

Abstract
The nuclear receptor corepressor (NCoR) and the related factor known as silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) are essential components of multiprotein complexes that mediate active repression by unliganded nuclear receptors. Recent studies suggest that NCoR and SMRT can interact with and exert repressive effects on several other classes of DNA-binding transcription factors, but the physiological importance of these interactions has not been established. Here, investigation of endogenous transcriptional programs regulated by NCoR in macrophages reveals that NCoR acts as a transcriptional checkpoint for activator protein (AP)-1-dependent gene networks that regulate diverse biological processes including inflammation, cell migration, and collagen catabolism, with loss of NCoR, resulting in derepression of AP-1 target genes. The NCoR corepressor complex imposes an active block of exchange of c-Jun for c-Jun/c-Fos heterodimers, with targeted deletion of the c-Jun locus, resulting in loss of NCoR complexes from AP-1 target genes under basal conditions. The checkpoint function of NCoR is relieved by signal-dependent phosphorylation of c-Jun, which directs removal of NCoR/HDAC3/TBL1/TBLR1 complexes through recruitment of a specific ubiquitylation complex, as a prerequisite to the default binding of c-Jun/c-Fos heterodimers and transcriptional activation. The requirement for a checkpoint function to achieve the appropriate dynamic range of transcriptional responses to inflammatory signals is likely to be used by other signal-dependent transcription factors that regulate diverse homeostatic and developmental processes.
Precise regulation of the transition from gene repression to activation is essential for development and function of mammalian organ systems. In the case of genes regulated by a subset of heterodimeric nuclear hormone receptors, exemplified by the retinoic acid and thyroid hormone receptors, binding of hormone effects a switch in receptor function from active repression to transcriptional activation (1-3). Investigation of mechanisms accounting for active repression by unliganded retinoic acid and thyroid hormone receptors led to the identification of the nuclear receptor corepressor (NCoR) and the highly related silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) (4, 5). NCoR and SMRT are essential components of multiprotein complexes that mediate active repression by means of associated enzymatic activities that include histone deacetylase (HDAC) activity, with biochemical purification of NCoR and SMRT corepressor complexes from HeLa cells, demonstrating that they both contain transducin β-like protein 1 (TBL1), TBL1-related protein (TBLR1), HDAC3, and GPS2 (6-8).
Interaction of NCoR and SMRT with unliganded nuclear receptors is mediated by a conserved extended helical motif, referred to as the CoRNR box, of consensus sequence LXXI/HIXXXI/L (9-13). Binding of hormone results in a conformational change in the ligand-binding domain that reduces affinity for NCoR/SMRT complexes while simultaneously enhancing affinity for coactivators that contain a conserved LXXLL interaction motif (1, 3, 13), thereby converting the receptor from a transcriptional repressor to an activator. Whereas ligand binding is sufficient to cause dissociation of NCoR and SMRT from retinoic acid and thyroid hormone receptors in vitro, recent studies indicate that an additional, ubiquitylation-mediated proteolytic step is required for removal of NCoR complexes from nuclear receptor target genes in vivo (14), perhaps due to the ability of the TBL1 and TBLR1 components of these complexes to interact directly with chromatin (15). In the case of retinoic acid receptor target genes, the TBLR1 component of the NCoR complex appears to function as an E3-ligase that directs ligand-dependent ubiquitylation and proteosome-mediated clearance of NCoR and HDAC3 (14).
Although initially identified as NCoRs, a number of studies suggest that NCoR and SMRT can interact with and exert repressive effects on several other classes of DNA-binding transcription factors, including activator protein (AP)-1 proteins (8, 14, 16), NF-κB factors (14, 16, 17), Eto (18, 19), homeodomain-containing proteins (20-22), and the E-26-transforming specific-domain proteins Tel (23, 24) and MEF2c (25). These findings suggest that NCoR and SMRT play diverse roles in mediating active transcriptional repression during development. Consistent with this hypothesis, deletion of the NCoR gene in the mouse resulted in embryonic lethality and severe developmental defects in the lymphocytic and erythropoietic lineages and the CNS (26). However, the specific target genes that account for these defects have not been identified and NCoR-dependent gene networks remain largely undefined.
In the present study, we explore the roles of NCoR in regulating tissue-specific programs of gene expression by using macrophages as a model. Whereas NCoR is not required for macrophage differentiation, genes involved in inflammation, chemotaxis, cell-cycle control, and collagen metabolism are up-regulated in NCoR-deficient macrophages, resulting in a partially activated phenotype. A significant fraction of the derepressed genes identified in NCoR-deficient macrophages are targets of AP-1 transcription factors, and several lines of evidence indicate that c-Jun functions as a required molecular beacon for recruitment of NCoR to these genes in the absence of an activating signal. Phosphorylation of the c-Jun N terminus initiates a TBLR1-dependent clearance of NCoR and HDAC3 from AP-1 target genes and permits the binding of c-Jun/c-Fos heterodimers. In concert, these findings indicate that the transcriptional checkpoint function of NCoR is required to establish the physiological dynamic range of signal-dependent gene expression in gene networks regulated by AP-1 transcription factors.
Materials and Methods
Cell Culture. Fetal liver-derived macrophages generated from embryonic day 14.5 embryo liver were plated and cultured in RPMI medium 1640 with 10% FBS plus L cell media for 7 days. Bone marrow-derived macrophages were obtained as described (27). Lipopolysaccharide (LPS) (Sigma) and 12-O-tetradecanoylphorbol-13-acetate (TPA) (Sigma) were used at a concentration of 100 ng/ml and 100 nM, respectively.
Materials and Reagents. Anti-TBL1 and anti-TBLR1 antibodies were generated as described (14). The following antibodies were obtained from Santa Cruz Biotechnology: anti-RARα, anti-HDAC3, anti-c-Jun, anti-c-Fos, and anti-p50. Additionally, anti-UbcH5 (Boston Biochem, Cambridge, MA), anti-SMRT (Affinity BioReagents, Neshanic Station, NJ) and anti-hemagglutinin (HA) (Covance, Richmond, CA) were also used in immunoprecipitation experiments.
Expression Array Profiling. RNA preparation, cRNA synthesis, and its hybridization to Affymetrix arrays were described (28). Hybridization data were collected from 36 independent hybridizations of Affymetrix U74A microarrays. Data were subjected to secondary analysis by using a multiplicative noise model (29), genespring software (Silicongenetics, Redwood City, CA) and the significance analysis of microarrays application (30).
Gene Ontology (GO) Analysis. Genes significantly up- or down-regulated in NCoR-/- macrophages and at least 1.5-fold above or below levels of expression in control macrophages were connected to biological process annotations provided by the GO Consortium. Based on the hierarchical structure of the GO annotations, the probability that each immediate daughter term (a P value) be linked to the number of selected genes by chance was calculated. This probability is
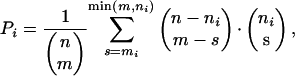 |
1 |
where n is the number of genes in the references list, m is the number of selected genes, ni is the number of reference genes connected to the term i, and mi is the number of selected genes connected to the term i. The link from the root to the daughter term with the lowest P value is removed and the process is started from the root again. In this step, the probabilities are calculated given the fact that one (most significant) daughter term is linked to a fixed number of genes. Eq. 1 is modified accordingly. When all links from the root are removed, the daughter terms become the root (one at a time), and so on, recursively, until all links in the subgraph are exhausted. We define the P value of a term as the smallest P value of a link that connects it with a parent (a term may have multiple parents).
Transient Transfection and Reporter Studies. Cells were transfected with 0.5 μg of the luciferase reporter plasmid and with 0.5 μg of NCoR expression plasmid by using Superfect transfection reagent (Qiagen, Valencia, CA). pRL-TK vector was also cotransfected as an internal control. After 12 h of incubation, cells were treated with TPA and were incubated for an additional 24 h. A luciferase assay was performed as described (31). The reporter gene constructs for matrix metalloproteinase (Mmp)12 (wild-type and AP-1 mutant), 3×AP-1, 5×UAS, and β-actin have been described (32-34). Gal-c-Jun and Gal-c-JunS63/73A were made by subcloning 1-252 aa of c-Jun and c-JunS63/73A into pCMX-Gal4 expression vector, respectively (35).
Chromatin Immunoprecipitation (ChIP) Analysis. A ChIp assay was conducted as described (14, 27). Briefly, macrophages and RAW264.7 cells were fixed with 1% formaldehyde and cross-linked adducts were resuspended and sonicated, resulting in DNA fragments of 300-900 bp. Protein-bound, immunoprecipitated DNA was reverse cross-linked at 65°C overnight and then purified by using a Qiagen PCR purification. A quantity of 4 μl from a 50-μl DNA extraction was used for PCR amplification (28-33 cycles).
Single-Cell Nuclear Microinjection Assay. The single-cell nuclear microinjection assays were performed as described (14). Subcloning the 1.0-kb proximal region of the Mmp12 promoter into the pGL2/LacZ reporter plasmid generated the Mmp12-LacZ. Specific short interfering RNA (siRNA) or nonspecific control siRNA (Dharmacon, Boston) was coinjected in each experiment.
Invasion of Basement Membrane Matrigel by Macrophages. Macrophages (5 × 10-4 cells per well) were placed on filters coated with an 8-μm-thick layer of Matrigel (Becton Dickinson). The lower compartment of the Boyden chamber contained the medium with murine monocyte chemotactic protein 1 (R & D Systems) at 10 ng/ml. After 4 days in culture, filters were washed and cells traversed to the undersurface of the filter were counted after a modified Wright stain.
Genetic Manipulation. The generation of a floxed allele for c-Jun has been described (36). c-Junf/f mice were bred to mice bearing a Cre recombinase transgene under transcriptional control of the Mx-1 promoter (37). Subsequent matings were performed to establish c-Junf/f × MxCre+ and c-Junf/f × MxCre- mice. Mice were injected with polyinosinic/polycytidylic acid to induce MxCre expression as described (38).
Results and Discussion
To explore roles of corepressor-dependent events in regulating tissue-specific programs of gene expression, macrophages were derived from wild-type and NCoR-/- embryos by culturing fetal liver hematopoietic progenitor cells in the presence of macrophage colony-stimulating factor (26). Although NCoR-/- macrophages were indistinguishable from wild-type macrophages with respect to morphology and expression of characteristic markers, including F4/80 and CD11b (data not shown), microarray analysis of ≈8,000 transcripts led to the identification of >150 genes exhibiting significantly different levels of expression in wild-type and NCoR-/- macrophages under basal conditions (Fig. 1A and Table 1, which is published as supporting information on the PNAS web site). Analysis of biological process annotations defined by the GO Consortium (39), associated with each differentially expressed gene, indicated statistically significant overrepresentation of genes involved in several categories, including cell-cycle control, inflammatory responses, and collagen catabolism. A subset of these genes is illustrated in Fig. 1B and an interactive directed acyclic graph for the entire set of differentially expressed genes is provided as Table 2, which is published as supporting information on the PNAS web site.
Fig. 1.

NCoR regulates programs of gene expression involved in inflammation and cell-cycle control. (A) Significance analysis of microarrays plot illustrating significant changes in gene expression in NCoR-/- macrophages as compared with wild-type macrophages. Expression data from four independent experiments were used for analysis. Points above the upper diagonal line represent significantly overexpressed genes and points below the diagonal line represent significantly underexpressed genes. Red and green points represent genes that are at least 1.5-fold over- or underexpressed in NCoR-/- macrophages, respectively. (B) Subsets of genes up-regulated in NCoR-/- macrophages with associated GO biological process annotations related to responses to external stimuli (including inflammatory response/chemotaxis annotations), cell proliferation (including M-phase annotations), and collagen catabolism. (C) Enhanced invasive activities of NCoR-/- macrophages. Wild-type and NCoR-/- macrophages were placed in the upper cell of a Boyden chamber that was separated from the lower cell by a porous filter and a collagen (Matrigel) matrix. Transmigration of macrophages through the matrix in response to murine monocyte chemotactic protein 1 was quantified 96 h later. hpf, high-power field. (Upper) Macrophages that are adherent to the undersides of filters.
This profile of gene expression suggested that the loss of NCoR resulted in functional characteristics of activated macrophages. Mmp12,-13, and -9 were among the genes overexpressed in NCoR-/- macrophages, with Mmp12 exhibiting a nearly 8-fold increase in expression, which was confirmed by Northern blotting experiments (Fig. 6, which is published as supporting information on the PNAS web site). The chemokine receptor Ccr1 and chemokines Ccl5, Ccl8, Cxcl1, and Cxcl14 were among the genes that were significantly derepressed in NCoR-/- macrophages, indicating the critical importance of corepressor imposed restriction in maintaining normal signal-dependent regulation. Macrophage migration through tissues in response to inflammatory stimuli is directed by chemokines and requires the expression of Mmps that degrade extracellular matrix components, including interstitial collagen (40, 41). Experiments were performed based on the predicted functional consequences of this derepression program to evaluate macrophage chemotaxis and invasiveness. These studies confirmed that the integrated functions required for macrophage migration through a reconstituted basement membrane were significantly enhanced in NCoR-/- macrophages (Fig. 1C).
Although NCoR was initially identified by its interaction with nuclear receptors, relatively few of the derepressed genes in NCoR-/- macrophages could be demonstrated to be nuclear receptor targets (data not shown). To explore the potential relationship between other signaling pathways that regulate macrophage function and NCoR target genes, microarray experiments were performed to characterize the transcriptional responses of wild-type and NCoR-/- macrophages to LPS and TPA. LPS activates Toll-like receptor 4 and regulates broad sets of genes involved in native and acquired immunity by stimulating the activities of transcription factors, including NF-κB and IFN regulatory factors (42), whereas TPA regulates gene expression in part by stimulating the activities of AP-1 transcription factors (43). Approximately 10% of the LPS-responsive genes and 30% of the TPA-responsive genes were derepressed under basal conditions in NCoR-/- macrophages, respectively (Fig. 2A). The expression profiles for the 30 most highly induced TPA-responsive genes indicated that approximately two-thirds of these genes were derepressed in NCoR-/- macrophages (Fig. 2B).
Fig. 2.

NCoR is required for active repression of a subset of TPA-inducible genes in macrophages. (A) Venn diagram indicating relationships between genes derepressed in NCoR-/- macrophages and genes induced by LPS and/or TPA. (B) Expression profiles for the 30 most highly induced TPA-responsive genes. Each column in the heat map represents the ratio of normalized expression for the two indicated conditions. Genes that exhibited increases in basal activity are red. (C) Expression profiles of target genes activated by TPA only (Mmp12), LPS only (Usp18), TPA or LPS (Cxcl1), or retinoic acid (Rarb). (D) ChIP assays of NCoR on target genes (Mmp12, Usp18, Cxcl1, or Rarb) in cells treated with TPA or LPS.
This pattern of gene expression suggested that signal-dependent relief of NCoR-dependent repression is achieved in a promoter-specific manner. To examine this possibility, ChIP experiments were performed in control, TPA-, or LPS-treated macrophages to correlate NCoR occupancy with transcriptional activation. Mmp12 was used as an example of a TPA-selective NCoR target gene; Usp18, as an example of a LPS-selective NCoR target gene; Cxcl1, as an example of an NCoR target gene induced by both TPA and LPS; and Rarb, as an example of an NCoR target gene not regulated by TPA or LPS (Fig. 2C). NCoR was bound under basal conditions to each of these genes, which was consistent with their being derepressed in NCoR-/- macrophages (Fig. 2D). TPA treatment resulted in loss of NCoR interaction from the Mmp12 and Cxcl1 promoters, but not from the Usp18 or Rarb promoters. Conversely, LPS treatment resulted in dissociation of NCoR from the Usp18 and Cxcl1 promoters, but not from the Mmp12 or Rarb promoters (Fig. 2D), revealing independent regulation of NCoR activity in a signal- and promoter-specific manner.
Many of the TPA-responsive genes derepressed in NCoR-/- macrophages are transcriptional targets of AP-1, as exemplified by Mmp12. To investigate the potential role of AP-1 as an essential target for NCoR activity, transient transfection studies of the Mmp12 promoter were performed in RAW264.7 macrophages (32). The Mmp12 promoter was strongly activated by TPA in these cells, and this response was blunted by overexpression of NCoR (Fig. 3A). The residual activity of an analogous Mmp12 promoter construct containing a mutated AP-1 site was not inhibited by overexpression of NCoR, suggesting that the AP-1 site was the critical target of NCoR-mediated repression. Overexpression of NCoR inhibited the activity of a minimal promoter containing 3xAP-1 sites, but not the β-actin promoter (Fig. 3A). Conversely, basal and TPA-dependent activities of the Mmp12 promoter in RAW264.7 cells were enhanced by cotransfection of siRNAs directed against NCoR that reduced its expression by >80%, but not by cotransfection with a control siRNA (Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 3.

c-Jun is a required molecular beacon for recruitment of NCoR to a subset of inflammatory response genes. (A) Activity of the wild-type Mmp12 promoter and an analogous Mmp12 promoter containing a mutated AP-1 site in RAW264.7 macrophages. Activities of a simple AP-1-responsive reporter gene (3xAP-1-Luc) and a β-actin-luciferase reporter gene in RAW264.7 macrophages were also demonstrated. Cells were cotransfected with an NCoR expression vector and treated with TPA as indicated. (B) Development of c-Jun-/- macrophages. Breeding of c-Junf/f mice and MxCre transgenic mice was performed to obtain mice with c-Junf/f × MxCre- or c-Junf/f × MxCre+ genotypes. Both genotypes were treated with polyinosinic/polycytidylic acid, with induction of Cre in MxCre+ mice, resulting in quantitative recombination of the c-Junf/f locus in bone marrow-derived macrophages. Western blotting indicates absence of c-Jun protein c-Junf/f × MxCre+ macrophages. Macrophages from three mice of each genotype were analyzed. (C) ChIP assays of three TPA-inducible NCoR target genes (Mmp9, Mmp12, and Mmp13) and two control genes (Csf3 and Alcam) in wild-type and c-Jun-/- macrophages. Specific antibodies against c-Jun, NCoR, or normal IgG were used to immunoprecipitate cross-linked chromatin derived from wild-type or c-Jun-/- macrophages. (D) ChIP assays of distal and promoter-proximal regions of the Mmp12 gene for NCoR, SMRT, c-Jun, and c-Fos in RAW264.7 cells before and 1 h after TPA treatment. (E) ChIP assays for c-Jun, c-Fos, NCoR, and HDAC3 on the Mmp12 promoter in wild-type and NCoR-/- macrophages. (F) ChIP assay documenting presence of NCoR, TBL1, TBLR1, and HCAC3 on Mmp12, Usp18, Cxcl1, and Rarb promoters under basal conditions in RAW264.7 macrophages.
These findings raised intriguing questions concerning the molecular mechanisms by which NCoR was recruited to AP-1 target genes, and the role of derepression in signal-dependent activation of AP-1 gene targets. c-Jun-deficient macrophages were developed by using MxCre to quantitatively disrupt floxed c-Jun alleles in bone marrow-derived macrophages (Fig. 3B and Fig. 8, which is published as supporting information on the PNAS web site). ChIP experiments were then performed, comparing wild-type and c-Jun-/- macrophages for three AP-1 target genes that were derepressed in NCoR-/- macrophages (Mmp9, -12, and -13) and two TPA-responsive genes that were not derepressed in NCoR-/- macrophages (Csf3 and Alcam). c-Jun was detected on the Mmp9, -12, and -13 promoters in wild-type macrophages, but not in c-Jun-/- macrophages, which was consistent with the absence of detectable c-Jun protein (Fig. 3C). Under basal conditions, NCoR was selectively present on the AP-1 target genes in wild-type macrophages, but not in c-Jun-/- macrophages (Fig. 3C), indicating an essential role of c-Jun in recruitment of NCoR to these genes.
TPA treatment caused dissociation of NCoR from the Mmp12 promoter, which is associated with coordinate recruitment of c-Fos to form c-Jun/c-Fos heterodimers (Fig. 3D), suggesting a mechanistic link between signal-dependent dissociation of NCoR and binding of c-Jun/c-Fos heterodimers. Although the related corepressor SMRT has been shown to interact with c-Jun and c-Fos and inhibit AP-1 transcriptional activity (16), relatively low levels of SMRT were observed on the Mmp12 promoter and were not altered by TPA treatment (Fig. 3D). Intriguingly, ChIP experiments examining the Mmp12 promoter in control and NCoR-/- macrophages revealed significantly increased occupancy by c-Fos in NCoR-/- cells in the absence of a signal (Fig. 3E). Although c-Fos expression is often dramatically induced in a signal-dependent manner, c-Fos is constitutively expressed in macrophages and the levels of its expression were not different in wild-type and NCoR-/- macrophages (data not shown). These findings thus reveal an active role of NCoR in inhibiting the formation of c-Jun/c-Fos heterodimers in the absence of an activating signal. In support of this, NCoR was found to inhibit the binding of c-Jun/c-Fos heterodimers to an AP-1 site in vitro (Fig. 9 and Supporting Text, which are published as supporting information on the PNAS web site).
Signal-specific dismissal of NCoR from the Mmp12, Usp18, Cxcl1, and Rarb genes raised the possibility that distinct NCoR complexes might be associated with differentially regulated target genes. However, ChIP experiments indicated that an apparently identical complex containing NCoR, TBL1, TBLR1, and HDAC3 (6-8) resides on the Mmp12, Usp18, Cxcl1, and Rarb genes under basal conditions (Fig. 3F). The finding that HDAC3 is recruited to endogenous AP-1 target genes is consistent with recent studies (44), indicating that HDAC3 suppresses signal-dependent activation of Gal4-c-Jun fusion proteins. Reduced occupancy of the Mmp12 promoter by HDAC3 in NCoR-/- macrophages (Fig. 3E) strongly suggests that it is at least partly recruited to AP-1 target genes in an NCoR-dependent manner.
The presence of a common NCoR complex on differentially regulated promoters suggested that signal-dependent dismissal of NCoR was determined by the specific transcription factors to which it bound. Signal-dependent activation of c-Jun requires phosphorylation of serines 63 and 73 (S63 and S73) by c-Jun N-terminal kinases or related kinases (45, 46) and is induced by TPA treatment of macrophages (data not shown). To specifically evaluate the role of c-Jun independent of potential heterodimeric partners, experiments were performed by using Gal4-c-Jun fusion proteins. Although Gal4-c-Jun fusion proteins typically function as signal-dependent activators when assessed on artificial promoters consisting of Gal4 (UAS)-binding sites linked to minimal promoter elements (44, 47), the use of a constitutively active UAS-TK-lacZ reporter gene revealed that Gal4-c-Jun functioned as a strong transcriptional repressor under basal conditions (Fig. 4A). The repression function of Gal4-c-Jun was relieved by TPA treatment or siRNA-mediated knockdown of NCoR expression (Fig. 4 A and B). In addition, siRNA-mediated knockdown of NCoR expression resulted in equivalent levels of derepression by Gal4-c-Jun and a Gal4-c-Jun chimera in which c-Jun S63 and S73 were mutated to alanine (Gal4-c-JunS63/73A, Fig. 4B).
Fig. 4.

Mechanism of signal-specific derepression of AP-1 target genes. (A) Gal-c-Jun represses transcription from a UAS-TK-LacZ reporter gene in HS68 cells. Cells were microinjected with the UAS-TK-lacZ reporter gene, GAL4-c-Jun expression plasmid, and NCoR siRNA, and treated with TPA as indicated. The percent of LacZ-positive cells was determined 12 h later. (B) Gal-c-Jun and Gal-c-JunS63/73A are equivalently derepressed after siRNA-mediated knockdown of NCoR expression. RAW264.7 cells were transfected with the indicated plasmids and NCoR siRNA and luciferase activity was measured 16 h later. (C) TPA-dependent dismissal of NCoR from the Mmp12 promoter requires phosphorylation of c-Jun S63/73. RAW264.7 macrophages were transfected with HA-c-Jun or HA-c-JunS63/73A and treated with TPA or control solvent. ChIP was performed by using anti-HA antibody followed by a second round of precipitation using anti NCoR IgG or control IgG. (D) ChIP assay indicates that UbcH5 is recruited to the Mmp12 promoter and NCoR and HDAC3 are dismissed within minutes of TPA treatment of RAW264.7 cells. (E) TPA induction of the Mmp12 promoter gene requires TBLR1. HS68 cells were microinjected with the Mmp12 promoter linked to lacZ and the indicated siRNAs. Cells were treated with control solvent or TPA and assayed for lacZ expression 12 h later. (F) TPA-dependent recruitment of UbcH5 to the Mmp12 promoter requires phosphorylation of c-Jun S63/73. RAW264.7 macrophages were transfected with HA-c-Jun or HA-c-JunS63/73A and treated with TPA or control solvent. ChIP was performed by using anti-HA antibody followed by a second round of precipitation using anti-UbcH5 IgG or control IgG.
To determine whether phosphorylation of c-Jun S63/73 is required for dismissal of NCoR from the Mmp12 promoter, RAW264.7 macrophages were transfected with HA-tagged wild-type c-Jun or HA-tagged c-JunS63/73A and were treated with vehicle or TPA. Sequential ChIP assays for HA-tagged c-Jun proteins and NCoR demonstrated that NCoR dissociated from the endogenous Mmp12 promoter bound by wild-type c-Jun within 5 min after TPA treatment, but not from the Mmp12 promoter bound by c-JunS63/73A (Fig. 4C), indicating an essential role of c-Jun N-terminal phosphorylation in promoting the events leading to NCoR dismissal.
Recent studies have suggested that that the TBLR1 component of the NCoR/TBL1/TBLR1/HDAC3 complex is required for clearance of NCoR from target genes by a proteosome-dependent mechanism involving the recruitment of a specific ubiquitylation complex containing the conjugating enzyme UbcH5, with corecruitment of the 19S proteasome (14). Consistent with these findings, UbcH5 was recruited to the Mmp12 promoter within 2 min of TPA treatment, with subsequent dismissal of NCoR (Fig. 4D). Furthermore, microinjection of siRNAs directed against TBLR1 blocked Mmp12 activation (Fig. 4E), suggesting that TPA-dependent activation of the Mmp12 promoter required TBLR1 with the associated ubiquitylation/19S proteasome complexes (14). This requirement was abrogated if NCoR/SMRT expression was knocked down by using specific siRNAs (data not shown), which was in agreement with the model that recruitment of the UbcH5-containing ubiquitylation complex by TBLR1 resulted in proteolytic clearance of NCoR complexes. Sequential ChIP assays for HA-tagged c-Jun proteins and UbcH5 demonstrated that UbcH5 was recruited to the Mmp12 promoter bound by wild-type c-Jun after TPA treatment, but not from the Mmp12 promoter bound by c-JunS63/73A (Fig. 4F), indicating an essential role of c-Jun N-terminal phosphorylation for recruitment of the proteolytic machinery required for NCoR dismissal.
These studies define an essential role of NCoR in physiological control of macrophage activation by acting as a checkpoint that controls signal-dependent exchange of c-Jun/corepressor complexes for c-Jun/c-Fos/coactivator complexes (Fig. 5). Although initially discovered as an NCoR, the identification of broad sets of NCoR target genes supports the emerging view that NCoR plays diverse roles in mediating active repression by a large number of additional sequence-specific regulated transcription factors. Whereas the full spectrum of these factors remains to be identified, the present studies establish c-Jun as an essential factor for the recruitment of NCoR to a significant subset of AP-1 target genes involved in the evolution of inflammatory responses. NCoR is thus likely to account, at least in part, for the cellular inhibitors of AP-1 initially identified nearly 15 years ago (47, 48). We propose that c-Jun N-terminal phosphorylation initiates a series of events, including recruitment of a specific ubiquitylation complex that initiates a proteolytic mechanism for removal of NCoR complexes from AP-1 target genes (Fig. 5), providing an explanation for signal-specific dismissal of NCoR from a subset of genes that are activated by TPA (Fig. 2 C and D). The observation of signal-independent binding of c-Jun/c-Fos heterodimers to the Mmp12 promoter in NCoR-/- macrophages reveals that a critical role for NCoR is to impose specific regulation on the DNA-binding activities of AP-1 components to determine gene repression or activation, with the signal-specific removal of NCoR being an apparent prerequisite to the binding of c-Jun/c-Fos heterodimers. The requirement for a corepressor-dependent checkpoint function to achieve the appropriate dynamic range of transcriptional responses to inflammatory signals is likely to be used by other signal-dependent transcription factors that regulate diverse homeostatic and developmental processes.
Fig. 5.

Model for relief of the NCoR-dependent checkpoint on AP-1 target genes. Signal-dependent phosphorylation of c-Jun results in TBLR1-dependent recruitment of UbcH5 and proteolytic removal of the NCoR/HDAC3 complex. This procedure allows binding of c-Jun/c-Fos heterodimers and transcriptional activation.
Supplementary Material
Acknowledgments
We thank K. A. Ohgi for technical assistance, A. Z. Howarth for figure preparation, M. Karin (University of California at San Diego, La Jolla, CA) for the c-Jun and c-JunS63/73A plasmids, and M. W. Feinberg (Brigham and Women's Hospital, Boston) for the Mmp12-Luc plasmid. M.G.R. is an investigator with the Howard Hughes Medical Institute. This work was supported in part by National Institutes of Health Grant 5 R01 CA52599, a grant from the Stanford Reynolds Center (to C.K.G.), a National Institutes of Health grant, and the Sandler Program for Asthma Research (to M.G.R.).
Abbreviations: NCoR, nuclear receptor corepressor; Mmp, matrix metalloproteinase; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor; AP, activator protein; HDAC, histone deacetylase; TBL1, transducin β-like protein 1; TBLR1, TBL1-related protein; LPS, lipopolysaccharide; TPA, 12-O-tetradecanoylphorbol-13-acetate; ChIP, chromatin immunoprecipitation; GO, Gene Ontology; siRNA, short interfering RNA; HA, hemagglutinin.
References
- 1.Rosenfeld, M. G. & Glass, C. K. (2001) J. Biol. Chem. 276, 36865-36868. [DOI] [PubMed] [Google Scholar]
- 2.Naar, A. M., Lemon, B. D. & Tjian, R. (2001) Annu. Rev. Biochem. 70, 475-501. [DOI] [PubMed] [Google Scholar]
- 3.McKenna, N. J. & O'Malley, B. W. (2002) Cell 108, 465-474. [DOI] [PubMed] [Google Scholar]
- 4.Horlein, A. J., Naar, A. M., Heinzel, T., Torchia, J., Gloss, B., Kurokawa, R., Ryan, A., Kamei, Y., Soderstrom, M., Glass, C. K., et al. (1995) Nature 377, 397-404. [DOI] [PubMed] [Google Scholar]
- 5.Chen, J. D. & Evans, R. M. (1995) Nature 377, 454-457. [DOI] [PubMed] [Google Scholar]
- 6.Li, J., Wang, J., Nawaz, Z., Liu, J., Qin, J. & Wong, J. (2000) EMBO J. 19, 4342-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guenther, M., Lane, W., Fischle, W., Verdin, E. & Lazar, M. (2000) Genes Dev. 14, 1048-1057. [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang, J., Kalkum, M., Chait, B. T. & Roeder, R. G. (2002) Mol. Cell 9, 611-623. [DOI] [PubMed] [Google Scholar]
- 9.Nagy, L., Kao, H., Love, J., Li, C., Banayo, E., Gooch, J., Krishna, V., Chatterjee, K., Evans, R. & Schwabe, J. (1999) Genes Dev. 13, 3209-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu, X. & Lazar, M. (1999) Nature 402, 93-96. [DOI] [PubMed] [Google Scholar]
- 11.Perissi, S., Staszewski, L. M., McInerney, E. M., Kurokawa, R., Kronnes, A., Rose, D. W., Lambert, M. H., Milburn, M. V., Glass, C. K. & Rosenfeld, M. G. (1999) Genes Dev. 13, 3198-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb, P., Anderson, C., Valentine, C., Nguyen, P., Marimuthu, A., West, B., Baxter, J. & Kushner, P. (2000) Mol. Endocrinol. 14, 1976-1985. [DOI] [PubMed] [Google Scholar]
- 13.Xu, H. E., Stanley, T. B., Montana, V. G., Lambert, M. H., Shearer, B. G., Cobb, J. E., McKee, D. D., Galardi, C. M., Plunket, K. D., Nolte, R. T., et al. (2002) Nature 415, 813-817. [DOI] [PubMed] [Google Scholar]
- 14.Perissi, V., Aggarwal, A., Glass, C. K., Rose, D. W. & Rosenfeld, M. G. (2004) Cell 116, 511-526. [DOI] [PubMed] [Google Scholar]
- 15.Yoon, H. G., Chan, D. W., Huang, Z. Q., Li, J., Fondell, J. D., Qin, J. & Wong, J. (2003) EMBO J. 22, 1336-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, S. K., Kim, J. H., Lee, Y. C., Cheong, J. & Lee, J. W. (2000) J. Biol. Chem. 275, 12470-12474. [DOI] [PubMed] [Google Scholar]
- 17.Baek, S. H., Ohgi, K. A., Rose, D. W., Koo, E. H., Glass, C. K. & Rosenfeld, M. G. (2002) Cell 110, 55-67. [DOI] [PubMed] [Google Scholar]
- 18.Wang, J., Hoshino, T., Redner, R. L., Kajigaya, S. & Liu, J. M. (1998) Proc. Natl. Acad. Sci. USA 95, 10860-10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutterbach, B., Westendorf, J., Linggi, B., Patten, A., Moniwa, M., Davie, J., Huynh, K., Bardwell, V., Lavinsky, R., Rosenfeld, M., et al. (1998) Mol. Cell. Biol. 18, 7176-7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu, L., Lavinsky, R. M., Dasen, J. S., Flynn, S. E., McInerney, E. M., Mullen, T. M., Heinzel, T., Szeto, D., Korzus, E., Kurokawa, R., et al. (1998) Nature 395, 301-306. [DOI] [PubMed] [Google Scholar]
- 21.Asahara, H., Dutta, S., Kao, H. Y., Evans, R. M. & Montminy, M. (1999) Mol. Cell. Biol. 19, 8219-8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saleh, M., Rambaldi, I., Yang, X. J. & Featherstone, M. S. (2000) Mol. Cell. Biol. 20, 8623-8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarti, S. R. & Nucifora, G. (1999) Biochem. Biophys. Res. Commun. 264, 871-877. [DOI] [PubMed] [Google Scholar]
- 24.Guidez, F., Petrie, K., Ford, A. M., Lu, H., Bennett, C. A., MacGregor, A., Hannemann, J., Ito, Y., Ghysdael, J., Greaves, M., et al. (2000) Blood 96, 2557-2561. [PubMed] [Google Scholar]
- 25.Wu, X., Li, H., Park, E. J. & Chen, J. D. (2001) J. Biol. Chem. 276, 24177-24185. [DOI] [PubMed] [Google Scholar]
- 26.Jepsen, K., Hermannson, O., Thandi, O., Gleiberman, A., Lunyak, V., Kurokawa, R., Kumar, V., Liu, F., Seto, E., Hedrick, S., et al. (2000) Cell 102, 753-763. [DOI] [PubMed] [Google Scholar]
- 27.Sawka-Verhelle, D., Escoubet-Lozach, L., Fong, A. L., Hester, K. D., Herzig, S., Lebrun, P. & Glass, C. K. (2004) J. Biol. Chem. 279, 17772-17784. [DOI] [PubMed] [Google Scholar]
- 28.Welch, J. S., Ricote, M., Akiyama, T. E., Gonzalez, F. J. & Glass, C. K. (2003) Proc. Natl. Acad. Sci. USA 100, 6712-6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasik, R., Calvo, E. & Corbeil, J. (2002) Bioinformatics 18, 1633-1640. [DOI] [PubMed] [Google Scholar]
- 30.Tusher, V. G., Tibshirani, R. & Chu, G. (2001) Proc. Natl. Acad. Sci. USA 98, 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogawa, S., Fujita, M., Ishii, Y., Tsurukami, H., Hirabayashi, M., Ikeda, K., Orimo, A., Hosoi, T., Ueda, M., Nakamura, T., et al. (2000) J. Biol. Chem. 275, 21372-21379. [DOI] [PubMed] [Google Scholar]
- 32.Feinberg, M. W., Jain, M. K., Werner, F., Sibinga, N. E., Wiesel, P., Wang, H., Topper, J. N., Perrella, M. A. & Lee, M. E. (2000) J. Biol. Chem. 275, 25766-25773. [DOI] [PubMed] [Google Scholar]
- 33.Ricote, M., Li, A. C., Willson, T. M., Kelly, C. J. & Glass, C. K. (1998) Nature 391, 79-82. [DOI] [PubMed] [Google Scholar]
- 34.Klappacher, G. W., Lunyak, V. V., Sykes, D. B., Sawka-Verhelle, D., Sage, J., Brard, G., Ngo, S. D., Gangadharan, D., Jacks, T., Kamps, M. P., et al. (2002) Cell 109, 169-180. [DOI] [PubMed] [Google Scholar]
- 35.Smeal, T., Binetruy, B., Mercola, D. A., Birrer, M. & Karin, M. (1991) Nature 354, 494-498. [DOI] [PubMed] [Google Scholar]
- 36.Palmada, M., Kanwal, S., Rutkoski, N. J., Gustafson-Brown, C., Johnson, R. S., Wisdom, R. & Carter, B. D. (2002) J. Cell. Biol. 158, 453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhn, R., Schwenk, F., Aguet, M. & Rajewsky, K. (1995) Science 269, 1427-1429. [DOI] [PubMed] [Google Scholar]
- 38.Akiyama, T. E., Sakai, S., Lambert, G., Nicol, C. J., Matsusue, K., Pimprale, S., Lee, Y. H., Ricote, M., Glass, C. K., Brewer, H. B., Jr., et al. (2002) Mol. Cell. Biol. 22, 2607-2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gene Ontology Consortium (2001) Genome Res. 11, 1425-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shipley, J. M., Wesselschmidt, R. L., Kobayashi, D. K., Ley, T. J. & Shapiro, S. D. (1996) Proc. Natl. Acad. Sci. USA 93, 3942-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brinckerhoff, C. E. & Matrisian, L. M. (2002) Nat. Rev. Mol. Cell Biol. 3, 207-214. [DOI] [PubMed] [Google Scholar]
- 42.Medzhitov, R. (2001) Nat. Rev. Immunol. 1, 135-145. [DOI] [PubMed] [Google Scholar]
- 43.Angel, P., Imagawa, M., Chiu, R., Stein, B., Imbra, R. J., Rahmsdorf, H. J., Jonat, C., Herrlich, P. & Karin, M. (1987) Cell 49, 729-739. [DOI] [PubMed] [Google Scholar]
- 44.Weiss, C., Schneider, S., Wagner, E. F., Zhang, X., Seto, E. & Bohmann, D. (2003) EMBO J. 22, 3686-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulverer, B. J., Kyriakis, J. M., Avruch, J., Nikolakaki, E. & Woodgett, J. R. (1991) Nature 353, 670-674. [DOI] [PubMed] [Google Scholar]
- 46.Eferl, R. & Wagner, E. F. (2003) Nat. Rev. Cancer 3, 859-868. [DOI] [PubMed] [Google Scholar]
- 47.Baichwal, V. R. & Tjian, R. (1990) Cell 63, 815-825. [DOI] [PubMed] [Google Scholar]
- 48.Auwerx, J. & Sassone-Corsi, P. (1991) Cell 64, 983-993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}