ring cyclization of the 2-aminopurine riboside analogue with bromoacetaldehyde. To elucidate the origin of this regiospecificity, we repeated the cyclization reaction (Scheme 4) at an elevated pH (pH 6.4) and compared our observations with related cyclization reactions (Table 1).
Table 1.
N1,N2 versus N2,N3-cyclization of 2-amino purine riboside analogues.
| Purine riboside C6-substituent |
Product formed at | |
|---|---|---|
| pH 4.5 | pH≥6.4 | |
| O | no reaction[a] | N1,N2-ε-guanosine (5; 8%)[a] |
| OR | N2,N3-ε-purine riboside[a] | N2,N3-ε-purine riboside (18%+by-products)[b] N1,N2-ε-purine riboside (55%)[c] |
| SR | N2,N3-ε-purine riboside | N1,N2-ε-purine riboside (17b; 47%) and N2,N3-ε-purine riboside (17a; 15%) |
| NHCH2Ar | N2,N3-ε-purine riboside | – |
| Cl | N2,N3-ε-purine riboside | – |
In our ring-closure reactions of various purine riboside derivatives with bromoacetaldehyde at pH 4.5 (Schemes 1–4), we observed that the nature of the C6 substituent, either Cl, NHCH2Ar, or SMe (as in 12, 13, and 16, respectively), plays no role in directing the ring closure. All these reactions occurred with complete regiospecificity, forming only the N2,N3-ε-purine riboside analogue.
Previous reports indicated that treatment of guanosine with chloroacetaldehyde at pH 4.5 resulted in no cyclization product.[19] However, at elevated pH (pH 6.5), the linear N1,N2-ε-G product 5 was formed in a low yield after several days (Table 1). To control the regiochemistry of this reaction, Leonard et al. selected O6-benzylguanine to direct the reaction with chloroacetaldehyde to guanosine N2 and N3 positions by sterically hindering N1.[19]
In a related report, Singer et al. treated 5′-phosphate-O6-benzyl-2′-deoxyguanosine with chloroacetaldehyde at pH~8. 2′-Deoxy-N2,N3-ε-G, 3, was obtained in only 18% yield, accompanied by several by-products.[33] In another reaction, the linear nucleotide, 2′-deoxy-N1,N2-ε-G, 5, was isolated in 55% yield at basic pH.[22]
Based on our observations, and those of others, we suggest that the regiospecificity of the reaction is controlled by pH, rather than by steric or electronic characteristics of the purine reactant. Specifically, at pH 4.5, the N1-position of analogues 13 and 16, which is the most basic position in this ring system,[34] is protonated and is not available for nucleophilic attack, thus making the N3 nitrogen the favored nucleophilic site. For the chloro analogue 12, probably no N1 protonation occurs at pH 4.5 due to the most significant electron-withdrawing effect of the adjacent chloro group.[35] However, the chloro substituent also reduces the nucleophilicity of N1; that is, the N3 nitrogen is the preferred nucleophilic site also in analogue 12.
Indeed, when we repeated the reaction of 16 and bromoacetaldehyde at pH 6.4, the major product was N1,N2-ε-6-SMe-purine riboside, 17b (47% yield), accompanied by some 17a (15% yield; Scheme 4). At an elevated pH, N1 in 16 is not protonated. Furthermore, electron donations from the two electron-rich substituents ortho to N1 result in higher electron density at N1 than at N3. Therefore, at pH 6.4 the major product is the N1,N2-ε-analogue.
The mechanism of ring cyclization to form etheno products 8, 14, and 17 involves, first, the displacement of the bromoacetaldehyde bromide by the pyrimidine N3 nitrogen, rather than the exocyclic nitrogen (Scheme 5). This step in the mechanism is supported by literature data on the reaction of 2-aminopyridines with α-haloketones.[36] The cyclic nitrogen is the favored nucleophilic site in 2-aminopyridine, since the positive charge that develops in the transition state is stabilized by a resonance interaction with the exocyclic amine, as shown by Hand.[36] The displacement of bromide by the pyrimidine N1 nitrogen (at pH 4.5) is impossible, as mentioned above.
Scheme 5.

Mechanism of regiospecific formation of 4a.
In the second step, the N2-exocyclic amine in 12, 13, and 16 reacts with the aldehyde moiety;[37] followed by elimination of a water molecule.
Spectral properties of N2,N3-ε-A
The absorption spectrum of compound 4a at pH 7.0 showed two bands at 234 and 274 nm. The absorption of 4a at pH 12.3 was almost identical to that at neutral pH (Table 2). However, at pH 1.6, there was a blue shift, due to protonation of 4a.
Table 2.
Absorption and emission data of 4a [4.85×10−5 M] at several pH values.
| pH | Absorption | Emission | ||
|---|---|---|---|---|
| λmax | εd×103 | λmax | ϕ | |
| 1.6 | 224 | 6.17 | 396 | 0.021 |
| 268 | 14.22 | |||
| 7.0 | 234 | 12.58 | 420 | 0.032 |
| 274 | 3.31 | |||
| 12.3 | 234 | 13.89 | 420 | 0.031 |
| 276 | 3.69 | |||
Upon excitation of 4a in water at 290 nm, an emission maximum was observed at 420 nm (quantum yield is 0.03). This value represents a red-shift compared to that of N2,N3-ε-G (emission maximum 410 nm, excitation at 280 nm).[19] At basic pH, the emission spectrum of 4a was identical to that at pH 7; however, at acidic pH, a blue-shift and reduced quantum yield were observed (Table 2).
In organic solvents, the emission spectrum of 4a is linearly dependent on the polarity of the medium (Table 3). Thus, increasing the solvent polarity from dioxane (εd=2.2) to DMSO (εd=47.6) resulted in increases of both λmax (from 434 to 458 nm) and ϕ (from 0.042 to 0.085). This polarity dependence of 4a is very different from that of the related N2,N3-ε-G. The wavelength of the emission maximum of the latter does not change significantly with decreasing solvent polarity. On the other hand, the quantum yields of N2,N3-ε-Gincreas e substantially with decreasing solvent polarity.[19]
Table 3.
Emission data of 4a at 4.85×10−5 M in various organic solvents.
| Solvent | εd | ϕ | λmax |
|---|---|---|---|
| DMSO | 47.6 | 0.085 | 458 |
| DMF | 37.6 | 0.082 | 448 |
| dioxane | 2.2 | 0.042 | 434 |
Spectral properties of N2, N3-ε-6-methylthio-purine riboside, 17a
Among the etheno intermediates 8, 14a, and 17a, the last analogue exhibited promising spectral properties. The absorption spectrum of compound 17a in water showed four bands at 227, 247, 307, and 345 nm (Table 4). The absorption of 17a at pH 12.3 was similar to that at neutral pH; however, at pH 1.6, there was a blue shift of the absorption spectrum due to protonation of 17a.
Table 4.
Absorption and emission data of 17a [4.85×10−5 M] at several pH values.
| pH | Absorption | Emission | ||
|---|---|---|---|---|
| λmax | εd×103 | λmax | ϕ | |
| 1.6 | 227 | 16.52 | 492 | 0.013 |
| 243 | 13.57 | 516 | 0.022 | |
| 301 | 7.39 | |||
| 331 | 7.13 | |||
| 7.0 | 231 | 16.09 | 494 | 0.046 |
| 247 | 13.25 | 518 | 0.107 | |
| 307 | 7.07 | |||
| 345 | 2.90 | |||
| 12.3 | 233 | 19.29 | 494 | 0.021 |
| 307 | 5.95 | 520 | 0.063 | |
Upon excitation of 17a in water at 330 nm, maximum emission was observed at 494 and 518 nm. At both acidic and basic pH (1.6 and 12.3), reduced quantum yield was observed (Table 4).
Biochemical evaluation of the novel probes
The biochemical relevance of the new fluorescent analogues was evaluated with respect to three different ATP-binding proteins. Nucleotide probes were evaluated either as ligands of the P2Y1 receptor or substrates/inhibitors of NTPDase1 and NTPDase2. For this purpose, nucleotide analogues of 18a–f and 19a–f were prepared (Scheme 6).
Evaluation of the probes as P2Y1-R agonists
The use of fluorescent ATP analogues in biochemical research has brought new insights into enzymatic mechanisms and protein–nucleotide interactions.[38–40] So far, only limited information is available about the interaction of such compounds with nucleotide receptors (P2 receptors). Therefore, several of the novel fluorescent ATP analogues were evaluated as potential P2Y1 receptor agonists.
Analogues 18a, d, and f were tested in a functional assay to induce an intracellular Ca2+ release in HEK293 cells stably transfected with rat brain P2Y1 receptor (P2Y1-R; Figure 2).[41] These P2Y1-R-transfected cells were shown to be suitable for pharmacological characterization.[42] Analogue 18a, with an EC50 of 3.90±0.31×10−7 M, was found to be almost equipotent with ATP, which has an EC50 value of 1.30±0.04×10−7 M. However, a benzyl substitution at N6 of the fluorescent ATP compounds, as in analogue 18d, reduced the ligand’s potency; this resulted in a clear drop by about two orders of magnitude in the EC50 value, which can only be estimated to be >1.00×10−5 M as concentrations higher than 10 μM were not investigated. A dimethoxybenzyl substituent at N6, as in analogue 18 f, resulted in a similar reduction of the potency of the ligand. For 18 f, the EC50 value was also estimated to be >1.00×10−5 M.
Figure 2.

Concentration-response curves for fluorescent ATP analogues. Intracellular [Ca2+] release in HEK293 cells stably expressing the rP2Y1 receptor was induced by the analogues. The potencies found for 18d (○), 18f (●), and 18a (▼) are compared to the potency of ATP (■) at this receptor. Measurements of the intracellular [Ca2+] release were made as described in the Experimental section. Data represent the mean values and standard error of the mean obtained from 60–100 cells, which were measured in at least 3 independent experiments.
Analysis of the Ca2+ responses in the presence of the selective P2Y1 receptor antagonist MRS2179 upon stimulation with 18a (1 μM) clearly showed interaction of the novel compound with the P2Y1 receptor. At a concentration of 100 μM (n=45), and even at 10 μM (n=33), MRS2179 completely abolished the response to 18a.
Whereas the well-established fluorescent probe N1,N6-etheno-ATP (ε-ATP) needs further modifications to gain an affinity for the binding site of P2Y1-R,[43] analogue 18a was found to be almost equipotent with ATP. Namely, the introduction of the 2,3-aminoetheno group to create a fluorophore has no negative effect on the potency of ATP at the P2Y1 receptor. The potency of analogues 18d and 18 f was weak; this emphasized the receptor’s requirement for H-bonding interactions with N6. In addition, the substituents at N6 might hinder the binding to P2Y1-R. The decreased potency due to benzyl substitution in 18d and f is consistent with a previous report for N6-alkyl or -chloro substituted adenine nucleotide at P2Y1-R.[44]
Next, we examined whether the specific binding of 18a to P2Y1-R has any influence on the fluorescence properties of the substrate. Firstly, we measured the fluorescence of 18a at a concentration (10 μM) that yielded a maximal response for agonist- induced Ca2+ release (see Figure 2). Then, we repeated the measurements in the presence of 1321N1 cells. These cells were found to be P2Y-R negative.[45,46] This should exclude any possible influence of binding of the substance to proteins of the cell surface other than P2Y-R.
Finally, we measured the fluorescence in the presence of HEK293 cells expressing the P2Y1-GFP receptor. A maximal response is obtained when all receptors are occupied by the ligand. Therefore, for measuring the effect of 18a binding to the P2Y1 receptor on its fluorescence spectrum, we used a concentration of the agonist that elicited a maximal response in the pharmacological assay. We can assume that, under these conditions, a significant amount of the ligand is bound to the receptor and that we would be able to detect any perturbation of its spectrum if binding to the receptor affected it. Importantly, the spectrum of 18a when bound to P2Y1-R was not significantly different from the one measured without cells or when incubated with 1321N1 cells. Nucleotide 18a displayed an almost superimposable spectrum in all cases with λmax=430 nm (data not shown). Therefore, we assume that the specific binding of the compound to the receptor has a minor effect on its fluorescence properties.
Evaluation of the probes as substrates/inhibitors of NTPDases 1 and 2
We first evaluated analogues 18a–f as potential substrates of human NTPDases 1 and 2. These two enzymes were selected due to their different substrate preferences. NTPDase1 hydrolyzes di- and triphosphonucleosides equally well to generate the corresponding monophosphonucleoside, while NTPDase2 prefers triphosphonucleosides.[47] All the analogues tested were hydrolyzed by NTPDases 1 and 2. Analogues 18a and 18c were hydrolyzed at about 80% of the rate of the parent compound, ATP or GTP, respectively (Figure 3). Apparently, N2,N3-etheno modification of the purine ring did not significantly affect the recognition of 18a or c by NTPDases 1 and 2. However, NTPDase1 was more affected by C6 substitutions (18b, 18d, and 18 f) on the nucleotides, as these analogues were hydrolyzed less efficiently by NTPDase1 than by NTPDase2. Only 18e, bearing dibenzyl substituents at N6 affected both enzymes equally, with hydrolysis of 27±2% and 22±1% for NTPDases 1 and 2, respectively (Figure 3). Taken together, these results suggest that the N2,N3-etheno modification of the adenine ring to generate fluorescent probes did not alter the interaction with NTPDases significantly.
Figure 3.

Hydrolysis of analogues 18a–f by human A) NTPDase1 and B) NTPDase2. Analogues 18a–f were incubated for 20 min at 37°C in the presence of lysates of COS-7 cells transfected with NTPDase1 or 2. Bars represent the mean±SEM of three independent experiments performed in triplicate, and significant differences compared to ATP (white bar), adenine nucleotide derivatives (light gray bars) or guanine nucleotide derivatives (dark gray bars) are indicated: *p<0.01 and **p<0.001; 100% of ATP hydrolysis= 0.44±0.07 μmol Pi min−1 mg−1 for NTPDase1 and 0.7±0.2 μmol Pi min−1 mg−1 for NTPDase2 protein extracts; 100% of GTP hydrolysis= 0.5±0.1 μmol Pi min−1 mg−1 for NTPDase1 and 0.23±0.06 μmol Pi min−1 mg−1 for NTPDase2.
Secondly, we also tested whether 18e and monophosphonucleoside analogues 19a–f could block NTPDase1 activity. Comparison of ATP hydrolysis with or without 18e showed that this nucleotide blocked ATPase activity of human NTPDase1 by 69±6% (p<0.05; Figure 4). The real inhibition might be underestimated, as 18e can also be hydrolyzed by NTPDase1 at 27% of the rate of ATP (Figure 3A) and as the malachite green assay does not discriminate phosphate origin. Analogue 19e also blocked human NTPDase1 activity by a significant 46±5% (p<0.05). However, none of the other monophosphonucleosides tested (AMP, GMP, and 19a–f) affected ATP hydrolysis by NTPDase1.
Figure 4.

Hydrolysis of ATP by human NTPDase1 in the presence of N2,N3-ε-purine-5′-monophosphates (19a, 19c–f)[*] or triphosphate (18e). Each analogue was incubated for 20 min at 37°C in the presence of the substrate ATP and lysates of COS-7 cells transfected with NTPDase1. Hydrolysis of ATP alone (0.56±0.06 μmol Pi min−1 mg−1 of NTPDase1-transfected protein extracts) was set as 100%. Bars represent the mean±SEM of three independent experiments each performed in triplicate. Only 18e and 19e significantly inhibited NTPDase1 (*p<0.01).
Next, we measured the fluorescence spectra of 18a, 18d, 18 f, and their monophosphate homologues, 19a, 19d, and 19 f, in the presence or absence of human NTPDase2. We chose NTPDase2 over NTPDase1 for these experiments as the former was not affected by the etheno modification on the adenine ring (Figure 3) and, more importantly, because NTPDase2 dephosphorylates triphosphonucleoside to the diphosphonucleoside derivative with little formation of monophosphate nucleoside.[47] This reaction should therefore be easier to interpret when compared to the more complex NTPDase1 reaction with triphosphonucleosides. For the three analogues tested (18a, 18d, and 18 f), we observed that the fluorescence intensity (Figure 5A) and emission spectra (Figure 5B) remained constant over the 20 min tested while they were hydrolyzed by human NTPDase2, as measured by the malachite green method (data not shown). Effectively, even with 54% hydrolysis, the fluorescence intensity of 18a did not change significantly (data not shown). This observation excludes a lower fluorescent intensity of formed product or a rapid intensity decay of the probe. Moreover, maximum emission for the free nucleotide does not shift upon the addition of the enzyme (Figure 5B); this implies that the catalytic site does not provide an acidic environment (cf. Table 2). These observations indicate that the fluorescence of the modified nucleotides does not appear to change in the presence of NTPDase2, at least not in a significant manner.
Figure 5.

Fluorescence properties of 18d in the presence of NTPDase2. A) Fluorescence was measured for 15 min during the hydrolysis of 18d by human NTPDase2. Inset: Pi released reveals that about 10% of 18d was hydrolyzed during the course of this reaction. B) Emission spectra of 18d, excitation at 315 nm before the addition of human NTPDase2 (◆) and in the presence of NTPDase2 after 0, 5, and 10 min hydrolysis. A full spectrum was measured in 160 s. The spectra were almost identical. For both assays, protein extracts of COS-7 cells transiently transfected with human NTPDase2 (3.1 μg) were added to 200 μM 18d in assay buffer (80 mM Tris-base, 5 mM CaCl2, pH 7.4).
Conclusion
We have developed three short, facile, and regiospecific syntheses of N2,N3-ε-A, 4a, starting from 2-amino-6-chloropurine riboside (6) or 2-amino-6-thiomethylpurine riboside (16). Our synthetic procedures also proved efficient for the known N2,N3-ε-G (3). In earlier syntheses, N2,N3-ε-G was achieved in a 25–48% yield,[23,48] whereas in our procedure (Scheme 1), this compound was obtained in an 86% yield. The best synthetic pathway for the preparation of 4a was a three-step synthesis starting from thioguanosine 15 (Scheme 4).
We have demonstrated the superior fluorescent properties of N2,N3-ε-A analogues compared to those of adenosine. Analogue 4a has λmax=420 nm in water (ϕ=0.03) and has similar fluorescence properties to those of N2,N3-ε-G (3) and N1,N6-ε-A (1).[3] However, unlike the latter, 4a exhibits significant fluorescence even at acidic pH. Analogue, N2,N3-ε-6-methylthiopurine riboside (17a) exhibits remarkable fluorescence (λmax=518 nm in water, ϕ=0.13).
N2,N3-ε-ATP (18a) proved to be nearly equipotent with ATP at P2Y1-R and NTPDases 1 and 2. Particularly, the extension at the ATP’s C2 and N3 positions is tolerated by both types of protein. Furthermore, 18a apparently preserves the H-bonding pattern of ATP, as the major interactions of these proteins with the adenine moiety are at the N1 and N6 positions. The importance of H-bonding interactions of the N6 amine with the protein binding pocket is demonstrated by the significant reduction of the affinities of 18d and 18 f to P2Y1-R and of 18e to NTPDase2. In addition, analogues bound to NTPDase2 and the P2Y1 receptor appeared to be fluorescent.
Based on the unique fluorescent and H-bonding properties, we propose the novel adenosine and ATP analogues 4a and 18a, respectively, as useful probes for studies of adenosine- and ATP-binding proteins. Furthermore, the novel analogues may be applied to the detection and exploration of nucleic acids. These applications will be reported in due course.
Experimental Section
General
New compounds were characterized and resonances were assigned by proton and carbon NMR spectroscopy on Bruker AC-200/DPX-300 and DMX-600 spectrometers. The HOD signal (4.78 ppm) was used as a reference for samples in D2O. Nucleoside derivatives were characterized by ESI/MS on a Q-TOF micro mass spectrometer (Micromass-Waters, Corp., UK). High-resolution mass spectra were obtained on an ApexIII Bruker mass spectrometer (ESI/MS).
Bromoacetaldehyde, used for the synthesis of the etheno derivatives, was prepared as follows: a mixture of bromoacetaldehyde diethyl acetal (5 mL), HCl (1 M, 15 mL), and ethanol (5 mL) was stirred for 3 days at 37°C. No phase separation occurred when the mixture was brought to room temperature. The final solution contained approximately 1.3 mmol bromoacetaldehyde in 1 mL. For the preparation of the etheno products, we used a NH4HCO3/HCl buffer (pH 4.5). This buffer proved superior to the commonly used NaOAc/HOAc, as the former can be removed by freeze-drying and enables simple and easy purification of the etheno product. Chlorination and amination reactions were performed in flame-dried flasks under Ar. Reagents were predried in a vacuum oven overnight. POCl3, acetonitrile and aromatic amines were freshly distilled. DMSO was dried over activated 4 Å molecular sieves. Compounds 17a and 4a were separated on an HPFC automated flash purification system (SP1, Biotage Inc.).
N2,N3-Ethenoguanosine 3
2-(4-(O-benzyl)-1H-imidazo[2,1-e]purine-1-yl)-tetrahydro-5-(hydroxymethyl) furan-3,4-diol
A solution of bromoacetaldehyde (1.3 M, 0.9 mL, 6 equiv) in NH4HCO3/HCl buffer (6 mL, pH 4.5) was added to a solution of 6-chloro-2-aminopurine riboside (6; 50 mg, 0.16 mmol) in EtOH (3 mL). The reaction mixture was stirred at 38°C for 30 h, then evaporated in vacuo to about half its original volume, and EtOH was removed. A few drops of aqueous sodium hydrogen carbonate (1 M) were added until pH 8 was attained. The mixture was diluted with water (100 mL) and freeze-dried. The product was purified on a silica-gel column and eluted with MeOH/CHCl3 (1:1). Product 3 was obtained as a white solid in 86% yield (44 mg). 1H NMR (600 MHz, D2O): δ=8.03 (s, 1 H), 7.40 (d, J=2.8 Hz, 1H), 7.21 (d, J=2.8 Hz, 1H), 5.89 (d, J=3.3 Hz, 1H), 4.75 (dd, J=3.3, 5.0 Hz, 1 H), 4.42 (t, J=5.0, 6.3 Hz, 1H), 4.23 (m, J=3.0, 3.6, 6.3 Hz, 1 H), 3.9 (dd, J=3.0, 11.8 Hz, 1 H), 3.83 ppm (dd, J=3.6, 11.8 Hz, 1H); 13C NMR (600 MHz, D2O): δ=153.1, 150.0, 145.5, 139.7, 115.8, 107.5, 117.6, 88.7, 85.9, 73.9, 71.2, 62.1 ppm; HRMS calcd for C12H13N5O5: 307.1150, found 307.09166.
2′,3′,5′-Tri-O-acetylated-N2,N3-ethenoguanosine 7
2-(4-(oxo)-1H-imidazo[2,1-e]purine-1-yl)-tetrahydro-2,3,5-tri-O-acetyl)-furan
A solution of 3 (200 mg, 0.58 mmol) in pyridine (0.8 mL) and acetic anhydride (124 μL, 8 equiv) was stirred at RT for 48 h, then the pyridine and acetic acid were evaporated in vacuo. The residue was washed with EtOH (4×), and the resulting solution was evaporated and purified on a silica-gel column by elution with MeOH/CHCl3 (1:1). Product 7 was obtained as yellow oil in 63% yield (178 mg). 1H NMR (600 MHz, CHCl3): δ=7.90 (s, 1H; H-8), 7.67 (d, J=2.4 Hz, 1H; etheno), 7.26 (d, J=2.4 Hz, 1H; etheno), 6.03 (d, J=4.8 Hz, 1H; H-1′), 6.10 (dd, J=4.8, 5.4 Hz, 1H; H-2′), 5.80 (dd, J=4.5, 5.4 Hz, 1H; H-3′), 4.42 (dd, J=3.0, 11.4 Hz, 1H; H-4′), 4.39 (dd, J=5.4, 11.4 Hz, 1H; H-5′), 4.32 ppm (m, J=3.0, 4.5, 5.4 Hz, 1H; H-5′); 13C NMR (600 MHz, CHCl3): δ=170.9 (CH3), 169.8 (CH3), 169.5 (CH3), 151.6 (C-6), 146.0 (C-4), 145.9 (C-2), 137.6 (C-8), 114.9 (C-5), 107.6 (etheno), 115.9 (etheno), 87.1 (C-1′), 79.8 (C-4′), 72.3 (C-2′), 70.6 (C-3′), 63.0 (C-5′), 20.7 (CO), 20.5 (CO), 20.4 ppm (CO); MS: m/z: 434 [M+H]+.
N2,N3-Etheno-6-chloro-2′,3′,5′-tri-O-acetylpurine riboside 8
2-(4-(chloro)-1H-imidazo[2,1-e]purine-1-yl)-tetrahydro-2,3,5-tri-O-acetyl) furan
Phosphoryl chloride (448 μL, 6 equiv) and N,N-dimethylaniline (101 μL, 1 equiv) were added to a solution of predried 7 (347 mg, 0.81 mmol) in dry acetonitrile (3 mL). The reaction flask was then placed in an oil bath preheated to 70 °C, and the solution was stirred and heated for 1 h. Volatile materials were then evaporated immediately in vacuo. The residue was dissolved in CHCl3 (5 mL), and the solution was stirred vigorously with crushed ice for 15 min. The layers were separated, and the aqueous phase was extracted with CHCl3 (3×5 mL). The combined organic phase was kept cold by the addition of crushed ice and washed with cold water (6×5 mL), 5% NaHCO3 (5 mL) and water (2×5 mL) to pH 7. The organic phase was dried over MgSO4 and filtered, and the solvent was evaporated in vacuo. Product 8 was obtained as a yellow oil in 45% yield (154 mg). 1H NMR (300 MHz, CDCl3): δ=7.97 (d, J=1.5 Hz, 1H; etheno), 7.26 (d, J=1.5 Hz, 1H; etheno), 5.98 (d, J=2.7 Hz, 1H; H-1′), 5.91 (dd, J=2.7, 5.4 Hz, 1H; H-2′), 4.42 (dd, J=5.4, 3.6 Hz, 1H; H-3′), 4.23 (ddd, J=2.4, 3.0, 3.6 Hz, 1H; H-4′), 3.9 (dd, J=2.4, 12.6 Hz, 1H; H-5′), 3.61 (dd, J=3.0, 12.6 Hz, 1H; H-5′), 2.10–2.12 ppm (s, 9H; CH3CO); 13C NMR (600 MHz, CDCl3): δ=170.3 (C-6), 169.5 (CO), 169.4 (CO), 169.2 (CO), 150.0 (C-4), 145.5 (C-2), 139.7 (C-8), 115.8 (C-5), 107.5 (etheno), 117.6 (etheno), 88.7 (C-1′), 85.9 (C-4′), 73.9 (C-2′), 71.2 (C-3′), 62.1 ppm (C-5′); HRMS calcd for C18H17N5O7Cl: 450.1450, found 450.08110.
Attempts to chlorinate compound 7 under drastic conditions
A) SOCl2 (1 mL) was added to a solution containing predried 7 (70 mg, 0.16 mmol) and DMF (1 drop). The reaction flask was placed in an oil bath preheated to 100°C for 3 h. Volatile materials were evaporated in vacuo. The residue was dissolved in CH2Cl2, and the solution was stirred vigorously with crushed ice. The layers were separated, and the organic phase was washed with 5% NaHCO3 (5 mL), dried over MgSO4, and filtered. The solvent was evaporated in vacuo. Product 9 was obtained in a quantitative yield. MS: m/z: 522 [M+H]+.
B) N,N-dimethylaniline (21 μL, 1 equiv) and phosphoryl chloride (91 μL, 6 equiv) were added to predried 7 (70 mg, 0.16 mmol) and predried tetraethylammonium chloride (53 mg, 2 equiv) at RT. The reaction flask was placed in an oil bath preheated to 120 °C, and the solution was heated under reflux with stirring for 3 h. Volatile materials were evaporated in vacuo. The residue was dissolved in CHCl3 (5 mL), and the solution was stirred vigorously with crushed ice for 15 min. The layers were separated, and the aqueous phase was extracted with CHCl3 (3×5 mL). The combined organic phase was kept cold by the addition of crushed ice. The organic phase was washed with cold water (6×5 mL), 5% NaHCO3 (5 mL), and water (2×5 mL) to pH 7. The organic phase was dried over MgSO4 and filtered, and the solvent was evaporated in vacuo. Product 10 was obtained in a quantitative yield. MS: m/z: 537 [M+H]+.
N2,N3-Etheno-6-chloro-2′,3′,5′-tri-O-acetylpurine riboside 8 by two-phase synthesis
A solution of bromoacetaldehyde in HCl(aq) and EtOH (1.3 M, 3.2 mL, 6 equiv) and tetrabutylammonium bromide (2 equiv) were added to 2′,3′,5′-triacetylated-2-amino-6- chloropurine riboside (12; 300 mg, 0.7 mmol) dissolved in CHCl3 (10 mL) and EtOH (15 mL). The pH of the mixture was adjusted to 4.5 throughout the entire reaction time by the dropwise addition of NH4HCO3 (1 M) and 10% HCl. The reaction mixture was stirred at 38°C for 48 h. At the end of the reaction, a few drops of saturated aqueous NaHCO3 were added until pH 8 was attained. Water (100 mL) was added to the reaction mixture, which was then freeze-dried and purified on a silica-gel column (elution with CHCl3). Product 8 was obtained in 38% yield (12 mg) as yellow oil. In addition, by-product 7 was isolated in 8% yield (2.4 mg) as a white solid.
2′,3′,5′-Tri-O-acetylated-6-chloro-2-aminopurine riboside 12
5-((2-amino-6-chloro-9H-purine-9-yl)-tetrahydro-2,3,5-tri-O-acetyl)furan
A) A solution of 6 (300 mg, 1 mmol) in pyridine (2 mL) and acetic anhydride (94 μL, 4 equiv) was stirred at RT for 48 h, then the pyridine and acetic acid were evaporated in vacuo. The residue was washed with EtOH (4×) and purified on a silica-gel column (elution with diethyl ether/EtOAc, 2.5:1). Product 12 was obtained as a white solid in 68% yield (287 mg). 1H NMR (200 MHz, CDCl3): δ=8.27 (s, 1H; H-8), 5.97 (d, J=5.5 Hz, 1H; H-1′), 4.4 (dd, J=5.2, 5.5 Hz, 1H; H-2′), 4.20 (dd, J=3.3, 5.2 Hz, 1H; H-3′), 5.40 (ddd, J= 3.0, 3.3, 5.6 Hz, 1H; H-4′), 4.35 (dd, J=3.0, 12.6 Hz, 1H; H-5′), 4.07 (dd, J=5.6, 12.6 Hz, 1H; H-5′), 2.10–2.12 ppm (s, 9H; CH3CO); MS: m/z: 428 [M+H]+.
B) Alternatively, product 12 was obtained by chlorination of per-acetylated guanosine 11. N,N-dimethylaniline (1.55 mL, 1 equiv) and phosphoryl chloride (6.83 mL, 6 equiv) were added to predried peracetylated guanosine (5 g, 0.012 mmol) in dry acetonitrile (25 mL). The flask was then placed in an oil bath preheated to 70°C, and the solution was heated with stirring for 1 h. Volatile materials were evaporated immediately in vacuo. The residue was dissolved in CHCl3 (50 mL), and the solution was stirred vigorously with crushed ice for 15 min. The layers were separated, and the aqueous phase was extracted with CHCl3 (5×). The combined organic phase was kept cold by the addition of crushed ice. It was then washed with cold water, 5% NaHCO3, and water to pH 7. The organic phase was dried over MgSO4 and filtered, and the solvent was evaporated in vacuo. The residue was purified on a silica-gel column (elution with EtOAc/diethyl ether, 7:3). Product 12 was obtained as a white solid in 49% yield (2.6 g).
6-Methylthio-2-aminopurine riboside 16
5-(2-amino-6-methylthio-9H-purine-9-yl)tetrahydro-2-(hydroxymethyl)- furan-3,4-diol
A) Sodium thiomethoxide (138 mg, 1.97 mmol, 3 equiv) was added to a solution of 6 (200 mg, 0.66 mmol) in dry DMSO (6 mL). The solution was stirred at 60 °C for 40 h. A new spot on TLC plates (CHCl3/MeOH 8:2) at Rf=0.53 indicated the formation of product 16. The solvent was evaporated in vacuo, and the crude residue was purified on a silica-gel column (elution with CHCl3/MeOH 95:5). Product 16 was obtained in 26% yield (53.6 mg) as a light yellow solid. 1H NMR (200 MHz, CDCl3): δ=8.19 (s, 1H; H-8), 5.87 (d, J=6.3 Hz, 1H; H-1′), 4.69 (t, J=6.3, 11.4 Hz, 1H; H-2′), 4.30 (m, 1H; H-3′), 4.12 (m, 1H; H-4′), 3.82 (dd, J=2.1, 12.3 Hz, 1H; H-5′), 3.78 (dd, J=2.4, 12.3 Hz, 1H; H-5′), 2.61 ppm (s, 3H; CH3); MS: m/z: HRMS calcd for C11H16N5O4S: 314.3444, found 314.0918.
B) 6-Thioguanosine (300 mg, 1 mmol) was treated with 0.4 M sodium hydroxide and methyl iodide, as described before for 6-thioinosine.[23] A new spot on TLC plates (CHCl3/MeOH 8:2) at Rf= 0.53 indicated the quantitative formation of product 16. The solution was freeze-dried, and the crude residue was purified on a silica-gel column (elution with CHCl3/MeOH 93:7). Product 16 was obtained in 84% yield (264.2 mg) as a white solid.
N2,N3-etheno-6-thiomethylpurine riboside 17a
Tetrahydro-2-(hydroxymethyl)-5-(4-methylthio-1H-imidazo[2,1-e]purin- 1-yl)furan-3,4-diol
Bromoacetaldehyde (1.3 M, 1.47 mL, 1.92 mmol, 6 equiv) was added to NH4HCO3/HCl buffer (pH 4.5), and this solution was added to 16 (0.1 g, 0.32 mmol) in EtOH (1.75 mL). The resulting solution was stirred at 60 °C for 8 h. TLC (CHCl3/MeOH 8:2) showed a new spot at Rf=0.46. A few drops of aqueous NaHCO3 (1 M) were added until pH 8 was attained. The mixture was then diluted with water (100 mL) and freeze-dried. Finally, the residue was purified on a silica-gel column and eluted with CHCl3/MeOH 75:25. Product 17a was obtained in 64% yield (69 mg) as a light yellow solid. 1H NMR (300 MHz, CD3OD): δ=8.62 (s, 1H; H-8), 7.82 (ABq, 1H; etheno), 7.71 (ABq, 1H; etheno), 6.01 (d, J=6.3 Hz, 1H; H-1′), 4.79 (dd, J=5.4, 6.3 Hz, 1H; H-2′), 4.41 (dd, J=4.8, 5.4 Hz, H-3′, 1 H), 4.16 (dd, J=2.4, 4.8 Hz, 1H; H-4′), 3.89 (dd, J=2.4, 12.3 Hz, 1H; H-5′), 3.85 (dd, J=4.8, 12.3 Hz, 1H; H-5′), 3.23 ppm (s, 3H; CH3); MS: m/z: HRMS calcd for C13H16N5O4S: 338.3664, found 338.0917.
N1,N2-etheno-6-thiomethylpurine riboside 17b
Tetrahydro-2-(hydroxymethyl)-5-(9-methylthio-3H-imidazo[1,2-f]furan- 3-ol
Product 17b was obtained by following the same conditions as for product 17a, starting from 16 (0.56 mmol). The pH of the reaction was kept at 6.4 throughout the entire reaction time. TLC (CHCl3/MeOH 8:2) showed a new spot at Rf=0.37 (17b). The reaction residue was purified on a silica-gel column by elution with CHCl3/MeOH (92:8). Product 17b was obtained in 47% yield (83.7 mg) as a light yellow solid, in addition some 17a (15% yield, 24 mg) was formed. 1H NMR (300 MHz, CD3OD): δ=8.49 (s, 1H; H- 8), 8.17 (d, J=1.5 Hz, 1H; etheno), 7.83 (d, J=1.5 Hz, 1H; etheno), 6.33 (d, J=5.1 Hz, 1H; H-1′), 4.67 (t, J=4.8, 5.1 Hz, 1H; H-2′), 4.43 (t, J=4.5, 4.8 Hz, 1H; H-3′), 4.28 (m, J=3.0, 3.6, 4.5 Hz, 1H; H-4′), 3.86 (dd, J=3.0, 12.3 Hz, 1H; H-5′), 3.82 (dd, J=3.6, 12.3 Hz, 1H; H- 5′), 3.35 ppm (s, 3H; CH3); MS: m/z: 338 [M+H]+.
N2,N3-Ethenoadenosine (4a)
Tetrahydro-2-(hydroxymethyl)-5-(4-amino-1H-imidazo[2,1-e]purin-1- yl)furan-3,4-diol
A) Product 8 (25 mg, 0.055 mmol) was dissolved in a solution of NH3 (2 M) in EtOH (10 mL) in a thick, sealed ampoule. The solution was stirred at 100°C for 24 h, and then at room temperature overnight. A new spot on TLC plates (MeOH) at Rf=0.63 indicated the formation of product 4a. The solvent was evaporated to dryness in vacuo, and the residue was separated on a silica-gel column (CHCl3/MeOH 1:1). Product 4a was obtained in 43% yield (7.3 mg) as a light yellow solid. 1H NMR (300 MHz, CD3OD): δ=8.37 (s, 1H; H-8), 7.78 (d, J=1.8 Hz, 1H; etheno), 7.26 (d, J= 1.8 Hz, 1H; etheno), 6.25 (d, J=4.8 Hz, 1H; H-1′), 4.63 (dd, J=4.8, 5.1 Hz, 1H; H-2′), 4.38 (dd, J=4.5, 5.1 Hz, 1H; H-3′), 4.25 (ddd, J= 3.0, 3.3, 4.5 Hz, 1H; H-4′), 3.85 (dd, J=3.0, 12.3 Hz, 1H; H-5′), 3.82 ppm (dd, J=3.3, 12.3 Hz, 1H; H-5′); 13C NMR (300 MHz, CD3OD): δ=191.7 (C-6), 130.6 (C-4), 136.9 (C-2), 137.9 (C-8), 118.2 (C-5), 125.6 (etheno), 109.8 (etheno), 87.9 (C-1′), 49.56 (C-4′), 76.7 (C-2′), 71.7 (C-3′), 62.1 ppm (C-5′); MS: m/z: HRMS calcd for C12H15N6O4: 307.2885, found 307.1149.
B) Alternatively, 17a (63 mg, 0.186 mmol) was dissolved in a solution of NH3 (2 M) in EtOH (15 mL) in a thick, sealed ampoule. The solution was stirred at 100°C for 14 h. A new spot on TLC plates (CHCl3/MeOH 8:2) at Rf=0.07 indicated the formation of product 4a. The solvent was evaporated to dryness in vacuo, and the crude residue was purified on a silica-gel column (elution with CHCl3/MeOH 6:4). Product 4a was obtained in 68% yield (38.5 mg) as a light yellow solid.
General procedure for the treatment of 2-amino-6-chloropurine riboside 12 with aromatic amines for the preparation of 13a–d
Aromatic amine analogue (6 equiv) was added to a solution of the 2-amino-6-chloropurine riboside 12 (0.33 mmol) in EtOH (10 mL). The reaction mixture was heated at 100°C for 18 h. Once TLC (CHCl3/MeOH 9:1) indicated that the reaction was completed, the mixture was concentrated under reduced pressure. The crude products were purified by flash column chromatography on silica gel with 90% CHCl3/MeOH. Yields: 13a: 92% (112.9 mg), 13b: 91% (167.8 mg), 13c: 91% (130.0 mg), and 13d: 90% (137.5 mg).
2-Amino-N6-benzylpurine riboside 13a
5-(2-amino-6-(N-benzyl)-9H-purine-9-yl)tetrahydro-2-(hydroxymethyl)- furan-3,4-ol
1H NMR (600 MHz, CDCl3): δ=7.39 (s, 1H; H-8), 7.29 (m, 5H; benzyl), 5.66 (d, J=9 Hz, 1H; H-1′), 5.09 (s, 2H; CH2), 4.86 (t, J=9, 18 Hz, 1H; H-2′), 4.30 (m, 1H; H-3′), 3.79 (m, 1H; H-4′), 3.64–3.57 ppm (m, 2H; H-5′); MS: m/z: 372 [M+H]+; HRMS calcd for C17H21N6O4: 373.3866, found 373.1624.
2′,3′,5′-Tri-O-acetylated-2-amino-N6-(2,3-dimethoxybenzyl)purine riboside 13b
5-(2-amino)-6-(2,3-dimethoxybenzyl)-9H-purin-9-yl)tetrahydro-2,3,5- (tri-O-acetyl)furan
1H NMR (200 MHz, CD3OD): δ=7.86 (s, 1H; H-8), 7.57 (m, 2H; benzyl), 7.30 (s, 1H; benzyl), 6.41 (d, J=5.0 Hz, 1H; H- 1′), 5.98 (t, J=5.0, 5.6 Hz, 1H, H-2′), 5.84 (t, J=5.1, 5.6 Hz, 1H; H-3′), 5.27 (s, 2H; CH2), 4.36 (m, 2H; H-4′, H-5′), 3.86 (s, 3H; OCH3), 3.77 (s, 3H; OCH3), 2.07–2.11 ppm (s, 9H, CH3); MS: m/z: HRMS calcd for C25H31N6O9: 559.5529, found 559.2147.
2-Amino-N6-(2,3-dimethoxybenzyl)purine riboside 13c
5-(2-amino-6-(2,3-dimethoxybenzyl)-9H-purine-9-yl)tetrahydro-2-(hydroxymethyl) furan-3,4-diol
1H NMR (300 MHz, CDCl3): δ=7.68 (s, 1H; H-8), 7.25 (m, 3H; benzyl), 6.05 (d, J=6.6 Hz, 1H; H-1′), 5.88 (s, 2H; CH2), 5.36 (m, 1H; H-2′), 5.19 (m, 1H; H-3′), 4.72 (m, 1H; H-4′), 4.51 (m, 2H; H-5′), 4.18 (s, 3H; CH3), 4.15 ppm (s, 3H; CH3); MS: m/z: 433 [M+H]+; HRMS calcd for C19H24N6O6: 432.4307, found 432.1757.
2-Amino-N6,N6-dibenzylpurine riboside 13d
5-(2-amino-6-(N-dibenzyl)-9H-purine-9-yl)tetrahydro-2-(hydroxymethyl)furan-3,4-diol
1H NMR (600 MHz, CDCl3): δ=7.83 (s, 1H; H-8), 7.39 (m, 5H; benzyl), 5.68 (d, J=7.5 Hz, 1H; H-1′), 4.93 (s, 2H; CH2), 4.38 (s, 2H; CH2), 5.18 (m, 1H; H-2′), 4.33 (m, 1H; H-3′), 3.95 (m, 1H; H-4′), 3.87 ppm (m, 2H; H-5′); MS: m/z: 463 [M+H]+; HRMS calcd for C24H26N6O4: 462.4012, found 462.2016.
Reaction of 6-aromatic amine-2-aminopurine riboside derivatives 13a–d with bromoacetaldehyde to produce N2,N3-etheno products 14a–d
Bromoacetaldehyde (1.3 M, 6 equiv) was added to a solution of 6-aromatic amine-2-aminopurine riboside (0.30 mmol) in a solution of H2O/NH4HCO3/HCl (6 mL, pH 4.5) and EtOH (3 mL). The reaction mixture was stirred at 38°C for 20 h. TLC showed the disappearance of starting material, and the presence of a new spot of the product at Rf=0.63, 0.35, 0.36, or 0.46, respectively. The mixture was evaporated in vacuo to about half its original volume to remove EtOH. A few drops of saturated aqueous NaHCO3 were added until pH 8 was attained. Then the mixture was diluted by adding water (100 mL) and freeze-dried overnight. Yields: 14a: 87% (103.6 mg), 14b: 73% (68.0 mg), 14c: 61% (89.1 mg), and 14d: 73% (99.8 mg).
2′,3′,5′-Tri-O-acetylated-N6-(2,3-dimethoxybenzyl)-N2,N3-ethenopurine riboside 14b
2-(4-(2,3-dimethoxybenzyl)-1H-imidazo[2,1-e]purin-1-yl)tetrahydro- 2,3,5-(tri-O-acetate)furan
1H NMR (300 MHz, CD3OD): δ=8.436 (s, 1H; H-8), 8.02 (d, J=3.9 Hz, 1H; etheno), 7.73 (d, J=3.9 Hz, 1H; etheno), 6.95 (m, 5H; benzyl), 6.14 (d, J=5.4 Hz, 1H; H-1′), 5.67 (2H; CH2), 4.96 (s, 3H; CH3), 4.80 (s, 3H; CH3), 4.71 (t, J=4.8, 5.4 Hz, 1H; H-2′), 4.43 (t, J=4.8, 5.1 Hz, 1H; H-3′), 4.17 (m, J=3.0, 3.3, 5.1 Hz, 1H; H-4′), 3.88 ppm (dd, J=3.0, 3.3, 15.3 Hz, 2H; H-5′); 13C NMR (300 MHz, CD3OD): δ=154.2 (C-6), 131.9 (C-4), 142.6 (C-2), 125.3 (C-8), 121.5 (C-5), 113.8 (etheno), 108.2 (etheno), 89.8 (C-1′), 62.8 (C-4′), 87.2 (C-2′), 75.7 (C-3′), 72.0 (C-5′), 56.3 (s, CH3), 49.6 (s, CH3) 44.7 (benzyl) 49.8–48.1 (m, benzyl), 129.2–128.6 ppm (benzyl); MS: m/z: HRMS calcd for C27H31N6O9: 583.5749, found 583.2147.
N6-benzyl-N2,N3-ethenopurine riboside 14a
2-(4-benzyl-1H-imidazo[2,1-e]purin-1-yl)tetrahydro-5-(hydroxymethyl)- furan-3,4-diol
1H NMR (600 MHz, CD3OD): δ=8.49 (s, 1H; H-8), 8.19 (ABq, 1H; etheno), 7.75 (ABq, 1H; etheno), 6.08 (d, J=2.4 Hz, 1H; H-1′), 5.52 (dd, J=7.5, 9 Hz, 2H; CH2), 4.70 (t, J=2.4, 5.4 Hz, 1H; H- 2′), 4.38 (t, J=5.4, 5.7 Hz, 1H; H-3′), 4.15 (m, 1H; H-4′), 3.84 (dd, J= 1.5, 6 Hz, 1H; H-5′), 3.76 ppm (dd, J=1.5, 6.3 Hz, 1H; H-5′); 13C NMR (600 MHz, CD3OD): δ=154.61 (C-6), 138.71 (C-4), 145.63 (C-2), 142.75 (C-8), 113.78 (C-5), 108.93 (etheno), 121.34 (etheno), 89.68 (C-1′), 44.30 (C-4′), 75.81 (C-2′), 71.85 (C-3′), 62.73 (C-5′), 48.49 (CH2), 128.32–130.09 ppm (benzyl); MS: m/z: 397 [M+H]+; HRMS calcd for C19H21N6O4: 397.2492, found 397.1642.
N6-dibenzyl-N2,N3-ethenopurine riboside 14d
2-(4-dibenzyl-1H-imidazo[2,1-e]purin-1-yl)tetrahydro-5-(hydroxymethyl)furan-3,4-diol
1H NMR (600 MHz, CD3OD): δ=8.46 (s, 1H; H-8), 8.09 (ABq, 1H; etheno), 7.63 (ABq, 1H; etheno), 6.27 (d, J=2.4 Hz, 1H; H-1′), 4.73 (dd, J=2.4, 5.1 Hz, 1H; H-2′), 4.38 (dd, J=2.4, 4.5 Hz, 1H; H-3′), 4.27 (dd, J=4.5, 6.0 Hz, 1H; H-4′), 3.81 (dd, J= 1.5, 6.3 Hz, 1H; H-5′), 3.75 ppm (dd, J=1.5, 6.0 Hz,1H; H-5′); 13C NMR (600 MHz, CD3OD): δ=154.31 (C-6), 137.42 (C-4), 144.82 (C-2), 136.71 (C-8), 119.51 (C-5), 119.32 (etheno), 111.12 (etheno), 90.88 (C-1′), 79.43 (C-4′), 76.38 (C-2′), 71.69 (C-3′), 61.97 (C-5′), 49.85–48.15 (2CH2), 128.63–129.20 ppm (benzyl); MS: m/z: 487 [M+H]+; HRMS calcd for C26H27N6O4: 487.53065, found 487.2094.
N6-(2,3-dimethoxybenzyl)-N2,N3-ethenopurine riboside 14c
2-(4-(2,3-dimethoxybenzyl)-1H-imidazo[2,1-e]purin-1-yl)tetrahydro-5- (hydroxymethyl)furan-3,4-diol
1H NMR (300 MHz, CD3OD): δ=8.436 (s, 1H; H-8), 8.02 (ABq, 1H; etheno), 7.73 (ABq, 1H; etheno), 6.95 (m, 5H; benzyl), 6.14 (d, J=5.5 Hz, 1H; H-1′), 5.67 (s, 2H; CH2), 4.96 (s, 3H; CH3), 4.80 (s, 3H; CH3), 4.71 (dd, J=4.5, 5.5 Hz, 1H; H-2′), 4.43 (dd, J=4.5, 5.8 Hz, 1H; H-3′), 4.17 (m, 1H; H-4′), 3.88 ppm (m, 2H; H-5′); 13C NMR (300 MHz, CD3OD): δ=154.18 (C-6), 131.99 (C- 4), 142.60 (C-2), 125.31 (C-8), 121.46 (C-5), 113.77 (etheno), 108.18 (etheno), 89.76 (C-1′), 62.82 (C-4′), 87.22 (C-2′), 75.75 (C-3′), 72.03 (C-5′), 56.30 (s, CH3), 49.56 (s, CH3) 44.71 (benzyl) 49.85–48.15 (m, benzyl), 128.63–129.20 ppm (benzyl); MS: m/z: 456 [M+H]+; HRMS calcd for C21H24N6O6: 456.4521, found 456.1757.
General procedure for the preparation of N2,N3-ethenonucleotides 18a–f
All phosphorylation reactions were carried out in flame-dried, argon-flushed, two-necked flacks sealed with rubber septa. Nucleosides were dried in vacuo for 2 days. The proton sponge was kept in a desiccator. Phosphorus oxychloride was distilled and kept under N2. Tri-n-butylammonium pyrophosphate solution was prepared as described before.[49]
A solution of N2,N3-ethenoguanosine (3; 0.1 g, 0.32 mmol) in dry trimethyl phosphate (1.0 mL) was cooled to 0°C, then proton sponge (0.140 g, 2 equiv) was added. After 20 min, phosphorus oxychloride (91 mL, 3 equiv) was added dropwise. Stirring was continued for 2 h at 0°C. TLC on a silica gel plate (propan-1-ol 28%/NH4OH/H2O 11:7:2) indicated the disappearance of starting material and the formation of a polar product (Rf=0.35). A mixture of Bu3N (0.31 mL) and (Bu3NH+)2P2O7H2 (1 M) in DMF (2.0 mL) was added at once. After 1 min, a solution of triethylammonium bicarbonate (TEAB) in water (0.2 M 15.45 mL) was added, and the clear solution was stirred at RT for 45 min. The solution was then freeze-dried overnight. The semisolid obtained after freeze-drying was chromatographed on an activated Sephadex DEAE-A25 column. The resin was washed with deionized water for 30 min and loaded with the crude reaction residue dissolved in a minimal volume of water. The separation was monitored by UV detection (ISCO, UA-6) at 280 nm. A buffer gradient of H2O (500 mL) to NH4HCO3 (0.5 M, 500 mL) was applied. The relevant fractions of the ATP and AMP derivatives were pooled and freeze-dried three times to yield a white solid. The final purification of the products was achieved on an HPLC system (Merck–Hitachi) by using a semipreparative reversed-phase LichroCART lichrospher 60, RP-select B column (Merck, Darmstadt, Germany) and a linear gradient of acetonitrile and 0.1 M triethylammonium acetate buffer (pH 7; solvent system I: linear gradient of acetonitrile/0.1 M TEAA buffer 15:85 to 45:55 over 25 min) with a flow rate of 6 mL min−1. These reaction conditions provided the nucleoside triphosphate products in yields ranging from 31 to 63%. The corresponding nucleoside monophosphate analogues (19a–f) were obtained as by-products in all reactions. Triethylammonium salts of mono- and trinucleotides, obtained after HPLC separation, were passed through Sephadex-CM C-25 and eluted with deionized water to obtain the corresponding sodium salts after freeze drying.
N2,N3-ethenoadenosine-5′-triphosphate (18 a)
2-(4-amino-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran- 5-triphosphate
Yield: 49% (24 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.51 (s, 1H; H-8), 7.78 (ABq, 2H; etheno), 7.65 (ABq, 2H; Ar), 6.12 (d, J=7.5 Hz, 1H; H-1′), H-2′, H-3′, and H-4′ are hidden by the water peak, 4.42 ppm (m, 2H; H-5′); 31P NMR (200 MHz, D2O, pH 6): δ=−9.91 (d), −10.92 (d), −22.58 ppm (t); tR=3.41 min, solvent system I, 2:98–65:35, purity >95%, tR=2.99 min, solvent system II: 5 mM tetrabutylammonium dihydrogenphosphate (TBAP) in MeoH/60 mM ammonium dihydrogenphosphate +5 mM TBAP in H2O/MeOH (90:10) 25:75 to 75:25 over 20 min, purity >95%; FAB (negative mode): m/z: 545 [M4−+3H]+; HRFAB calcd for C12H16N6O13P3: 544.9988, found 544.9993.
6-SMe-N2,N3-ethenopurine riboside-5′-triphosphate 18b
2-(4-thiomethyl-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran- 5-triphosphate
Yield: 58% (62 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.90 (s, 1H; H-8), 8.01 (ABq, 2H; etheno), 7.91 (ABq, 2H; Ar), 6.18 (d, J=2.1 Hz, 1H; H-1′), H-2′ and H-3′ are hidden by the water peak, 4.48 (m, 1H; H-4′), 4.33 (m, 2H; H-5′), 3.12 ppm (s, 3H; SMe); 31P NMR (200 MHz, D2O, pH 6): δ=−9.85 (d), −10.85 (d), −22.32 ppm (t), tR=9.47 min, solvent system I, 2:98–65:35, purity >94%, tR=2.72 min, solvent system II, purity >95%. FAB (negative mode): m/z: 577 [M4−−3H]+.
N2,N3-ethenoguanosine-5′-triphosphate (18 c)
2-(4-oxo-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran-5- triphosphate
Yield: 31% (19 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.34 (s, 1H; H-8), 8.89 (ABq, 2H; etheno), 7.42 (ABq, 2H; Ar), 6.12 (d, J=9.0 Hz, 1H; H-1′), 5.01 (dd, J=7.2, 9.0 Hz, 1H; H-2′), H-3′ and H-4′ are hidden by the water peak, 4.43 ppm (m, 2H, H-5′); 31P NMR (200 MHz, D2O, pH 6): δ=−5.43 (d), −10.39 (d), −21.15 ppm (t); tR=3.71 min, solvent system I: 2:98–65:35, purity >95%, tR=2.24 min, solvent system II, purity >97%; FAB (negative mode): m/z: 546 [M4−−H]+; HRFAB calcd for C12H15N5O14P3: 545.9828, found 545.9857.
N6-Benzyl-N2,N3-ethenoadenosine-5′-triphosphate (18 d)
2-(4-benzyl-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran- 5-triphosphate
Yield: 63% (28 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.18 (s, 1H; H-8), 7.65 (ABq, 2H; Ar), 7.32 (ABq, 2H; Ar), 6.12 (d, J=8.1 Hz, 1H; H-1′), 4.72 (m, 1H; H-2′), 4.60 (m, 1H; H-3′), 4.43 (m, 1H; H-4′), 4.21 ppm (m, 2H; H-5′); 31P NMR (200 MHz, D2O, pH 6): δ=−6.23 (d), −10.44 (d), −21.24 ppm (t); tR=4.11 min, solvent system I, 15:85–45:55, purity >98%, tR=6.56 min, solvent system II, purity >95%; FAB (negative mode): m/z: 546 [M4−−3H]+; HRFAB calcd for C19H22N6O13P3: 635.33172, found 635.0463.
N6,N6-Dibenzyl-N2,N3-ethenoadenosine-5′-triphosphate (18 e)
2-(4-dibenzyl-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran- 5-triphosphate
Yield: 59% (33 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.42 (s, 1H; H-8), 8.00 (ABq, 2H; Ar), 7.78 (ABq, 2H; Ar), 7.35 (m, 10H; benzyl), 6.30 (d, J=8.3 Hz, 1H; H-1′), 5.27 (s, 4H; benzyl), H-2′ and H-3′ are hidden by the water peak, 4.50 (m, 1H; H-4′), 4.25 ppm (m, 2H; H-5′); 31P NMR (200 MHz, D2O, pH 6): δ= −5.37 (d), −10.67 (d), −21.27 ppm (t); tR=9.79 min, solvent system I, purity >99 %, 25:75–40:60, tR=13.44 min, solvent system II, 40:60–90:10, purity >95 %; FAB (negative mode): m/z: 725 [M4−+3H]+; HRFAB calcd for C26H28N6O13P3: 725.3391, found 725.0932.
N6-2,3-Dimethoxybenzyl-N2,N3-ethenoadenosine-5′-triphosphate (18 f)
2-(4-(2,3-dimethoxybenzyl)-1H-imidazo[2,1-e]purin-1-yl)-3,4-dihydroxytetrahydrofuran- 5-triphosphate
Yield: 58% (42 mg); 1H NMR (200 MHz, D2O, pH 6): δ=8.30 (s, 1H; H-8), 7.71 (ABq, 2H; Ar), 7.57 (ABq, 2H; Ar), 6.87 (m, 3H; benzyl), 6.00 (d, J=8.1 Hz, 1H; H-1′), 5.24 (s, 2H; benzyl), H-2′, H-3′, and H-4′ are hidden by the water peak, 4.26 (m, 2H; H-5′), 3.78 (s, 3H; OCH3), 3.72 ppm (s, 3H; OCH3); 31P NMR (200 MHz, D2O, pH 6) δ=−6.59 (d), −10.40 (d), −21.28 ppm (t); tR=6.13 min, solvent system I, purity >99 %, 15:85–45:55, tR=11.87 min, solvent system II, purity >95 %; FAB (negative mode): m/z: 695 [M4−+3H]+; HRFAB calcd for C21H26N6O15P3: 695.0668 found, 695.06713.
Absorption spectra
Absorption spectra of compounds 4a and 17a were determined in dilute HCl or NaOH over the pH range 1.6–12.3. The concentration of samples of 4a and 17a in aqueous solutions and in organic solvents was of the order of 4.85×10−5 M.
Emission spectra
The measurement conditions of compounds 4a and 17a included 740 V sensitivity and a 4- or 2-nm slit for the emission spectra in aqueous or organic medium, respectively. Emission spectra were corrected by the subtraction of the medium’s emission spectrum. Spectra were determined in dilute HCl or NaOH over the pH range 1.6–12.3. The concentration of the aqueous or organic samples was of the order of 4.85×10−5 M. All organic solvents were freshly distilled prior to emission measurements. Measurements of the emission of the nucleoside analogues were conducted with subtraction of the solvent emission. The quantum yield of each compound was calculated from the observed absorbance at 290 nm and the area of the fluorescence emission band. Quinine sulfate was used as reference compound, and a quantum yield value of 0.55 was assumed.[50]
Fluorescence studies of the 18a–NTPDase2 complex
Emission spectra of 18a, 18d, 18 f, and their monophosphate homologues (19a, 19d, 19 f) were measured in the presence of human NTPDase2 by using an Aminco–Bowman series 2 Luminescence Spectrometer. Measurements were done at 37°C at various excitation wavelengths (18a: 290 nm, 18d: 315 nm, 18 f: 334 nm) and 740–780 V sensitivity. The concentration of the samples was 2×10−4 M in NTPDase incubation mixture (5 mM CaCl2 and 80 mM Tris/HCl, pH 7.4). Measurements of the emission spectra (18a: 425 nm, 18d: 445 nm, 18 f: 442 nm) were performed in fluorescent nucleosides (1.5 mL, 200 μM) without enzyme. A sample of NTPDase2-transfected COS-7 cells lysate (3.6 μg) was added, and then emission spectra were taken at 5 min intervals. Samples of the reaction mixture (200 μL) were taken before each emission measurement and added to malachite green reagent (50 μL) to estimate Pi released (see NTPDases activity assay).
Fluorescence studies of P2Y1-R-bound 18a
Fluorescence spectra were taken of 18a in the presence of P2Y-receptor-expressing and P2Y-receptor-negative cells. Samples were excited at 330 nm, and the fluorescence intensity was detected from 340 to 520 nm by using a Perkin–Elmer LS50B fluorimeter. 18a (10 μM) was first measured in physiological buffer (pH 7.4), then in the presence of HEK293 cells expressing the P2Y1-GFP receptor. Cells were suspended in the buffer (NaHBS), which was used for the physiological experiments (100000 cells per mL). Finally, the fluorescence spectrum of 18a was measured in the presence of 1321N1 cells (100000 cells per mL), which are known to lack endogenous P2Y receptor expression. Buffer and cells showed no fluorescence in the range determined (data not shown).
[Ca2+]i measurements
The HEK293 cells expressing the rat P2Y1 receptor at high density[41] were plated on PLL-coated plates, and single cell measurements were made after 3 days, when the cells were 40–60% confluent. The changes in free intracellular Ca2+ concentration ([Ca2+]i) were measured, as described before,[51, 52] after preincubation of the cells with fura-2AM (2 μM) and with varying concentrations of MRS2179 (for the test of the P2Y1-receptor specificity of the fluorescent analogues) for 30 min at 37 °C in NaHBS (HEPES buffered saline solution: 145 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 25mM glucose, and 20 mM HEPES/Tris pH 7.4). Then the cells were stimulated under continuous superfusion of prewarmed NaHBS at 37°C with different concentrations of the different agonists and MRS2179 in 1 min agonist pulses at 37°C. Fluorescence intensity was recorded alternately at 340 and 380 nm excitation and 520 nm emission. Changes were monitored in single cells bathed in a perfusion chamber with a volume of 500 μL which was placed on the microscope stage of a fluorescence imaging system from TILL Photonics with a 40X/NA 1.30 oil immersion objective and a flow rate of 1 mL min−1.[53]
NTPDases activity assay
Protein extracts were obtained from COS-7 cells transiently transfected with human NTPDase1 or 2, as previously described.[47] Contaminating activity of intracellular ATPases or other ectonucleotidases was minimal; untransfected cells had no detectable ADPase activity and low intrinsic ATPase activity (less than 5% compared to transfected COS-7 cells lysate). The enzymatic assays were performed for 20 min at 37 °C in CaCl2 (5 mM) and Tris/HCl (80 mM) incubation mixture (200 μL, pH 7.4) with the protein extracts in the presence of ATP, GTP, or N2,N3-ε-adenosine triphosphates (100 μM) or an equal concentration of ATP and N2,N3-ε-adenosine monophosphates (100 μM each) as indicated. For hydrolysis assay, reaction was started with ATP, GTP, or N2, N3-ε-adenosine triphosphates (10 μL) and stopped with malachite green reagent (50 μL). For inhibition assays, enzyme extracts were preincubated for 3 min with monophosphate (19a–f) or 18e, and reaction was started with ATP (100 μM) and stopped with malachite green reagent. Inorganic phosphate (Pi) liberated during the enzymatic reaction was quantified according to Baykov et al.[54] Enzymatic activity was expressed as micromoles of Pi released per min per mg of protein. Protein concentration was estimated by Bradford microplate assays with bovine serum albumin as a standard of reference.[55]
Statistical analysis
All measurements were made in three independent experiments, each performed in triplicate. The mean and the standard error of the mean (SEM) were calculated from the mean specific activity of enzyme in presence of nucleotides. Data were compared for difference by Student’s t-test. All analyses were performed according to the Microsoft Excel software. p values of less than 0.01 were considered to be statistically significant.
Supplementary Material
Acknowledgments
This work was supported in part by the Marcus Center for Medicinal Chemistry (B.F.), by grants from the Canadian Institutes of Health Research (CIHR; to J.S), and grant 01ZZ0407 from the Bundesministerium für Bildung und Forschung (G.R.). We thank D. Terhardt for expert technical assistance with the cell culture and calcium measurements, and Prof. P. Schönfeld for support with the fluorescence measurements (G.R.). B.F. thanks Lior Kremer for the optimization of the synthesis of 4a. S.A.L. was a recipient of a scholarship from the “Fonds de la Recherche en Santé du Québec” and J.S. received a New Investigator award from the CIHR.
Footnotes
A US patent application was filed on 24 January, 2005.
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Callis PR. Annu Rev Phys Chem. 1983;34:329. [Google Scholar]
- 2.Thomas RW, Leonard NJ. Heterocycles. 1976;5:839. [Google Scholar]
- 3.Secrist JA, III, Barrio JR, Leonard NJ, Weber G. Biochemistry. 1972;11:3499. doi: 10.1021/bi00769a001. [DOI] [PubMed] [Google Scholar]
- 4.Leonard NJ. CRC Crit Rev Biochem. 1984;15:125. doi: 10.3109/10409238409102299. [DOI] [PubMed] [Google Scholar]
- 5.Leonard NJ. Chemtracts: Biochem Mol Biol. 1992;3:273. [Google Scholar]
- 6.Secrist JA, III, Barrio JR, Leonard NJ. Science. 1972;175:646. doi: 10.1126/science.175.4022.646. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama H, Yamaga T. Biophys Chem. 1998;75:1. doi: 10.1016/s0301-4622(98)00194-x. [DOI] [PubMed] [Google Scholar]
- 8.Lee CH, Wetmur JG. Biochem Biophys Res Commun. 1973;50:879. doi: 10.1016/0006-291x(73)91327-2. [DOI] [PubMed] [Google Scholar]
- 9.Meyer RB, Jr, Shuman DA, Robins RK, Miller JP, Simon LN. J Med Chem. 1973;16:1319. doi: 10.1021/jm00270a001. [DOI] [PubMed] [Google Scholar]
- 10.Tolman GL, Barrio JR, Leonard NJ. Biochemistry. 1974;13:4869. doi: 10.1021/bi00721a001. [DOI] [PubMed] [Google Scholar]
- 11.Gualix J, Abal M, Pintor J, Miras-Portugal MT. FEBS Lett. 1996;391:195. doi: 10.1016/0014-5793(96)00732-6. [DOI] [PubMed] [Google Scholar]
- 12.Harvey SC, Cheung HC. Biochem Biophys Res Commun. 1976;73:865. doi: 10.1016/0006-291x(76)90201-1. [DOI] [PubMed] [Google Scholar]
- 13.Worthington RA, Hansen MA, Bennett MR, Barden JA, Balcar VJ. Biochem Biophys Res Commun. 1998;249:166. doi: 10.1006/bbrc.1998.8968. [DOI] [PubMed] [Google Scholar]
- 14.Caiolfa VR, Gill D, Parola AH. Biophys Chem. 1998;70:45. doi: 10.1016/s0301-4622(97)00106-3. [DOI] [PubMed] [Google Scholar]
- 15.Schram KH, Townsend LB. Tetrahedron Lett. 1974;15:1345. [Google Scholar]
- 16.Zhou J, Gao JG, Fischer B. Youji Huaxue. 2004;24:498. [Google Scholar]
- 17.Eritja R, Kaplan BE, Mhaskar D, Sowers LC, Petruska J, Goodman MF. Nucleic Acids Res. 1986;14:5869. doi: 10.1093/nar/14.14.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochetkov NK, Shibaev VW, Kost AA. Tetrahedron Lett. 1971;12:1993. [Google Scholar]
- 19.Sattsangi PD, Leonard NJ, Frihart CR. J Org Chem. 1977;42:3292. doi: 10.1021/jo00440a020. [DOI] [PubMed] [Google Scholar]
- 20.Kusmierek JT, Jensen DE, Spengler SJ, Stolarski R, Singer B. J Org Chem. 1987;52:2374. [Google Scholar]
- 21.Kusmierek JT, Singer B. Chem Res Toxicol. 1992;5:634. doi: 10.1021/tx00029a007. [DOI] [PubMed] [Google Scholar]
- 22.Boryski J. Nucleosides Nucleotides. 1990;9:803. [Google Scholar]
- 23.Kusmierek JT, Jensen DE, Spengler SJ, Stolarski R, Singer B. Tetrahedron Lett. 2000;41:1695. [Google Scholar]
- 24.Kikugawa K, Kawashima T. Chem Pharm Bull. 1971;19:2629. [Google Scholar]
- 25.Arun K, Mandel P, Shrotri A, Ghogare D. Synth Commun. 1986;16:221. [Google Scholar]
- 26.Campbell J, Greene E, Lavagnino D, Gardner D, Pike A, Snoddy J. J Heterocycl Chem. 1986;23:669. [Google Scholar]
- 27.Nguyen C, Emile B. Synth Commun. 1984;14:765. [Google Scholar]
- 28.Simon R, Vinod K. Tetrahedron. 1988;44:6367. [Google Scholar]
- 29.Guillermo G, Angeles B, Nuria R, Teresa G, Rosario G. Heterocycles. 2002;56:501. [Google Scholar]
- 30.Michael J, Jean M. Helv Chim Acta. 1998;81:1921. [Google Scholar]
- 31.John A, Anita S, Lee B, John A. Nucleosides Nucleotides. 1994;13:1017. [Google Scholar]
- 32.Jack JW, Alexander H, Iris L. J Am Chem Soc. 1958;80:1669. [Google Scholar]
- 33.Kusmierek JT, Folkman W, Singer B. Chem Res Toxicol. 1989;2:230. doi: 10.1021/tx00010a003. [DOI] [PubMed] [Google Scholar]
- 34.Lebruska LL, Kuzmine II, Fedor MJ. Chem Biol. 2002;9:465. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 35.Major DT, Laxer A, Fischer B. J Org Chem. 2002;67:790. doi: 10.1021/jo0107554. [DOI] [PubMed] [Google Scholar]
- 36.Hand SM, Paudler WW. Tetrahedron. 1982;38:49. [Google Scholar]
- 37.Beland FA, Fullerton NF, Heflich RH. J Chromatogr. 1984;308:121. doi: 10.1016/s0021-9673(01)87539-7. [DOI] [PubMed] [Google Scholar]
- 38.Barrio JR, Secrist JA, III, Chien Y-h, Taylor PJ, Robinson JL, Leonard NJ. FEBS Lett. 1973;29:215. doi: 10.1016/0014-5793(73)80022-5. [DOI] [PubMed] [Google Scholar]
- 39.Conibear PB, Jeffreys DS, Seehra CK, Eaton RJ, Bagshaw CR. Biochemistry. 1996;35:2299. doi: 10.1021/bi951824+. [DOI] [PubMed] [Google Scholar]
- 40.Rosenfeld SS, Taylor EW. J Biol Chem. 1984;259:11 920. [PubMed] [Google Scholar]
- 41.Vöhringer C, Schäfer R, Reiser G. Biochem Pharmacol. 2000;59:791. doi: 10.1016/s0006-2952(99)00390-1. [DOI] [PubMed] [Google Scholar]
- 42.Zündorf G, Schäfer R, Vöhringer C, Halbfinger E, Fischer B, Reiser G. Biochem Pharmacol. 2001;61:1259. doi: 10.1016/s0006-2952(01)00593-7. [DOI] [PubMed] [Google Scholar]
- 43.Sharon E, Zündorf G, Levesque SA, Beaudoin AR, Reiser G, Fischer B. Bioorg Med Chem. 2004;12:6119. doi: 10.1016/j.bmc.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 44.Jacobson KA, Moro S, Hoffmann C, Kim YC, Kim HS, Ravi RG, Harden TK, Boyer JL. Farmaco. 2001;56:71. doi: 10.1016/s0014-827x(01)01023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazarowski ER, Watt WC, Stutts MJ, Boucher RC, Harden TK. Br J Pharmacol. 1995;116:1619. doi: 10.1111/j.1476-5381.1995.tb16382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gendron FP, Neary JT, Theiss PM, Sun GY, Gonzalez FA, Weisman GA. Am J Physiol Cell Physiol. 2003;284:C571. doi: 10.1152/ajpcell.00286.2002. [DOI] [PubMed] [Google Scholar]
- 47.Kukulski F, Lévesque SA, Lavoie EG, Lecka J, Bigonnesse F, Knowles AF, Robson SC, Kirley TL, Séigny J. Purinergic Signalling. 2005;1:193. doi: 10.1007/s11302-005-6217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khazanchi R, Yu PL, Johnson F. J Org Chem. 1993;58:2552. [Google Scholar]
- 49.Fischer B, Boyer JL, Hoyle CHV, Ziganshim AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA. J Med Chem. 1993;36:3937. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsin ATC, Pedrozo-Fernandez HA, Gallas JM, Chambers JP. Life Sci. 1988;43:1379. doi: 10.1016/0024-3205(88)90304-9. [DOI] [PubMed] [Google Scholar]
- 51.Ubl JJ, Reiser G. Glia. 1997;21:361. doi: 10.1002/(sici)1098-1136(199712)21:4<361::aid-glia3>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 52.Ubl JJ, Sergeeva M, Reiser G. J Physiol. 2000;525:319. doi: 10.1111/j.1469-7793.2000.00319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tulapurkar ME, Laubinger W, Nahum V, Fischer B, Reiser G. Br J Pharmacol. 2004;142:869. doi: 10.1038/sj.bjp.0705859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baykov AA, Evtushenko OA, Avaeva SM. Anal Biochem. 1988;171:266. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 55.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.