

Abstract
To understand why the classical two-state allosteric model of Monod, Wyman, and Changeux explains cooperative oxygen binding by hemoglobin but does not explain changes in oxygen affinity by allosteric inhibitors, we have investigated the kinetic properties of unstable conformations transiently trapped by encapsulation in silica gels. Conformational trapping reveals that after nanosecond photodissociation of carbon monoxide a large fraction of the subunits of the T quaternary structure has kinetic properties almost identical to those of subunits of the R quaternary structure. Addition of allosteric inhibitors reduces both the fraction of R-like subunits and the oxygen affinity of the T quaternary structure. These kinetic and equilibrium results are readily explained by a recently proposed generalization of the Monod-Wyman-Changeux model in which a preequilibrium between two functionally different tertiary, rather than quaternary, conformations plays the central role.
The two-state allosteric model of Monod, Wyman, and Changeux (1) represented a conceptual breakthrough in explaining the cooperative and regulated behavior of multisubunit proteins, with application to a wide range of biological systems (2-5). Monod, Wyman, and Changeux proposed that ligands control protein function by altering a preexisting equilibrium between high (R) and low (T) reactivity conformations that differ in intersubunit bonding (quaternary structure) and not by inducing conformational changes that are propagated to neighboring subunits as in a sequential model (6, 7). Enzyme activation, for example, results from preferential binding of ligands to the R quaternary structure, whereas inhibitors preferentially bind to T. However, a long-known serious deficiency in the application of the Monod-Wyman-Changeux (MWC) model to hemoglobin, the paradigm of allosteric proteins, is that inhibitors may also change oxygen (O2) affinity without a change in quaternary structure (8-11). To understand this phenomenon, we have investigated the ligand binding kinetics and equilibria of hemoglobin encapsulated in silica gels in either the T or R quaternary structure (Fig. 1).
Fig. 1.

Schematic structures of hemoglobin made with the program ribbons. Shown are deoxyhemoglobin in the T quaternary structure (a) (Protein Data Bank ID code 4HHB) and oxyhemoglobin in the R quaternary structure (b) (Protein Data Bank ID code 1HHO). The quaternary conformational change consists of a 15° relative rotation of the αβ dimers that are interchanged by the twofold symmetry (C2) axis. (c) A comparison of the tertiary structures, using the invariant interface between α and β subunits of the αβ dimer as a reference (37), is shown.
Previous studies of hemoglobin encapsulated in silica gels showed greatly simplified equilibrium properties, compared with those in solution, because quaternary conformational changes are markedly slowed by the constraints of the surrounding cross-linked polymer (12-16). In sharp contrast to hemoglobin free in solution, O2 binding to gel-encapsulated hemoglobin, like O2 binding to the hemoglobin crystal (17-19), is noncooperative (Fig. 2). Encapsulation as the fully deoxygenated molecule traps hemoglobin in the low-affinity T quaternary structure, whereas encapsulation as the fully oxygenated molecule traps hemoglobin in the high-affinity R structure (12). Moreover, the affinity of the deoxy-encapsulated molecule is lowered by inhibitor ligands (called negative heterotropic allosteric effectors) such as protons, chloride ions, inositol hexaphosphate, and bezafibrate (Fig. 2) in the same way as found for the T quaternary structure in solution (13, 15). The finding of noncooperative binding in both the R and T quaternary structures confirms the fundamental prediction of the MWC model that cooperativity arises from a shift in the population from T to R as successive molecules of O2 (the homotropic ligand) bind and not from direct subunit-subunit interactions as in a sequential model (20, 21) (see Supporting Text, which is published as supporting information on the PNAS web site). However, according to the MWC model, heterotropic effectors lower O2 affinity by preferential binding to the T quaternary structure and do not affect the affinity of either T or R, in contrast to experimental observations (8-11, 22).
Fig. 2.

O2-binding curves. Shown is a Hill plot of O2-binding curves for T- gel (100 mM Hepes buffer/1 mM EDTA, pH 7.0; cyan circles); T+ gel (100 mM Hepes buffer/10 mM inositol hexaphosphate/2 mM bezafibrate/200 mM chloride ions/1 mM EDTA, pH 7.0; blue circles); and single crystal in T quaternary structure, red dashed line. Data is from Mozzarelli et al. (19). y, fractional saturation of the hemes. The continuous lines through the data yield p50 = 26 ± 1 torr and n = 0.95 ± 0.03 for T-, and p50 = 134 ± 5 torr and n = 0.93 ± 0.03 for T+. The corresponding values for the crystal are p50 = 135 ± 1 torr and n = 0.97 ± 0.03. (Inset) Fraction of liganded r subunits in T quaternary structure (T- gels) from oxygen-binding curves [f(O2)] versus fraction of liganded r subunits in T quaternary structure from CO photolysis experiments [f(CO)], determined at different pH values and in the presence or absence of chloride ions. The former is calculated from f(O2) = {1 - [p50(T-)/p50(T+)]}. The p50 of the T+ gels is independent of pH between 6.6 and 7.6.
The O2 binding experiments suggested that if encapsulation also traps the CO complex of hemoglobin in the T quaternary structure, it should be possible to use ligand rebinding kinetics after photolysis (23, 24) to probe more deeply into the origin of the alteration in the affinity of the T quaternary structure. We therefore studied the nanosecond-to-millisecond CO rebinding kinetics of hemoglobin encapsulated in silica gels under different conditions. To trap the T quaternary structure, the fully deoxygenated molecule was encapsulated in the gel in the presence or in the absence of allosteric effectors. Then, CO was added after the gel was fully formed. To study the R quaternary structure, CO was added before encapsulation. Oxygen-binding curves were also measured for encapsulated deoxyhemoglobin (without addition of CO) to compare the O2 affinity and kinetics under the same conditions.
Materials and Methods
Sample Preparation. Human hemoglobin was purified as described in ref. 18. Encapsulation of deoxyhemoglobin in silica gels was performed with a slight modification of the protocol reported by Bruno et al. (15) in the presence and absence of allosteric effectors. For gels, which we call T-, a solution containing 10 mM Hepes/1 mM EDTA (pH 6.2) was added to an equal volume of tetramethyl orthosilicate and vortexed for a few minutes. The mixture was then deoxygenated by bubbling humidified helium for 90 min at 4°C. A solution containing 1.0% (wt/vol) deoxyhemoglobin A, 10 mM Hepes, 30 mM sodium dithionite, and 1 mM EDTA (pH 6.2) was added. The final hemoglobin concentration was ≈300 μM in heme. Gelification occurred in ≈10 min at room temperature. When the gel was formed, a solution containing 100 mM Hepes (at different pHs between 6.6 and 7.6), 30 mM sodium dithionite, and 1 mM EDTA was layered on it. The protocol for what we call T+ gels was the same except that before gelification allosteric effectors were added to produce a final concentration of 10 mM inositol hexaphosphate, 2 mM bezafibrate, and 0.2 M chloride ions in all solutions. Gels were inserted in a 1 × 1-cm gas-tight cuvette filled with a solution equilibrated with 1 atm CO (1 atm = 101.3 kPa).
Equilibrium and Kinetic Measurements. O2-binding curves were measured by using a microspectrophotometer (Zeiss) and O2 electrode (Yelow Springs Instruments) as described in refs. 13 and 15. For the kinetic studies, photolysis was carried out with the second harmonic (532 nm) of a Q-switched Nd:YAG laser (Continuum, Santa Clara, CA), with the 488-nm line of an Ar+ laser (Uniphase, San Jose, CA) as a probe source and an avalanche photodiode (Hamamatsu, Middlesex, NJ) for detection. The instrument is described in detail in ref. 25. All equilibrium and kinetic measurements were carried out at 15°C.
Results and Discussion
The rebinding kinetics of hemoglobin encapsulated as the CO complex, and therefore in the R quaternary structure, is shown in Fig. 3a. The kinetics are very similar to what is expected from solution studies (26, 27), with two major phases: a large-amplitude phase (≈50%) at ≈100 ns, corresponding to rebinding of ligands that have returned to the same heme from which they were photodissociated (geminate rebinding), followed by bimolecular rebinding at ≈100 μs (Fig. 3a). Because the gel has prevented the R-to-T quaternary change, there is no slower bimolecular phase, as observed in solution, corresponding to rebinding to molecules that have switched from the R to the T quaternary structure (26, 27). Importantly, identical kinetic progress curves are observed over the entire pH range of 6.6-7.6 (Fig. 3a).
Fig. 3.

Ligand rebinding kinetics after photodissociation of the CO complex. (a) Kinetic progress curves are shown. Fraction of deoxyhemes [f(deoxy)] versus time on a logarithmic scale for T+ (Upper) and R (Lower) gels at different pHs but otherwise the same conditions as in Fig. 2 (yellow, pH 6.6; red, pH 7.0; green, pH 7.3; cyan, pH 7.6). (b) Fraction of deoxyhemes versus time on a logarithmic scale for T- gels (100 mM Hepes buffer/1 mM EDTA) at pH 7.6 (cyan), pH 7.0 (red), and pH 7.0 plus 200 mM potassium chloride (magenta). The black curves superimposed on the T- curves are linear combinations of the T+ and R curves. (c) Change of geminate yield for T- gel at pH 7.0 is shown. The geminate yield (φ) is plotted versus the time (on a logarithmic scale) after adding CO in the sample preparation to produce either 100% (cyan circles) or 50% (black squares) saturation. The cyan curve is a stretched exponential fit to the data with a time constant of 770 min, a stretching exponent of 0.5, a geminate yield at t = 0 of 0.25, and a geminate yield at t = ∞ of 0.49.
When encapsulation was carried out in the deoxy form in the presence of the allosteric effectors inositol hexaphosphate and bezafibrate (T+ molecules) and followed by the addition of CO, the kinetics were again very similar to what was expected for hemoglobin in the T quaternary structure from studies on molecules free in solution (28). There is no geminate phase (<1%) and only a single bimolecular phase at ≈10 ms (Fig. 3a). As with R-encapsulated hemoglobin, the kinetics for the T+ gels are independent of pH.
The surprise came with measurements on molecules encapsulated as deoxy in the absence of any allosteric effectors or in the presence of chloride, which is a weaker allosteric effector (T- molecules) (Fig. 3b). Although these molecules are assumed to remain in the T quaternary structure, the time course of ligand rebinding looks very much like a mixture of R and T+ kinetics, with a substantial geminate phase and both rapid and slow bimolecular phases. Remarkably, the T- curve is closely reproduced under all conditions with a simple linear combination of R and T+ progress curves (Fig. 3a). That is, there is no change in any of the relaxation rates, only in the relative proportions of R and T+ (see Table 1, which is published as supporting information on the PNAS web site). The most straightforward interpretation of these results is that under all conditions a major fraction of the subunits in the T quaternary structure has the kinetic properties of the R quaternary structure; changing solution conditions simply alters this fraction, which varies from ≈0.4 to 0.8 (Fig. 2 Inset).
The obvious possibility to be ruled out is that R-like kinetic properties appear in the T- gels because the molecules have switched to the R quaternary structure in the 10- to 15-min interval between adding CO and recording the kinetics. To exclude this possibility, we took advantage of the known enormous sensitivity of the rate of the T-to-R transition on fractional saturation with ligand, which, in solution, decreases by -108-fold when the number of bound ligands decreases from 4 to 0 (27, 29, 30). The change in the geminate yield with time was therefore compared on T- gels initially 100% or 50% saturated with CO. When the initial saturation was 100%, a slow increase in the amplitude of the geminate phase was observed with a half-time of ≈360 min (Fig. 3c), which is ≈108-fold slower than the T-to-R transition for the fully liganded molecule in solution (14, 27, 29, 30). However, with an initial saturation of 50%, there is no detectable change in the geminate yield after 800 min (Fig. 3c), consistent with the expectation that the rate of the T-to-R quaternary change at partial saturation with CO would be dramatically slowed. This result provides convincing evidence that the R-like geminate rebinding is occurring in the T quaternary structure.††
Our discovery that a major fraction of the subunits in the T quaternary structure have R-like properties is obviously inconsistent with the MWC model. Because the fraction is independent of the initial saturation (Fig. 3c), the results cannot be explained by a sequential model because this model requires the functional properties to depend on the number of ligands bound to the tetramer (7). Our results can, however, be readily explained by the recently proposed tertiary two-state (TTS) allosteric model (30), in which there is incomplete coupling between the tertiary and quaternary structures. The TTS model is one of the limiting cases of the more general model of Herzfeld and Stanley (31, 32), which encompasses both the MWC and Koshland-Nemethy-Filmer (KNF) models, as well as all possible combinations of the two. Tertiary conformational changes also play a central role in the Szabo and Karplus model, based on the Perutz stereochemical mechanism (8, 10, 22, 33).
In the TTS model, shown schematically in Fig. 4a, there are only two functionally different conformational states, as in the MWC model. However, these states correspond to r and t tertiary conformations of individual subunits rather than the R and T quaternary conformations of the MWC two-state model. Ligation and the R quaternary structure bias a subunit toward the high-affinity r tertiary conformation, whereas deligation, the T quaternary structure, and negative allosteric effectors favor the low-affinity t tertiary conformation. The TTS model preserves the central idea of MWC, that ligand binding without a change in quaternary structure is noncooperative, but, unlike the MWC model, it allows for variation in the affinity of the R and T quaternary structures. According to the TTS model, in both crystals of T hemoglobin and in deoxygenated gels encapsulated in the presence of allosteric effectors, all four liganded subunits remain in the t tertiary conformation upon ligand binding, explaining the striking finding of identical affinities under the two conditions (Fig. 2) (30). In the absence of allosteric effectors (T-), the affinity is higher because ligation switches some fraction of subunits from t to r, but the quaternary structure remains T (Fig. 4b).
Fig. 4.

Interpretation of gel equilibrium and kinetic experiments in terms of the TTS allosteric model. The TTS partition function is (30) 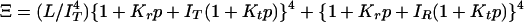 . (a) Schematic description of the model. Open symbols, unliganded subunit; filled symbols, liganded subunit; circles, r tertiary conformation; squares, t tertiary conformation. Kt and Kr, affinities of the subunits in the t and r conformations in which the liganded subunits remain in t and r, respectively; lT, ratio of t-to-r populations of the unliganded subunits in the T quaternary structure; lR, corresponding equilibrium constant in the R quaternary structure; L, ratio of the T-to-R populations in which all of the subunits of T are unliganded t and all of the subunits of R are unliganded r. See Supporting Text for additional properties of the partition function. (b) O2 equilibrium binding experiments. Analysis of the O2-binding curves with the TTS model shows that, at pH 7.3, 80% of the oxygenated subunits switch from t to r, whereas the CO photolysis experiments indicate that 65% of the oxygenated subunits switch from t to r. For O2 binding to the R quaternary structure in the absence of allosteric effectors, all liganded subunits of R are in the r conformation. Fitting the solution kinetic data for CO rebinding with the TTS model predicts that ≈40% of the unliganded subunits in the R quaternary structure are in the t tertiary conformation (lR = 0.7) (30). Only one representative species from the most probable microstate, defined by the number of liganded and unliganded r and t subunits, is shown for each ligation state. (c) CO photolysis experiments on hemoglobin encapsulated in the gel. CO rebinding is complete in milliseconds before either tertiary or quaternary conformations change.
. (a) Schematic description of the model. Open symbols, unliganded subunit; filled symbols, liganded subunit; circles, r tertiary conformation; squares, t tertiary conformation. Kt and Kr, affinities of the subunits in the t and r conformations in which the liganded subunits remain in t and r, respectively; lT, ratio of t-to-r populations of the unliganded subunits in the T quaternary structure; lR, corresponding equilibrium constant in the R quaternary structure; L, ratio of the T-to-R populations in which all of the subunits of T are unliganded t and all of the subunits of R are unliganded r. See Supporting Text for additional properties of the partition function. (b) O2 equilibrium binding experiments. Analysis of the O2-binding curves with the TTS model shows that, at pH 7.3, 80% of the oxygenated subunits switch from t to r, whereas the CO photolysis experiments indicate that 65% of the oxygenated subunits switch from t to r. For O2 binding to the R quaternary structure in the absence of allosteric effectors, all liganded subunits of R are in the r conformation. Fitting the solution kinetic data for CO rebinding with the TTS model predicts that ≈40% of the unliganded subunits in the R quaternary structure are in the t tertiary conformation (lR = 0.7) (30). Only one representative species from the most probable microstate, defined by the number of liganded and unliganded r and t subunits, is shown for each ligation state. (c) CO photolysis experiments on hemoglobin encapsulated in the gel. CO rebinding is complete in milliseconds before either tertiary or quaternary conformations change.
The explanation of the CO rebinding kinetics in gels is very similar. In the T+ gels, the subunits liganded with CO remain in the t conformation, whereas in the T- gels CO binding converts a substantial fraction of the subunits to the r conformation (Fig. 4c). After photodissociation, the r subunits in the T quaternary structure remain in the r conformation because the kinetic constraints of the gel prevent any r-to-t transition before the millisecond completion of CO rebinding. The lack of exact superposition of the T- curves with linear combinations of R and T+ curves can be explained by a lack of complete averaging among the conformational substates of r and t during rebinding (Fig. 3b) (30). The T quaternary structure is expected to bias the distribution of liganded r toward the more slowly reacting r substates to produce a smaller geminate yield and therefore also a slower bimolecular rate [the bimolecular rate is the product of the rate of ligand entry into the heme pocket and the geminate yield (30)], whereas the heterotropic effectors would bias the liganded t distribution in T toward the more slowly rebinding t substates.
To more quantitatively understand the effect of slowing tertiary conformational changes by the gel, we carried out kinetic simulations by using the TTS model with varying slowing factors, σ, on the rate coefficients. The kinetic scheme of the TTS model for each quaternary structure is (30)
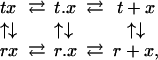 |
1 |
where tx and rx are liganded subunits and t.x and r.x are subunits with the ligand dissociated from the heme but inside the protein. Almost all of the rate coefficients of this scheme have been well determined, thanks to decades of research on the kinetics of hemoglobin ligand binding and dissociation (20, 34). Fig. 5 shows the results of numerically integrating the coupled differential equations of the above scheme with varying values of σ that slow the tertiary conformational changes in T- gels. For simplicity, we have treated the tertiary conformational kinetics as proceeding with a simple exponential time course rather than the stretched exponential time course observed in experiments (Fig. 7 and Table 1, which are published as supporting information on the PNAS web site) (27, 30, 35, 36). Because the tertiary changes are apparently slowed less by the gel than the quaternary change, which is presumably related to the difference in the extent of the conformational changes (compare quaternary and tertiary changes in Fig. 1), we can estimate an upper limit of ≈108 for the slowing factor by comparing the half-time of 360 min for the fully liganded T- gel with the predicted rate in solution (27, 29, 30). For σ = 1 (i.e., no gel constraints), the r-to-t conformational change is so fast that there is very little geminate rebinding, and the rebinding kinetics are virtually identical to those of the T+ gel (Fig. 3a) in which all of the liganded subunits have the t conformation (Fig. 5 a and b). For σ ≥ 106, the rebinding curves are identical and reflect the relative populations of r and t before photolysis because CO rebinding is complete before any tertiary conformational changes can occur (Fig. 5 a and d). At intermediate σ (106 > σ > 1), the kinetics become more complex, but for σ ≥ 103, the geminate yield does not change and is therefore an accurate measure of the fraction of liganded subunits in the T quaternary structure that are initially in the r tertiary conformation (Fig. 5a).
Fig. 5.

Simulation of kinetics after photodissociation. Table 2, which is published as supporting information on the PNAS web site, gives the rate coefficients used in simulating the kinetic and equilibrium experiments in the T- gel, together with those obtained from nanosecond photolysis studies of the CO complex (30) and the rate coefficients obtained for the overall binding and dissociation rates for CO and O2 (42). Difference in solution conditions of temperature and pH required only small adjustments to obtain rate coefficients that reproduced the gel photolysis and oxygen equilibrium data. Shown are the fraction of deoxyhemes, f(deoxy), as a function of time after photodissociation of T- molecules with varying values of the slowing factor σ (a) and the fraction of each of the six species in the T quaternary structure as a function of time after photodissociation of T- molecules with σ = 1(b), σ = 103 (c), and σ = 106 (d). The differential equations were solved by using the program mlab.
An important test of our interpretation of the results in terms of the TTS model is the self-consistency of the CO-rebinding kinetics (Fig. 3b) and the O2 equilibrium binding curves (Fig. 2). Kinetic simulations (Fig. 6) show that oxygen dissociation is so much faster than CO dissociation that equilibrium can be achieved in oxygen-binding experiments even if the tertiary conformational changes are slowed by as much as the factor of 108-fold that we estimate for the T-to-R quaternary conformational change (Fig. 1). With frozen tertiary and quaternary conformations in photolysis experiments and tertiary but not quaternary equilibrium in the oxygen-binding measurements, the TTS model predicts that the fraction of liganded subunits in the r conformation in the T quaternary structure measured in photolysis experiments is related to the ratio of O2 affinities in the absence (T-) and presence (T+) of saturating concentrations of allosteric effectors (Fig. 2) by the following: f ≈ {1 - [p50(T-)/p50(T+)]}. The data in Fig. 2 Inset show that the fractions calculated from the oxygen-binding curves in the absence and presence of chloride ions and as a function of pH are in good agreement with the values derived from the CO kinetic experiments under identical conditions. The slightly higher fractions in the oxygen-binding experiments may reflect differences in the ratio of r and t affinities for the two ligands.
Fig. 6.

Simulation of kinetics of equilibration. Shown is the fraction of liganded t and liganded r conformations as a function of time after the addition of ligand to the T- gel with varying values of the slowing factor σ defined in Table 2. (a) Addition of CO at 760 torr CO (1 torr = 133 Pa) (1.13 mM at 15°C). (b) Addition of O2 at the p50 of 26 torr. Even for the maximum σ of 108, equilibrium with respect to oxygen binding at the p50 of 26 torr is achieved in ≈15 min, which is the typical time required for equilibration of the system (gas lines, flow cell, suspending medium, and gels). Because of the much slower dissociation rate for CO compared with O2, the amount of time required to achieve equilibrium after adding CO is more than 100 times greater. If Kt/Kr are the same for CO and O2, this result suggests, as does the possible long time for equilibration after CO rebinding in the photolysis experiments (Fig. 5c), that the fraction of r subunits in the liganded T- molecules determined in kinetic experiments will always be equal to or less than the fraction observed in the oxygen-binding experiments. This kinetic effect provides an alternative explanation for the lower fraction of r subunits obtained from CO kinetic experiments, compared with the O2 equilibrium experiments, and also suggests that the variation in rebinding kinetics previously observed for different encapsulation protocols is most probably associated with different values of σ (24).
In summary, we have shown that a simple but important extension of the MWC model straightforwardly explains our discovery of R-like subunits in the T quaternary structure and also provides a coherent and quantitative explanation of the kinetic and equilibrium properties of hemoglobin in crystals, solution, and gels. The change in oxygen affinity caused by heterotropic effectors without a change in quaternary structure is readily explained by the TTS model as resulting from an alteration in the equilibrium between low-affinity t and high-affinity r tertiary conformations. Because lattice constraints prevent the tertiary conformational changes that occur in solution (17-19, 37), solution NMR spectroscopy (38) will most probably be required to identify the functionally relevant structural differences between r and t. Finally, the results of our study suggest that it will be interesting to apply the TTS model to other systems in which neither the MWC nor sequential models adequately describe allosteric behavior (39, 40)
Supplementary Material
Acknowledgments
We thank Attila Szabo for many helpful discussions and suggestions, James Hofrichter and Gian Luigi Rossi for helpful discussion, and Dan Garrett for invaluable assistance in preparing Fig. 1. This work was supported by the Italian Ministry of Instruction, University, and Research for Grants; the Fondo per gli Investimenti della Ricerca di Base Project on Nanotechnology (C.V. and A.M.); the Italian National Institute for the Physics of Matter; and National Institutes of Health Program Project Grant GM58890.
Abbreviations: MWC, Monod-Wyman-Changeux; TTS, tertiary two-state.
Footnotes
This result also rules out the possibility of dimer formation, which is also very sensitive to the fractional saturation with ligand and occurs only at much lower protein concentrations than those employed in our experiments (41).
References
- 1.Monod, J., Wyman, J. & Changeux, J.-P. (1965) J. Mol. Biol. 12, 88-118. [DOI] [PubMed] [Google Scholar]
- 2.Perutz, M. F. (1989) Q. Rev. Biophys. 22, 138-236. [DOI] [PubMed] [Google Scholar]
- 3.Tibbs, G. R., Goulding, E. H. & Siegelbaum, S. A. (1997) Nature 386, 612-615. [DOI] [PubMed] [Google Scholar]
- 4.Changeux, J.-P. & Edelstein, S. J. (1998) Neuron 21, 959-980. [DOI] [PubMed] [Google Scholar]
- 5.Ma, J. P. & Karplus, M. (1998) Proc. Natl. Acad. Sci. USA 95, 8502-8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauling, L. (1935) Proc. Natl. Acad. Sci. USA 21, 186-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koshland, D. E., Nemethy, G. & Filmer, D. (1966) Biochemistry 5, 365-385. [DOI] [PubMed] [Google Scholar]
- 8.Szabo, A. & Karplus, M. (1972) J. Mol. Biol. 72, 163-197. [DOI] [PubMed] [Google Scholar]
- 9.Minton, A. P. & Imai, K. (1972) Proc. Natl. Acad. Sci. USA 71, 1418-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee, A. K. M. & Karplus, M. (1983) Proc. Natl. Acad. Sci. USA 80, 7055-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yonetani, T., Park, S., Tsuneshige, A., Imai, K. & Kanaori, K. (2002) J. Biol. Chem. 277, 34508-34520. [DOI] [PubMed] [Google Scholar]
- 12.Shibayama, N. & Saigo, S. (1995) J. Mol. Biol. 251, 203-209. [DOI] [PubMed] [Google Scholar]
- 13.Bettati, S. & Mozzarelli, A. (1997) J. Biol. Chem. 272, 32050-32055. [DOI] [PubMed] [Google Scholar]
- 14.Shibayama, N. & Saigo, S. (1999) J. Am. Chem. Soc. 121, 444-445. [Google Scholar]
- 15.Bruno, S., Bonaccio, M., Bettati, S., Rivetti, C., Viappiani, C., Abbruzzetti, S. & Mozzarelli, A. (2001) Protein Sci. 10, 2401-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibayama, N. & Saigo, S. (2001) FEBS Lett. 492, 50-53. [DOI] [PubMed] [Google Scholar]
- 17.Mozzarelli, A., Rivetti, C., Rossi, G. L., Henry, E. R. & Eaton, W. A. (1991) Nature 351, 416-418. [DOI] [PubMed] [Google Scholar]
- 18.Rivetti, C., Mozzarelli, A., Rossi, G. L., Henry, E. R. & Eaton, W. A. (1993) Biochemistry 32, 2888-2906. [DOI] [PubMed] [Google Scholar]
- 19.Mozzarelli, A., Rivetti, C., Rossi, G. L., Eaton, W. A. & Henry, E. R. (1997) Protein Sci. 6, 484-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eaton, W. A., Henry, E. R., Hofrichter, J. & Mozzarelli, A. (1999) Nat. Struct. Biol. 6, 351-358. [DOI] [PubMed] [Google Scholar]
- 21.Shulman, R. G. (2001) IUBMB Life 51, 351-357. [DOI] [PubMed] [Google Scholar]
- 22.Szabo, A. & Karplus, M. (1976) Biochemistry 15, 2869-2877. [DOI] [PubMed] [Google Scholar]
- 23.Juszczak, L. J. & Friedman, J. M. (1999) J. Biol. Chem. 274, 30357-30360. [DOI] [PubMed] [Google Scholar]
- 24.Khan, I., Shannon, C. F., Dantsker, D., Friedman, A. J., Perez-Gonzalez-de-Apodaca, J. & Friedman, J. M. (2000) Biochemistry 39, 16099-16109. [DOI] [PubMed] [Google Scholar]
- 25.Abbruzzetti, S., Viappiani, C., Bruno, S. & Mozzarelli, A. (2001) Chem. Phys. Lett. 346, 430-436. [Google Scholar]
- 26.Hofrichter, J., Sommer, J. H., Henry, E. R. & Eaton, W. A. (1983) Proc. Natl. Acad. Sci. USA 80, 2235-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henry, E. R., Jones, C. M., Hofrichter, J. & Eaton, W. A. (1997) Biochemistry 36, 6511-6528. [DOI] [PubMed] [Google Scholar]
- 28.Murray, L. P., Hofrichter, J., Henry, E. R., Ikeda-Saito, M., Kitagishi, K., Yonetani, T. & Eaton, W. A. (1988) Proc. Natl. Acad. Sci. USA 85, 2151-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eaton, W. A., Henry, E. R. & Hofrichter, J. (1991) Proc. Natl. Acad. Sci. USA 88, 4472-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henry, E. R., Bettati, S., Hofrichter, J. & Eaton, W. A. (2002) Biophys. Chem. 98, 149-164. [DOI] [PubMed] [Google Scholar]
- 31.Herzfeld, J. & Stanley, H. E. (1974) J. Mol. Biol. 82, 231-265. [DOI] [PubMed] [Google Scholar]
- 32.Bansil, R., Herzfeld, J. & Stanley, H. E. (1976) J. Mol. Biol. 103, 89-126. [DOI] [PubMed] [Google Scholar]
- 33.Perutz, M. F. (1970) Nature 228, 726-739. [DOI] [PubMed] [Google Scholar]
- 34.Antonini, E. & Brunori, M. (1971) Hemoglobin and Myoglobin in Their Reaction with Ligands (North-Holland, Amsterdam).
- 35.Hagen, S. J. & Eaton, W. A. (1996) J. Chem. Phys. 104, 3395-3398. [Google Scholar]
- 36.Hagen, S. J., Hofrichter, J. & Eaton, W. A. (1995) Science 269, 959-962. [DOI] [PubMed] [Google Scholar]
- 37.Perutz, M. F., Wilkinson, A. J., Paoli, M. & Dodson, G. G. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 1-34. [DOI] [PubMed] [Google Scholar]
- 38.Lukin, J. A., Kontaxis, G., Simplaceanu, V., Yuan, Y., Bax, A. & Ho, C. (2003) Proc. Natl. Acad. Sci. USA 100, 517-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helmstaedt, K., Krappmann, S. & Braus, G. H. (2001) Microbiol. Mol. Biol. Rev. 65, 404-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karpen, J. W. & Ruiz, M. L. (2002) Trends Biochem. Sci. 27, 401-409. [DOI] [PubMed] [Google Scholar]
- 41.Ackers, G. K. (1998) Adv. Protein Chem. 51, 185-253. [DOI] [PubMed] [Google Scholar]
- 42.Unzai, S., Eich, R., Shibayama, N., Olson, J. S. & Morimoto, H. (1998) J. Biol. Chem. 273, 23150-23159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}