

Abstract
Current structural genomics projects are yielding structures for proteins whose functions are unknown. Accordingly, there is a pressing requirement for computational methods for function prediction. Here we present phunctioner, an automatic method for structure-based function prediction using automatically extracted functional sites (residues associated to functions). The method relates proteins with the same function through structural alignments and extracts 3D profiles of conserved residues. Functional features to train the method are extracted from the Gene Ontology (GO) database. The method extracts these features from the entire GO hierarchy and hence is applicable across the whole range of function specificity. 3D profiles associated with 121 GO annotations were extracted. We tested the power of the method both for the prediction of function and for the extraction of functional sites. The success of function prediction by our method was compared with the standard homology-based method. In the zone of low sequence similarity (≈15%), our method assigns the correct GO annotation in 90% of the protein structures considered, ≈20% higher than inheritance of function from the closest homologue.
Keywords: functional residue, function prediction, structural genomics
Increasingly, protein structures are being determined without a knowledge of the function of the molecule. Proteins with unassigned function began to accumulate in the Protein Data Bank (PDB) (1) 6 years ago, and their number is growing exponentially (see supporting information, which is published on the PNAS web site). Today, there are ≈500 proteins annotated as “hypothetical” in PDB (roughly 1 per 50 entries). This gap is expected to increase dramatically as a consequence of structural genomics projects in which high-throughput methods are applied to determine the conformations of numerous proteins in a genome-wide strategy (e.g., ref. 2). One major motivation of structural genomics projects is that the determination of the structure of a protein provides insight into its molecular function, which is a step toward understanding its cellular function. The current structure–function gap clearly shows that more powerful bioinformatics techniques for function prediction are urgently needed (3–5). Recently, several groups have developed algorithms to identify functionally important residues often employing sequence conservation and/or structural information (see below). However, identification of function residues is distinct from actually assigning a function to the protein. Here we present an automated structure-based method for function prediction.
The complexity of protein function makes the establishment of any functional classification problematic (6, 7). Today, an extensively used functional classification is derived from the Gene Ontology (GO) project (8). By means of GO, one can establish a functional hierarchy that progresses from general functions to more specific functions. As exemplified in GO, protein function ranges from the very general (e.g., enzyme activity) through broad terms (e.g., hydrolase) down to more specific terms (e.g., hydrolysis of O-glycosyl compounds). The aim of function prediction is to cover as many of these GO levels as possible.
There are several sequence-based approaches for function prediction. A simple and widely used strategy is the identification of a high sequence similarity between proteins of known and unknown function that is then used to transfer the specific function. However, as shown by several general analyses (9–11), lower levels of sequence similarity can only be used to transfer general functions, and, even then, this approach is not reliable. Other widely used sequence-based methods employ specific profiles and related hidden Markov models with several prominent strategies now available from the InterPro resource (12). An alternate approach, developed by Hannenhalli and Russell (13), extracts subfamily-specific functional sites and then uses these sites to assign proteins to functional subclasses (see Discussion). In addition, there are methods that do not rely directly on sequence similarity (e.g., refs. 14–16).
Structural information provides valuable insight into protein function (17). But just recognizing that two proteins have similar 3D folds in the absence of clear sequence identity does not imply similar function (18–21). One must identify a similarity both in sequence and spatial location of the key functional residues between the proteins of known and unknown function. The alternative scenario of convergent evolution that results in the key functional residues being hosted on different folds (e.g., the serine proteases subtilisin and trypsin) is rare (e.g., ref. 20). Based on these observations, a number of approaches successfully use 3D templates (a set of residues in a 3D layout) known to be associated to functions (i.e., enzyme active sites) to scan new structures against the profile library (22–24). A drawback of these methods is their inability to locate 3D profiles automatically. In their current form, these approaches can be applied only to functional templates already described in the literature; moreover, they restrict the sequence variation allowed in the templates.
Given the difficulties in actually predicting function, as a step toward this goal several groups have focused on the identification of functional sites and regions. Early work considered fully conserved residues (25). This approach was extended to the detection of family-specific conservation (residues responsible for specificity) (26–30). 3D structural information has also been used to detect functionally important residues and regions, either alone (31–34) or in combination with sequence information (35–39). Recently, a method has been developed (40) that can extract 3D profiles automatically from a set of 3D structures without the need of a structural alignment. This method has been successfully applied to a number of proteins. However, the approach cannot handle hydrophobic residues and imposes restrictions both on the number of positions in the profiles and on the number of residue types at each position. This method has not yet been tested on function prediction.
Here we propose phunctioner, a new automated method for the prediction of function based on the identification of functional sites. Our approach is based on several concepts. First, structural alignments of homologous proteins can more accurately establish sequence alignments between proteins than would be obtained by sequence-based methods alone. Second, the GO classification provides a powerful approach to group proteins with similar function. Third, the GO classification provides hierarchical and extensive classification of functions. Fourth, residues contributing to function for an entire homologous family will tend to be conserved throughout the family, whereas residues responsible for the specificity of a subfamily will only be conserved within that subfamily.
We tested the ability of the method to predict function and to locate functional sites and compared these with standard sequence-based approaches.
Methods
Fig. 1 shows a schema of the method. Proteins with the same GO annotation (8) are extracted from the FSSP (Families of Structurally Similar Proteins) database of structural alignments (41). Conserved residues in these alignments are extracted and used to generate position-specific scoring matrices (PSSMs). A structure of unknown function would be scanned against the library of PSSMs, and a confident match is used to assign function to the structure.
Fig. 1.

Schema of the method. (A) Initial filtered structural alignment. This alignment contains proteins with different functional annotations (depicted here with different colors). Ideally, such an alignment would contain only a few conserved positions because of structural constraints. (B) Splitting of the initial structural alignment into function-specific subalignments according to the functional annotations of the proteins. Conservation patterns due to functional reasons become visible in those subalignments. The conserved residues in those subalignments, which can be interpreted as the function-determinant subalignments and mapped into the 3D structure, are used to construct PSSMs. (C) These PSSMs are used to assign a new structure to the corresponding function by scoring the sequence against the profiles in the search for the one where it fits better (thick arrow).
Extraction of the “Function-Determining” Residues. Structural alignments were taken from the FSSP database (41) in October of 2001. Each FSSP structural alignment is filtered by removing proteins with ≥35% sequence identity with any other in the alignment, proteins with structural similarity <6.0 (FSSP Z score), and proteins annotated as “mutant.”
The resulting alignment is split into different subalignments (Fig. 1), one for each GO term. The sets of PDB chains in these subalignments may overlap because a chain with more than one GO term can be present in more than one subalignment. The mapping between GO (June 2003) and PDB is taken from the GO Annotation project (42). Subalignments with fewer than four sequences are omitted.
The conservation for position i (Ci) in one of these subalignments is calculated as
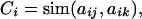 |
where j and k run for all pairs between the sequences in the subalignment; sim(aij, aik) is the similarity between the residues of sequences j and k in position i in accordance with the McLachlan substitution matrix (43).
To assess how conserved a position is with respect to the whole subalignment, the Z score of its conservation is calculated as
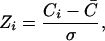 |
where C̄ is the average conservation value of all of the positions in the subalignment, and σ is the standard deviation. Positions with Zi higher than a given threshold (Zcut) are taken as the conserved positions. Positions with Zi in the whole original FSSP alignment (before splitting) higher than another cutoff (Zocut) are excluded from this set because they are conserved through all subalignments possibly because of structural (not functional) requirements. This set of positions (profile), extracted automatically, can be considered as the “function determinant” of a given GO “function” in a given structural family (Fig. 1). To exclude profiles with low conservation, we impose an additional filter by calculating the average entropy of the whole set of positions and allowing only profiles with an average entropy (AvS) lower than a given cutoff AvScut. The average entropy for a profile of length n is
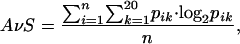 |
where pik is the fraction of sequences in position i having the residue type k.
Construction of the PSSM. Each profile is converted to a PSSM (Fig. 1) by using the Gribskov–Luethy–Eisenberg method (44). For a profile composed by n positions, a matrix of n positions by 20 residue types is constructed where the entry i, j is given by
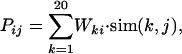 |
where k runs over the 20 residue types. sim(k, j) is the similarity (43) between the residue type k and the one represented by the jth column of the profile. Wki represents the fraction of residue type k in position i in the subalignment determined by logarithmic weighting (44),
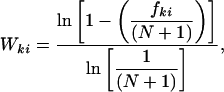 |
where fki is the absolute frequency of residue type k in position i of the subalignment. The score of a set of n residues against one of these profiles is calculated as
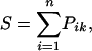 |
where k is the residue type in the ith position. This score is converted to a Z score by shuffling the n positions 5,000 times, rescoring against the profile, and calculating the corresponding average (S̄) and standard deviation (σs). The phunctioner Z score provides a measure of the reliability of the prediction, with a high positive score indicating a confident prediction:
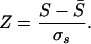 |
Testing the Method. For each sequence in a given filtered FSSP structural alignment, we removed it from the alignment, rebuilt the subalignments and corresponding PSSMs without it, and calculated the Z score of this sequence against each one of these PSSMs.
We compared this method with a simple assignment by sequence identity. For each sequence in the FSSP file, we calculated the percentage of sequence identity with all of the others. We converted these values to Z scores by shuffling the query sequence 2,000 times and obtaining the average and standard deviation of the percentage of identity. So, for a given sequence, we obtained a ranked list of PSSMs (representing their corresponding GO terms), and/or a ranked list of sequences. To compare both methods on the same set of proteins, we evaluated the cases in which at least one profile and one sequence in the corresponding lists match the query sequence. For these cases, we evaluated how frequently the high-scoring subalignments actually correspond to functions performed by the query protein and how frequently the high-scoring sequences have at least one function in common with the query protein. More than 50% of the lists contain four profiles or more, 13% contain three profiles, 20% contain two profiles, and 17% contain only one profile.
Our method is intended primarily to be used when there is no strong sequence similarity with proteins of known function (otherwise alternate methods for function transfer can be used). To simulate this scenario, we repeated that experiment and removed proteins with a sequence identity higher than a given threshold with the query sequence. To assess how both methods work for the different levels of function specificity, we repeated the tests and considered only GO terms belonging to a level in the GO “hierarchy” higher than a certain value.
With this procedure, we obtained 4,753 subalignments (profiles) comprising 121 different GO terms in different levels of the GO hierarchy. We tested the method on sets of proteins ranging from 2,011 to 6,168. Because of the organization of the FSSP database and the eventual presence of more than one structural domain, a protein can be in more than one structural alignment and hence be evaluated more than once, which allows for the assignment of different GO terms (different top scores) to a single protein.
Results
Structure-Based Function Prediction. Fig. 2 shows the accuracy of phunctioner and seqid in predicting the correct GO term for different sets of parameters. We quantify the percentage of cases in which the first hit predicted by a method is correct (Fig. 2 A). For most sets of parameters, phunctioner outperforms seqid. The accuracy of phunctioner ranges from 75% to >90%, depending on the parameters, whereas the accuracy of seqid ranges from 60% to 90%. The results show that phunctioner can reliably assign function in zones of low sequence identity, where seqid fails. A “sign test” (45) demonstrates that this method is significantly better than seqid at <20% sequence identity (data not shown). As expected, the accuracy of seqid improves as we permit hits with more similar sequences (15–30%). For the 30% sequence identity cutoff, the accuracy of both methods is comparable, and seqid outperforms phunctioner for some sets of parameters. The accuracy of phunctioner improves as we restrict the test set to more informative profiles (AvScut from 1.4 to 1.0; see supporting information) and as we discard very general (unspecific) functions (GO level from 2 to 3) at the expense of obtaining predictions for less proteins.
Fig. 2.

Evaluation of the phunctioner method and seqid in function prediction. (A) Percentage of correct predictions in the first position of the ranking lists of both methods. The black bars represent the phunctioner method, and the gray bars represent the seqid method. The sets of columns represent different values for the sequence identity and GO-level cutoffs (values are shown on the left). In all of the cases, the Zcut parameter is 2, no Zocut cutoff is applied, and the AvScut cutoff is 1.0. See supporting information for extended versions showing other sets of parameters. (B) Receiver–operator-characteristic curves of both methods for the ≤15% sequence identity test set. (Parameters: Zcut, 2; Zocut, none; GO level, 3; AvScut, 1.4.) The corresponding phunctioner Z score for some point is also indicated. TP, true positives; FN, false negatives; TN, true negatives; FP, false positives. (C) Regions of sequence identity (seqid.; between the query sequence and the found hit) where both methods found a correct hit in the first position. The percentage of cases for which both methods found a correct hit in the first position at each level of sequence identity is shown. (D) Relationship between the phunctioner Z score of the first hit and the number of true (black) and false (gray) positives. (Parameters: Zcut,2; Zocut, none; GO level, 2; AvScut, 1.4.)
For a user, it is important to have an estimation of the likely number of true- and false-positive predictions. Formally this can be represented by a receiver operator characteristic curve (ROC) that plots (1-specificity) against the sensitivity (see Fig. 2B). A perfect method, able to recover all true hits without any false positive would be represented by a point in the upper left corner of the graph, whereas a random method that produces equal numbers of true- and false-positive predictions uniformly distributed across all scores would be a diagonal from (0,0) to (1,1). We calculated the ROC curves with the scores of the first hit for the cases when there are no hits with appreciable sequence identity (≤15%). This curve shows the ability of the method in discriminating true from false hits for various Z-score cutoffs. ROC curves can also be used to compare methods, and Fig. 2B shows that phunctioner is better than seqid for any cost ratio (sensitivity versus specificity).
A user of the method would examine the Z score to obtain an indication of the reliability of the prediction. We calculated the relation between the Z score of the first hit and the number of true and false positives (Fig. 2D). The number of false positives in the first position is lower than the number of true ones, and the false positives are shifted to lower scores. So, as we restrict Z scores to higher values, the proportion of true positives increases. For example, if we restrict to Z scores >6.0, 88% of the cases are correct (i.e., 88% of the predictions are true positives), and if we Z restrict scores to ≥10.0, 95% are correct (but at expense of having lower coverage). The corresponding values for coverage are 57.0% of the proteins for a Z score ≥6.0 and 11.5% for a Z score ≥10.0).
Example of Structure-Based Functional Assignation. We applied our method to a structural genomics target, the hypothetical protein MTH538 (PDB ID code 1EIW) (46). This protein has the structure of two-component system receiver domains (CheY), nucleotide triphosphate (NTP)-binding proteins, and flavodoxins. Experiments show that it binds Mg2+ but not flavin mononucleotide. A sequence search with psi-blast (47) finds the COG (48) family COG1618 (“putative ATPases or kinases”), although the protein does not have the classical NTP binding motifs. Our method assigns this protein to the “two-component response regulatory activity” (GO term GO:0000156; Z score = 4.96) and to the “Mg2+ ion binding activity” (GO term GO:0000287; Z score = 7.07) annotations. Interestingly, the residues automatically detected by the method as responsible of the Mg2+ binding activity (residues 13, 19, 70, and 71) map in similar regions to the ones whose resonances shift after Mg2+ titration in NMR experiments (46): residues 13, 15, 55–56, 77, 92–95, and 99. Our method could not test the protein against flavin-related GO terms because profiles could not be generated. The method clearly “rejects” the assignment of the protein to NTP binding functions (GTP or ATP) by producing very bad Z scores for them (0.80 for GTP binding and 1.71 for ATP binding). Thus, our method clearly detects that the protein does not fit in the NTP-binding motifs, despite the global sequence signals detected by psi-blast. Although these features were already identified through extensive manual inspection of sequences and structures, our method identified them automatically. Our method also predicts three additional functions that do not match what is known so far about the protein: Ca2+ binding (Z score = 5.18), transcription factor [Z score = 3.44; because of profiles derived from receiver domains of transcription factors (a false positive)], and transferase [Z score = 2.10 (another possible false positive)]. Refer to Fig. 2 for expected ratios of false positives associated with these scores.
We include four additional examples of function prediction for hypothetical proteins in supporting information. The Z scores range from 1 to 4 when one would expect roughly equal numbers of true and false predictions. Inspection of the information about the function of these proteins suggests that our method is providing several reasonable functional annotations.
Extracted Function-Determinant Residues. In addition to the prediction of function, our method automatically detects sets of residues (3D profiles) associated to GO terms (Fig. 1). Fig. 3A shows the residues automatically extracted by this method for the GO term “GTP-binding activity” (GO:0005525) in the 1ctqA.fssp structural alignment, mapped on the structure of the Ras oncogene (PDB ID code 1CTQ). All of the residues lie around the bound nucleotide accounting for the GTP-binding activity function. This profile is highly specific as indicated by the average value of 7.1 for the phunctioner Z score for all proteins in the alignment with GTP-binding activity. In contrast, the average Z score for proteins without GTP-binding activity (including the highly similar ATP-binding activity) against this profile is only 1.1.
Fig. 3.

Different ways to extract functional residues associated with the GTP-binding activity GO annotation (GO:0005525). The nucleotide (GTP) is shown in green. (A) Residues extracted with the method described here. The residues are represented in spacefill and in red. (B) Conserved residues in the sequence alignment of Ras, a protein annotated with GTP-binding activity. In decreasing order of conservation, positions with VAR ≤ 10 are shown in black, positions with 10 < VAR ≤ 15 are shown in dark-blue, and positions with 15 < VAR ≤ 20 are shown in light-blue. (C) Family-dependent conserved residues extracted from the same alignment as in B. Residues predicted by the sequencespace program (26) are shown in blue, residues predicted by the mtreedet program (29) are shown in red, and residues predicted by both methods (like the well studied Glu-37, which is marked with an arrow) are shown in purple. Other views of the structures are available in supporting information.
To illustrate the differences between this and other methods, we calculated other sets of functional residues one would obtain for this GTP-binding function by using different strategies:
Conserved residues in the sequence alignment of a family of proteins having that GTP-binding function (Ras). We obtained the multiple sequence alignment for this family from the HSSP (Homology-Derived Secondary Structure of Proteins) database (49), removing redundant sequences (>95% sequence identity). We used three cutoffs of conservation according to the HSSP VAR parameter (sequence variability on a scale of 0–100). These conserved residues largely map to the nucleotide-binding site, but they are also in other parts of the protein, reflecting other functions of Ras beside the GTP-binding in which we are interested (Fig. 3B).
Family-dependent conserved residues were obtained from the same multiple sequence alignment by using two different methods: sequencespace (26) (selecting positions present in more than three subfamilies) and mtreedet, which implements the Mutational Behavior method (29) (using a correlation cutoff of 0.6). These methods identified residues that are to some extent located in the nucleotide binding site, but mainly in the regions conferring specificity to the different subfamilies. Most of the residues clearly identify the surface known to be implicated in the interaction with different Ras effectors (down in Fig. 3C) (50). Again, those positions are only slightly related to the GTP-binding activity GO term, and not exclusively to it.
Conserved residues for all of the proteins annotated with that GO term. In general, it is impossible to align the large number of proteins associated to a given GO term because they comprise very divergent sequences.
This example illustrates the kind of functional positions our method is detecting and the differences with the existing methods. The alternate methods will generally be unable to extract the residues exclusively responsible for a general GO function except when it is highly specific.
Discussion
We have developed a method (phunctioner) for the prediction of protein function and the concomitant identification of functionally important residues. The approach is fully automated and considers the entire spectrum of function in terms of specificity. The method is based on the widely used GO classification, and it employs structural superpositions to extract the profiles in the library and thus can be applied to proteins for which purely sequenced-based methods would not be applicable. Our benchmark shows that the approach is more effective than sequence–homology-based methods, especially in the twilight zone of function annotation.
There are several limitations in the current implementation of the method. First, it cannot generate a profile for a function hosted on more than one fold as a result of convergent evolution. But convergent evolution of function is rare because the evolutionary relationship between function and structure is mainly the result of divergence (e.g., ref. 20). Second, the requirement of structural information plus the need of four or more homologous or analogous structures to build a profile limits the coverage of the method. Third, the method is restricted to identifying functions associated with proteins of known structure.
In terms of accuracy, many false negatives of the method are due to profiles with low information content. Actually, the results improve when restricting the test set to more specific GO levels or low entropy profiles (see Results) but with a corresponding decrease in coverage. There are also false positives, some of them with very clear profiles, for which we do not have an explanation.
Our approach has similarities to some recently proposed strategies. The method of Hannenhalli and Russell (13) is also based on the extracted subfamily-specific residues to assign proteins to subfamilies. However, their method can only assign subclasses to proteins already known to belong to a class (thus requiring high sequence identity). So, their method is intended to work at high levels of function specificity. However, an advantage of their method is that it does not require structural information.
Other methods for function prediction are based on the matching of structures to amino acid 3D profiles (22–24). The advantages of our method are the ability to locate previously unreported 3D profiles automatically and the richer representation of their sequence composition with PSSMs. A step toward the automatic generation of 3D profiles without the requirement of being previously reported comes from the method of Wangikar et al. (40). However, their approach is restricted in the profile size, its inability to handle any kind of residue (including hydrophobic), and the lack of flexibility associated with the PSSM representation of the profiles. Moreover, their method has not been tested in function prediction but only in the generation of 3D profiles.
An important consequence of our work is the generation of “3D templates” (profiles) for a large number of GO terms, some of them without previously associated “functional residues.” These profiles could be used with the other methods described for matching 3D structures and 3D profiles not based in structural alignments (22, 24, 40).
As with most predictions, the accuracy values obtained are not perfect; hence, any result of this method has to be taken as a hypothesis for further investigation. Our method will benefit in both accuracy and coverage from the expansion of functional and structural databases. Because the method is fully automatic, it could be coupled to the pipeline of structural genomics projects to have automatic prediction of function and functional sites for the outgoing structures when no clear sequence information is available.
Supplementary Material
Acknowledgments
We thank Lawrence Kelley, Juan A. García-Ranea, and Alfonso Valencia for interesting discussion. We also thank the maintainers of the databases used in this work and the two anonymous referees for constructive comments. F.P. is supported by Department of Trade and Industry Beacon Award QCBB/C/012/00003.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PDB, Protein Data Bank; GO, Gene Ontology; PSSM, position-specific scoring matrix.
References
- 1.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christendat, D., Yee, A., Dharamsi, A., Kluger, Y., Savchenko, A., Cort, J. R., Booth, V., Mackereth, C. D., Saridakis, V., Ekiel, I., et al. (2000) Nat. Struct. Biol. 7, 903–909. [DOI] [PubMed] [Google Scholar]
- 3.Goldsmith-Fischman, S. & Honig, B. (2003) Protein Sci. 12, 1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinoshita, K. & Nakamura, H. (2003) Curr. Opin. Struct. Biol. 13, 396–400. [DOI] [PubMed] [Google Scholar]
- 5.Norin, M. & Sundström, M. (2002) Trends Biotech. 20, 79–84. [DOI] [PubMed] [Google Scholar]
- 6.Shrager, J. (2003) Bioinformatics 19, 1934–1936. [DOI] [PubMed] [Google Scholar]
- 7.Jeffery, C. J. (2003) Trends Genet. 19, 415–417. [DOI] [PubMed] [Google Scholar]
- 8.Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. (2004) Nucleic Acids Res. 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson, C. A., Kreychman, J. & Gerstein, M. (2000) J. Mol. Biol. 297, 233–249. [DOI] [PubMed] [Google Scholar]
- 10.Devos, D. & Valencia, A. (2000) Proteins 41, 98–107. [PubMed] [Google Scholar]
- 11.Rost, B. (2002) J. Mol. Biol. 318, 595–608. [DOI] [PubMed] [Google Scholar]
- 12.Mulder, N. J., Apweiler, R., Attwood, T. K., Bairoch, A., Barrell, D., Bateman, A., Binns, D., Biswas, M., Bradley, P., Bork, P., et al. (2003) Nucleic Acids Res. 31, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannenhalli, S. S. & Russell, R. B. (2000) J. Mol. Biol. 303, 61–76. [DOI] [PubMed] [Google Scholar]
- 14.Clare, A. & King, R. D. (2003) Bioinformatics 19, Suppl. 2, II42–II49. [DOI] [PubMed] [Google Scholar]
- 15.Jensen, L. J., Gupta, R., Staerfeld, H. H. & Brunak, S. (2003) Bioinformatics 19, 635–642. [DOI] [PubMed] [Google Scholar]
- 16.Sjölander, K. (2003) Bioinformatics 20, 170–179. [DOI] [PubMed] [Google Scholar]
- 17.Thornton, J. M., Todd, A. E., Milburn, D., Borkakoti, N. & Orengo, C. (2000) Nat. Struct. Biol. 7, 991–994. [DOI] [PubMed] [Google Scholar]
- 18.Todd, A. E., Orengo, C. A. & Thornton, J. M. (2001) J. Mol. Biol. 307, 1113–1143. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett, G. J., Borkakoti, N. & Thornton, J. M. (2003) J. Mol. Biol. 331, 829–860. [DOI] [PubMed] [Google Scholar]
- 20.Shakhnovich, B. E., Dokholyan, N. V., DeLisi, C. & Shakhnovich, E. I. (2003) J. Mol. Biol. 326, 1–9. [DOI] [PubMed] [Google Scholar]
- 21.Orengo, C. A., Todd, A. E. & Thornton, J. M. (1999) Curr. Opin. Struct. Biol. 9, 374–382. [DOI] [PubMed] [Google Scholar]
- 22.Di Gennaro, J. A., Siew, N., Hoffman, B. T., Zhang, L., Skolnick, J., Neilson, L. I. & Fetrow, J. S. (2001) J. Struct. Biol. 134, 232–245. [DOI] [PubMed] [Google Scholar]
- 23.Wallace, A. C., Borkakoti, N. & Thornton, J. M. (1997) Protein Sci. 6, 2308–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark, A. & Russell, R. B. (2003) Nucleic Acids Res. 31, 3341–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuckerkandl, E. & Pauling, L. (1965) in Evolving Genes and Proteins, eds. Bryson, V. & Vogel, H. J. (Academic, New York), pp. 97–166. [DOI] [PubMed]
- 26.Casari, G., Sander, C. & Valencia, A. (1995) Nat. Struct. Biol. 2, 171–178. [DOI] [PubMed] [Google Scholar]
- 27.Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996) J. Mol. Biol. 257, 342–358. [DOI] [PubMed] [Google Scholar]
- 28.Aloy, P., Querol, E., Aviles, F. X. & Sternberg, M. J. E. (2001) J. Mol. Biol. 311, 395–408. [DOI] [PubMed] [Google Scholar]
- 29.del Sol Mesa, A., Pazos, F. & Valencia, A. (2003) J. Mol. Biol. 326, 1289–1302. [DOI] [PubMed] [Google Scholar]
- 30.Lichtarge, O. & Sowa, M. E. (2002) Curr. Opin. Struct. Biol. 12, 21–27. [DOI] [PubMed] [Google Scholar]
- 31.Luque, I. & Freire, E. (2000) Proteins 4, Suppl. 4, 63–71. [DOI] [PubMed] [Google Scholar]
- 32.Elcock, A. (2001) J. Mol. Biol. 312, 885–896. [DOI] [PubMed] [Google Scholar]
- 33.Shulman-Peleg, A., Nussinov, R. & Wolfson, H. J. (2004) J. Mol. Biol. 339, 607–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ondrechen, M. J., Clifton, J. G. & Ringe, D. (2001) Proc. Natl. Acad. Sci. USA 98, 12473–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landgraf, R., Xenarios, I. & Eisenberg, D. (2001) J. Mol. Biol. 307, 1487–1502. [DOI] [PubMed] [Google Scholar]
- 36.de Rinaldis, M., Ausiello, G., Cesareni, G. & Helmer-Citterich, M. (1998) J. Mol. Biol. 284, 1211–1221. [DOI] [PubMed] [Google Scholar]
- 37.Ota, M., Kinoshita, K. & Nishikawa, K. (2003) J. Mol. Biol. 327, 1053–1064. [DOI] [PubMed] [Google Scholar]
- 38.Gutteridge, A., Bartlett, G. & Thornton, J. M. (2003) J. Mol. Biol. 330, 719–734. [DOI] [PubMed] [Google Scholar]
- 39.Innis, C. A., Anand, A. P. & Sowdhamini, R. (2004) J. Mol. Biol. 337, 1053–1068. [DOI] [PubMed] [Google Scholar]
- 40.Wangikar, P. P., Tendulkar, A. V., Ramya, S., Mali, D. N. & Sarawagi, S. (2003) J. Mol. Biol. 326, 955–978. [DOI] [PubMed] [Google Scholar]
- 41.Holm, L. & Sander, C. (1994) Nucleic Acids Res. 22, 3600–3609. [PMC free article] [PubMed] [Google Scholar]
- 42.Camon, E., Magrane, M., Barrell, D., Lee, V., Dimmer, E., Maslen, J., Binns, D., Harte, N., Lopez, R. & Apweiler, R. (2004) Nucleic Acids Res. 32, D262–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLachlan, A. D. (1971) J. Mol. Biol. 61, 409–424. [DOI] [PubMed] [Google Scholar]
- 44.Gribskov, M., Luethy, R. & Eisenberg, D. (1990) Methods Enzymol. 183, 146–159. [DOI] [PubMed] [Google Scholar]
- 45.Bland, M. (1987) An Introduction to Medical Statistics (Oxford Univ. Press, London).
- 46.Cort, J. R., Yee, A., Edwards, A. M., Arrowsmith, C. H. & Kennedy, M. A. (2000) J. Mol. Biol. 302, 189–203. [DOI] [PubMed] [Google Scholar]
- 47.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278, 631–637. [DOI] [PubMed] [Google Scholar]
- 49.Sander, C. & Schneider, R. (1993) Nucleic Acids Res. 21, 3105–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer, B., Mirey, G., Vetter, I. R., Garcia-Ranea, J. A., Valencia, A., Wittinghofer, A., Camonis, J. H. & Cool, R. H. (1999) J. Biol. Chem. 274, 17763–17770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}