

Abstract
Background
Gene knockouts are a common tool used to study gene function in various organisms. However, designing gene knockouts is complicated in viruses, which frequently contain sequences that code for multiple overlapping genes. Designing mutants that can be traced by the creation of new or elimination of existing restriction sites further compounds the difficulty in experimental design of knockouts of overlapping genes. While software is available to rapidly identify restriction sites in a given nucleotide sequence, no existing software addresses experimental design of mutations involving multiple overlapping amino acid sequences in generating gene knockouts.
Results
Pyviko performed well on a test set of over 240,000 gene pairs collected from viral genomes deposited in the National Center for Biotechnology Information Nucleotide database, identifying a point mutation which added a premature stop codon within the first 20 codons of the target gene in 93.2% of all tested gene-overprinted gene pairs. This shows that Pyviko can be used successfully in a wide variety of contexts to facilitate the molecular cloning and study of viral overprinted genes.
Conclusions
Pyviko is an extensible and intuitive Python tool for designing knockouts of overlapping genes. Freely available as both a Python package and a web-based interface (http://louiejtaylor.github.io/pyViKO/), Pyviko simplifies the experimental design of gene knockouts in complex viruses with overlapping genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-016-0920-3) contains supplementary material, which is available to authorized users.
Keywords: Virus, Knockout virus, Mutation, Overprinting, Bioinformatics, Cloning, Viral mutant, Knockout, Pyviko
Background
Gene knockouts are an important tool used to study gene function in viruses [1], bacteria [2], and other organisms, including model organisms such as mice [3]. Although the principle of removing a gene in an attempt to discern its cellular role is not new, the recent development of CRISPR/Cas9 as a tool for knocking out genes in vivo has revolutionized the field of genome editing and underscores the importance of using knockouts as a tool to study gene function [4]. A common experimental approach to knocking out a gene is to simply excise the gene of interest from the target organism’s genome. Viruses, however, often contain DNA sequences that code for multiple protein products in separate reading frames, called overprinted genes (Fig. 1a) [5, 6]. For, example, human immunodeficiency virus type 1 (HIV-1) contains 8 instances of gene overprinting [7], as shown in Fig. 1b. This phenomenon is widespread among different viral families [5] and precludes the excision strategy of knocking out a viral gene.
Fig. 1.

Examples of gene overprinting in viruses. a Outline showing different types of gene overprinting and associated nucleotide and amino acid sequences. Gene B overprints into gene A in the same direction but a different frame. Genes C and D showcase overprinting in different frames and directions. Start codons in nucleotide sequences are shown in green. b Genome of human immunodeficiency virus type I [7] with annotated genes. tat and rev splicing is indicated by a solid line
An alternate approach to excising genes is to mutate a sequence in order to insert a premature stop codon in the target gene, which results in a non-functional truncation of the final translated protein. Throughout this work, “target gene” refers to the gene we wish to mutate in order to insert a premature stop codon. In Fig. 1a, gene A is the target (overprinted) gene, and gene b is the overlapping gene. To ensure that this truncation retains no function of the wild-type gene, the stop codon should occur as early in the gene sequence as possible. However, the amino acid sequence of the overlapping gene must be preserved in order to experimentally discriminate between changes in phenotype due to changes in the overlapping and target genes. Such mutations are possible due to the degeneracy of the genetic code—several amino acids can be coded for by more than one nucleotide sequence [1]. However, designing such mutants by hand is slow and non-trivial, especially when mutating many clones or strains of viruses. Additionally, changes in restriction enzyme recognition sites that can be used to trace the newly introduced mutations are important experimentally in planning molecular cloning and mutagenesis protocols [8].
Restriction enzymes are a class of bacterial endonucleases that specifically cleave DNA at a 4–8 nucleotide recognition sequence. Mutagenesis protocols are frequently designed to add or remove a restriction site as tracers, so that resulting constructs can be analyzed inexpensively for the presence of the desired mutation without having to sequence multiple clones [8]. Given the diversity and number of restriction enzymes commercially available [9], searching for potential restriction enzyme recognition sites in a given DNA sequence by hand is unfeasible. Methods for rapid searching of nucleotide sequences for restriction sites have been previously published [10] and are not discussed here. In this work, we introduce Pyviko—a tool which automates the process of designing knockout viruses while taking into account changes in restriction enzyme recognition sites and the integrity of the overlapping gene.
Implementation
Pyviko was implemented in Python 2.7. The Python regex module [11] is optional and can be installed to augment the base functionality of Pyviko, but is not required to use the software. Source code is freely available [12] under the MIT license and is thoroughly unit tested prior to each release. Releases are available from the Python Package Index [13]. A client-side web interface in JavaScript is available for making single-gene knockouts without installing Pyviko [14]. Extensive documentation for Pyviko is available online [15] and as comments in the source code. Online documentation is build directly from comments in the source using pdoc [16] and is available without installing Pyviko. Bug reports should be submitted on the project’s GitHub page [12].
The basic functionality of Pyviko is divided into three modules: core, restriction, and mutation. Functions for basic nucleic acid sequence manipulation, including reading from and writing to Fast-All (FASTA) files, are included within the core module. The restriction module contains functions to analyze sequences for restriction sites and find changes in restriction sites that result from sequence changes. The mutation module includes functions to find favorable mutations as well as the Mutant and OverGene classes. The Mutant class brings together the three modules and identifies favorable knockout mutants in sequences of interest.
Results
Use in interactive and stand-alone scripts
Pyviko was created to streamline the process of planning viral mutageneses. Each module is designed to be intuitive and extensible to facilitate its use in a variety of applications. Figure 2a–b shows the generation of knockouts from a single target and overlapping gene pair in an interactive fashion in a Python interpreter. Using the sequences of an input gene and its overprinted counterpart, the overlapping sequence is automatically detected and all possible knockouts matching the input parameters are displayed. Thus, in the example shown in Fig. 2a, the target gene can be knocked out by mutating the initiation codon (index 0) to ACG without changing the coding capacity of the overlapping gene (TAT and TAC both code for tyrosine). Alternatively, the TCA (serine) codon at index 3 of the target gene can be mutated to a stop codon (TGA or TAA) without changing the coding sequence of the overlapping gene (CTC, CTA and CTG all encode leucine).
Fig. 2.

Usage of Pyviko in stand-alone scripts and web interface. a Schematic of sample target (Y) and overlapping (X) genes used in code examples. b Minimal Python commands for generating gene knockouts. In this interactive example, “>>>” denotes input into a Python interpreter and output is displayed directly below the input on an unindented line. c Pyviko input interface [14] including sample gene sequences. d Results of analysis for input shown in (c)
To reach a wider, non-computational audience, this single-gene pair knockout script has also been implemented as a graphical interface in JavaScript [14]. This interface supports generation of knockouts for a gene or pair of genes under various parameters, including requirements for start codon mutants or restriction site changes. Figure 2c shows the input interface and 2D shows the output of the analysis for the gene pair shown in Fig. 2a. The full mutant sequence for each mutation is shown together with a link to an interface to design further restriction site changes for a particular point mutant.
For experiments requiring the generation of many mutants, Pyviko provides a rapid, high-throughput approach to viral mutagenesis. Additionally, Pyviko supports reading from and writing to files in the universal FASTA format to maximize inter-application compatibility. Additional file 1 contains a Python script showcasing the ability of Pyviko to generate mutants in a batched fashion, taking input from FASTA files and writing mutagenesis results to FASTA. Additional file 1 also shows many of the options available when searching sequences, including filtering for mutants that add or remove a restriction site and including or excluding mutants that perturb the start codon. Mutating a gene’s start codon is another feasible option to prevent production of a specific protein. Some viruses, however, have been shown to use alternate start codons [17]. Care should be taken to biologically validate knockouts made using this strategy.
Integration with existing tools and extensibility
Recent trends toward open-source software in bioinformatics have led to the development of a variety of new Python tools for analyzing nucleotide sequences, including Biopython [10] and Pydna [18]. While Biopython and Pydna both include functions for restriction site identification, neither is designed to analyze restriction site changes as a result of nucleotide sequence mutations. Pyviko is designed to solve the problem of mutating overprinted genes, which are most common in viruses but do occur in other organisms, including bacteria [19], mice, and humans [20]. Unlike other software, Pyviko is optimized to analyze multiple overlapping sequences concurrently, examining changes in translated amino acid sequence and restriction sites resulting from changes in nucleotide sequence.
While Pyviko is not dependent on existing software for its basic nucleic acid manipulation and restriction site identification functionalities, it is not designed to supersede other general nucleotide sequence analysis programs. Rather, Pyviko can be used in conjunction with software like Pydna and Biopython to complement Pyviko’s approach to viral mutagenesis. Additional file 2 contains the script used to collect sequences from GenBank for the large-scale analysis discussed in the following section. This script leverages Biopython’s Entrez module to retrieve viral genomes to be analyzed by Pyviko. Additional file 3 is a Python script that retrieves a viral genome sequence (HIV-1 NL4-3 [21]) from the NCBI Nucleotide database [22] via Biopython, generates a knockout for a target overprinted gene (vpr) with Pyviko, then uses Pydna to design primers for molecular cloning.
Although Pyviko was designed to generate knockouts of viral overprinted genes, the software is generally applicable to any mutagenesis of overprinted genes. Additional file 4 is a Python script containing a variety of examples of mutagenesis design involving overprinted genes, including: mutagenesis of hydrophobic to non-hydrophobic amino acid residues in the target gene, identification of all mutations in the overprinted region that do not change the polypeptide sequence of the overlapped gene, and generating a sequence that scrambles the amino acids of a target gene without changing the amino acids of the overlapping gene.
Large-scale functional testing
To validate the approach of Pyviko in knocking out overprinted viral genes, we performed a large-scale analysis of annotated viral genomes deposited in the National Center for Biotechnology Information (NCBI) Nucleotide database. Using the script included as Additional file 2, we collected 48,770 sequences annotated as complete viral genomes from the NCBI Nucleotide database. From these genomes, 248,777 pairs of overprinted gene pairs were identified and analyzed by Pyviko. Note that individual genes may appear in more than one pair as a gene may overlap with more than one other gene (e.g. vif gene in Fig. 1b).
Pyviko was able to identify point mutants that added a premature stop codon in the target gene without changing the amino acid sequence of the overlapped gene in 96.5% of all genes analyzed. Many of these mutations added or removed a restriction enzyme recognition site [9], and 96.2% of all target genes analyzed could be knocked out as above with the additional constraint of a restriction site change. However, the location of the premature stop codon is important for the efficacy of the knockout—a “premature” stop codon close to the end of the target gene could still result in a gene product with some level of function. Thus, we decided to further judge the efficacy of Pyviko knockouts by examining the first possible premature stop codon in each target gene identified by Pyviko.
To quantify the effectiveness of Pyviko knockouts, we calculated the percentage of stop codons that could be added within the first 20 codons of the target gene. While there is evidence that polypeptides shorter than 20 amino acids could have intracellular functions [23], it is highly unlikely that a protein truncated to 20 amino acids or less would retain its original function. Pyviko identified potential premature stop codons within the first 20 codons in 93.2% of all target genes (Fig. 3a). Requiring a restriction site change, Pyviko identified a premature stop codon within the first 20 codons in 76.4% of target genes (Fig. 3b). These data show that, while requiring a restriction site change does not greatly change the percentage of target genes that can be knocked out, the distribution of first premature stop codons is much wider when restriction site changes are required (Fig. 3a–d). It is also possible that the relative truncated length of a knockout is important—for example, a truncated gene coding for 20 amino acids is 20% of the final polypeptide length of a 100 amino acid protein, but only 2% of a 1000 amino acid protein. However, a heat map of first-codon knockouts along a unit gene show that over 90% of knockouts are within the first 10% of the coding sequence both without (Fig. 3c) and with (Fig. 3d) restriction site change constraints. This is consistent with the conclusion that the vast majority of Pyviko-designed gene knockouts will not result in a functional protein product.
Fig. 3.
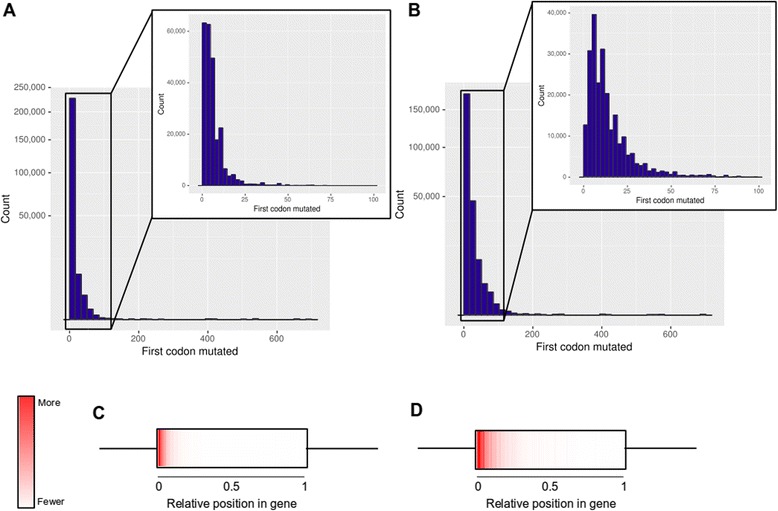
Large-scale analysis of viral overprinted genes from the NCBI Nucleotide database. a and b Show counts of the first position at which a directed point mutation can insert a premature stop codon in the overprinted gene without changing the amino acid sequence of the overlapped gene in each gene pair surveyed. Full-size graphs show mutations over the full length of all genes surveyed with a log2 scale to ensure visibility of bins with low counts. Insets show the first 100 codons of each gene with a linear y axis. a Shows counts without requiring a restriction site change and (b) shows counts with a required restriction site change. c and d Show positions of the first stop codon mutants from (a) and (b) expressed as the ratio of the position of the first codon relative to the total length of the gene in codons. c, as in (a), shows counts without requiring a restriction site change and (d), as in (b), shows counts with a required restriction site change
Conclusions
In this work, we introduce Pyviko, an intuitive and extensible Python tool for designing viral knockouts. While the software is platform-independent and does not require any external modules other than Python itself, Pyviko can also be used with existing Python tools such as Biopython and Pydna to extend its base functionality as shown in the example scripts. Pyviko performed well on a test set of over 240,000 gene pairs collected from viral genomes deposited in the NCBI Nucleotide database, identifying a point mutation that could be inserted within the first 20 codons of the target gene in 93.2% of all tested gene-overprinted gene pairs. This shows that Pyviko can be used successfully in a wide variety of contexts to facilitate the molecular cloning and study of viral overprinted genes. The complete source code and quick-start guide are included as Additional files 5 and 6, respectively.
Availability and requirements
Project name: Pyviko
Project home pages: https://github.com/louiejtaylor/pyViKO, https://pypi.python.org/pypi/pyviko
Operating system(s): Platform independent
Programming language: Python
Other requirements: Python 2.7 or higher
License: MIT license
Acknowledgements
Thanks to J. Haber of the University of California at Berkeley for help with unit tests, D. G. Fisher, M. M. Addison and N. Kadry of the University of Pennsylvania for critical reading of the manuscript and to members of the Strebel lab for discussions and suggestions.
Funding
This work was supported by the Intramural Research Program of the NIH, NIAID (KS: 1 Z01 AI000669) and by an NIH Post-baccalaureate Fellowship (LJT).
Availability of data and materials
All data and analysis scripts are included as supplementary files. The Pyviko web interface is available at http://louiejtaylor.github.io/pyViKO/, and the code is hosted on GitHub at https://github.com/louiejtaylor/pyViKO.
Authors’ contributions
LJT and KS conceived and tested the software. LJT designed and implemented the software, unit tests, and functional tests. LJT and KS drafted and edited the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Per the National Institutes of Health’s Ethical Guidelines & Regulations, no formal ethics approval was required for this study.
Abbreviations
- DNA
Deoxyribonucleic acid
- FASTA
Fast-All (file format)
- HIV-1
Human immunodeficiency virus type 1
- NCBI
National Center for Biotechnology Information
- Pyviko
Python viral knockouts
- RNA
Ribonucleic acid
Additional files
A Python script showcasing the ability of Pyviko to generate mutants in a batched fashion, taking input from FASTA files and writing mutagenesis results to FASTA. Additional file 1 also shows many of the options available when searching sequences, including filtering for mutants that add or remove a restriction site and including or excluding mutants that perturb the start codon. (PY 787 bytes)
The Python script used to collect sequences from GenBank for the large-scale analysis in Fig. 3. This script leverages Biopython’s Entrez module to retrieve viral genomes to be analyzed by Pyviko. (PY 4 kb)
A Python script that retrieves a viral genome sequence (HIV-1 NL4-3) from the NCBI Nucleotide database via Biopython, generates a knockout for a target overprinted gene (vpr is overprinted by vif) with Pyviko, then uses Pydna to design primers for molecular cloning. (PY 1 kb)
A Python script containing a variety of examples of mutagenesis design involving overprinted genes: mutagenesis of hydrophobic to non-hydrophobic amino acid residues in the target gene, identification of all mutations in the overprinted region that do not change the polypeptide sequence of the overlapped gene, and generating a sequence that scrambles the amino acids of the target gene without changing the amino acids of the overlapped gene. (PY 4 kb)
The Pyviko Web User Interface Quick Start Guide, also available at [24]. This document explains the use of the Pyviko web user interface. (PDF 461 kb)
The Pyviko source code, version 1.0.1.1. Current release available from the Python Package Index [13]. (GZ 7 kb)
Contributor Information
Louis J. Taylor, Email: louist@upenn.edu
Klaus Strebel, Email: kstrebel@nih.gov.
References
- 1.Crick FH, Barnett L, Brenner S, Watts-Tobin RJ. General nature of the genetic code for proteins. Nature. 1961; doi:10.1038/1921227a0. [DOI] [PubMed]
- 2.Mori H, Baba T, Yokoyama K, Takeuchi R, Nomura W, Makishi K, Otsuka Y, Dose H, Wanner BL. Identification of Essential Genes and Synthetic Lethal Gene Combinations in Escherichia coli K-12. Gene Essentiality. 2015; doi:10.1007/978-1-4939-2398-4. [DOI] [PubMed]
- 3.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998; doi:10.1006/meth.1998.0593. [DOI] [PubMed]
- 4.Ran FA, Hsu PPD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; doi:10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed]
- 5.Pavesi A, Magiorkinis G, Karlin DG. Viral Proteins Originated De Novo by Overprinting Can Be Identified by Codon Usage: Application to the “Gene Nursery” of Deltaretroviruses. PLoS Comput Biol. 2013; doi:10.1371/journal.pcbi.1003162. [DOI] [PMC free article] [PubMed]
- 6.Firth AE, Brown CM. Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics. 2006; doi:10.1186/1471-2105-7-75. [DOI] [PMC free article] [PubMed]
- 7.Strebel K. HIV accessory proteins versus host restriction factors. Curr Opin Virol. 2013; doi:10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed]
- 8.Roberts RJ. How restriction enzymes became the workhorses of molecular biology. Proc Natl Acad Sci USA. 2005; doi:10.1073/pnas.0500923102. [DOI] [PMC free article] [PubMed]
- 9.Alphabetized List of Recognition Specificities. New England BioLabs, Inc. 2016. https://www.neb.com/tools-and-resources/selection-charts/alphabetized-list-of-recognition-specificities. Accessed 4 Jan 2016.
- 10.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; doi:10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed]
- 11.Barnett, M. regex module. Python Software Foundation. 2015. https://pypi.python.org/pypi/regex. Accessed 11 Nov 2015.
- 12.Taylor, LJ. Pyviko source. 2016. https://github.com/louiejtaylor/pyViKO. Accessed 1 Jan 2016.
- 13.Taylor, LJ. Pyviko releases. 2016. https://pypi.python.org/pypi/pyviko. Accessed 2 March 2016.
- 14.Taylor, LJ. Pyviko web user interface. 2016. http://louiejtaylor.github.io/pyViKO/. Accessed 5 Jan 2016.
- 15.Taylor, LJ. Pyviko documentation. http://louiejtaylor.github.io/pyViKO/doc/. Accessed 1 Jan 2016.
- 16.BurntSushi. pdoc source. https://github.com/BurntSushi/pdoc. Accessed 1 Jan 2016.
- 17.Andreev DE, Fernandez-Miragall O, Ramajo J, Dmitriev SE, Terenin IM, Martinez-Salas E, Shatsky IN. Differential factor requirement to assemble translation initiation complexes at the alternative start codons of foot-and-mouth disease virus RNA. RNA. 2007; doi:10.1261/rna.469707. [DOI] [PMC free article] [PubMed]
- 18.Pereira F, Azevedo F, Carvalho Â, Ribeiro GF, Budde MW, Johansson B. Pydna: a simulation and documentation tool for DNA assembly strategies using python. BMC Bioinformatics. 2015; doi:10.1186/s12859-015-0544-x. [DOI] [PMC free article] [PubMed]
- 19.Delaye L, Deluna A, Lazcano A, Becerra A. The origin of a novel gene through overprinting in Escherichia coli. BMC Evol Biol. 2008; doi:10.1186/1471-2148-8-31. [DOI] [PMC free article] [PubMed]
- 20.Sanna CR, Li W-H, Zhang L. Overlapping genes in the human and mouse genomes. BMC Genomics. 2008; doi:10.1186/1471-2164-9-169. [DOI] [PMC free article] [PubMed]
- 21.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler DL. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2004; doi:10.1093/nar/gki062. [DOI] [PMC free article] [PubMed]
- 23.Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014; doi:10.1038/nrg3520. [DOI] [PubMed]
- 24.Taylor LJ. Pyviko Web UI Quick Start Guide. http://louiejtaylor.github.io/pyViKO/doc/Pyviko_quick-start.pdf. Accessed 2 Apr 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and analysis scripts are included as supplementary files. The Pyviko web interface is available at http://louiejtaylor.github.io/pyViKO/, and the code is hosted on GitHub at https://github.com/louiejtaylor/pyViKO.