

Phosphatidylinositol 4-phosphate undergoes striking multiphasic changes during phagosome formation and maturation. The molecular mechanisms underlying these changes and the role of phosphatidylinositol 4-phosphate in phagolysosome formation are investigated.
Abstract
We analyzed the distribution, fate, and functional role of phosphatidylinositol 4-phosphate (PtdIns4P) during phagosome formation and maturation. To this end, we used genetically encoded probes consisting of the PtdIns4P-binding domain of the bacterial effector SidM. PtdIns4P was found to undergo complex, multiphasic changes during phagocytosis. The phosphoinositide, which is present in the plasmalemma before engagement of the target particle, is transiently enriched in the phagosomal cup. Soon after the phagosome seals, PtdIns4P levels drop precipitously due to the hydrolytic activity of Sac2 and phospholipase C, becoming undetectable for ∼10 min. PtdIns4P disappearance coincides with the emergence of phagosomal PtdIns3P. Conversely, the disappearance of PtdIns3P that signals the transition from early to late phagosomes is accompanied by resurgence of PtdIns4P, which is associated with the recruitment of phosphatidylinositol 4-kinase 2A. The reacquisition of PtdIns4P can be prevented by silencing expression of the kinase and can be counteracted by recruitment of a 4-phosphatase with a heterodimerization system. Using these approaches, we found that the secondary accumulation of PtdIns4P is required for proper phagosomal acidification. Defective acidification may be caused by impaired recruitment of Rab7 effectors, including RILP, which were shown earlier to displace phagosomes toward perinuclear lysosomes. Our results show multimodal dynamics of PtdIns4P during phagocytosis and suggest that the phosphoinositide plays important roles during the maturation of the phagosome.
INTRODUCTION
Phagocytosis, the ingestion of particulate matter, is essential for the elimination of invading pathogens, serving as a first line of defense of the immune system (Levin et al., 2016). It also plays a key role in tissue homeostasis and remodeling, disposing of apoptotic bodies and debris (Elliott and Ravichandran, 2010). The efficient and timely removal of effete cells is necessary to prevent secondary necrosis and unwanted inflammation.
Phagocytosis is initiated by the engagement of surface receptors that trigger extensive remodeling of the plasma membrane (PM) and the actin cytoskeleton (Levin et al., 2016), culminating with the extension of pseudopods that surround and engulf the target. The nascent phagosome then undergoes profound changes, acquiring microbicidal and degradative properties that enable it to process its contents. This is accomplished through a series of fission and fusion events with other endomembrane compartments, a process globally termed phagosome maturation (Fairn and Grinstein, 2012).
Phagosome formation and maturation are both temporally and spatially restricted; as such, they are subject to exquisite control, relying on highly coordinated signals. Phosphatidylinositol derivatives are central to the signaling sequence (Swanson, 2014; Levin et al., 2015). A combination of biochemical determinations and imaging using genetically encoded biosensors showed that phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) accumulates at the phagosomal cup, whereas phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) is acutely depleted (Botelho et al., 2000; Marshall et al., 2001). These changes orchestrate actin remodeling. Soon after sealing, the phagosome accumulates phosphatidylinositol 3-phosphate (PtdIns3P), which signals the initiation of the maturation process (Vieira et al., 2001).
Unlike these phosphoinositides, little is known about phosphatidylinositol 4-phosphate (PtdIns4P) in phagocytosis, despite its abundance. This is due, at least partly, to the lack of appropriate tools to detect this lipid with sufficient sensitivity and specificity. This hurdle, however, was recently overcome by the development of a reliable biosensor to monitor PtdIns4P in live cells. Hammond et al. (2014) developed a novel PtdIns4P-sensing probe using a domain of the Legionella pneumophila PtdIns4P-interacting effector SidM (Brombacher et al., 2009) that recognizes the head group of the phospholipid stereospecifically. Expression of fluorescently tagged versions of this biosensor in mammalian cells not only highlighted the previously identified pools of PtdIns4P in the Golgi and plasma membrane, but it also revealed the presence of PtdIns4P in Rab7-positive late endosomes and lysosomes (Hammond et al., 2014).
We took advantage of this newly developed probe to analyze the fate of PtdIns4P during phagosome formation and maturation. Our results revealed the occurrence of striking triphasic changes in the content of PtdIns4P of the nascent and maturing phagosome. Of importance, we detected a previously unappreciated wave of PtdIns4P regeneration that is critical for the transition from early to late phagosomes, enabling luminal acidification and the acquisition of the Rab7-interacting lysosomal protein (RILP).
RESULTS
Detection of PtdIns4P in macrophages
We initially assessed the ability of a chimeric construct consisting of the P4M domain of SidM linked to green fluorescent protein (GFP-P4M) to detect PtdIns4P in macrophages. When transiently expressed in RAW264.7 cells—a murine line of monocyte/macrophage origin—GFP-P4M was most prominently localized to the Golgi apparatus/trans-Golgi network (TGN), with less prominent accumulation at the PM and in cytoplasmic puncta (Figure 1Ai). However, a large fraction of the probe was unbound (cytosolic), lowering the contrast and making the organellar pools difficult to discriminate and track during phagocytosis. We therefore tested a second probe consisting of two P4M domains fused in tandem and tagged with GFP (GFP-2xP4M), which is expected to bind to PtdIns4P-containing membranes with greater avidity. GFP-2xP4M labeled not only the Golgi elements but also the PM (Figure 1Aii) and endosomal structures that were Rab7 positive (unpublished data). We routinely observed far less cytosolic GFP signal in cells expressing the tandem probe compared with the single P4M domain, consistent with the notion that GFP-2xP4M binds PtdIns4P with higher avidity. Of note, the morphology of the Golgi apparatus, assessed using mCherry (mCh)-sialyltransferase as marker, was not affected in cells expressing low to medium levels of the tandem biosensor (Supplemental Figure S1). In addition, we observed no perturbations in the distribution of a PtdIns(4,5)P2-binding probe (the PH domain of phospholipase Cδ [PLCδ]), suggesting that the metabolism of this lipid was not altered. We therefore concluded that, when expressed at moderate levels, the GFP-2xP4M probe did not interfere noticeably with overall phosphoinositide metabolism or cell function.
FIGURE 1:

PtdIns4P undergoes triphasic changes during phagocytosis. (A) Confocal section of RAW264.7 cells transiently expressing (i) GFP-P4M or (ii) two P4M domains fused in tandem tagged with GFP (GFP-2xP4M). (B) Specificity of the P4M domain. A hydrophobic membrane where 100-pmol samples of the indicated lipids had been spotted was overlaid with purified recombinant GST-GFP-P4M. Bound protein was detected with an anti-GFP antibody followed by an HRP-conjugated secondary antibody. (C) Recruitment of Sac1 to the PM through a rapamycin heterodimerization system consisting of a membrane-targeted FRB domain (Lyn11-FRB) and mCherry-tagged, FKBP-attached Sac1(mCh-FKBP-Sac1). (D) RAW264.7 cells transiently coexpressing GFP-2xP4M, mCh-FKBP-Sac1, and Lyn11-FRB imaged before (left) and after (right) the addition of rapamycin (1 μM); insets, mCh-FKBP-Sac1 (F-Sac1) fluorescence (black and white are inverted for clarity). (E) Time course of changes in the ratio of intensities of plasmalemmal to Golgi GFP-2xP4M after recruitment of Sac1 to the PM. Time 0 represents values before the addition of rapamycin. Values are means ± SEM of four independent experiments. (F) Time-lapse gallery of confocal micrographs acquired during phagocytosis of TAMRA-labeled IgG-SRBCs (red) by RAW264.7 cells transiently expressing GFP-2xP4M (green). At the bottom is the time elapsed from the moment the target was engaged. Insets, magnifications of the area delimited by dotted white boxes. (G) Summary of changes in PtdIns4P content of phagosomes after engagement of IgG-SRBCs. The intensity of GFP-2xP4M in phagosomes was quantified and normalized to that of the plasmalemma. Three phases were defined: I) an initial transient increase (0–2 min), II) a subsequent disappearance (2–10 min), and III) a reappearance and gradual increase (10–30 min). Data are expressed relative to the maximum value attained during phase I. Values are means ± SEM of 25 phagosomes from 15 independent experiments. (H) PtdIns4P reappearance in phagosomes was often accompanied by formation of 2xP4M-positive tubules. Scale bars, 5 μm.
The specificity of 2xP4M for PtdIns4P, and hence the validity of the determinations, was tested next. To this end, we constructed a glutathione S-transferase (GST)–GFP-P4M plasmid to generate and purify recombinant protein. We used hydrophobic membranes spotted with different lipid species to test the interaction of the purified protein with biologically relevant lipids. As shown in Figure 1B, GST-GFP-P4M bound PtdIns4P with exquisite specificity. This finding is consistent with the observations made using full-length SidM (Brombacher et al., 2009), implying that selectivity for PtdIns4P was retained after excision of the P4M domain and attachment to GFP. Next we assessed the specificity of the sensing probe in cells. To verify the specificity of the probe when expressed in macrophages, we depleted PtdIns4P from the PM in a controlled manner, using a heterodimerization system to recruit the PtdIns4P-specific phosphatase Sac1 using rapamycin (Figure 1C). Cells were triple transfected with the chimera mCh-FKBP-Sac1, a construct containing FRB targeted to the PM through the N-terminal 11 amino acids of Lyn kinase (Lyn11-FRB), and with GFP-2xP4M. As illustrated in Figure 1D, a significant fraction of GFP-2xP4M localized to the PM before the addition of rapamycin, whereas mCh-FKBP-Sac1 remained soluble in the cytosol (Figure 1D, left). On addition of rapamycin, mCh-FKBP-Sac1 rapidly translocated to the PM, catalyzing the hydrolysis of PtdIns4P and causing the release of the GFP-2xP4M probe within seconds; the biosensor released from the PM then accumulated on the Golgi and endosomal membranes (Figure 2, D, right, and E, and Supplemental Video S1). Taken together, these results validate 2xP4M as a specific and reliable biosensor for PtdIns4P in macrophages.
PtdIns4P dynamics during phagosome formation and maturation
We proceeded to determine the dynamics of PtdIns4P during phagocytosis. We chose FcγR-mediated phagocytosis, the most extensively characterized phagocytic system, as an experimental model. Immunoglobulin G (IgG)–opsonized sheep red blood cells (IgG-SRBCs) were used as phagocytic targets, and the dynamics of the PtdIns4P biosensor was monitored by time-lapse video microscopy. As illustrated in Figure 1F and Supplemental Video S2 and quantified in Figure 1G, when RAW264.7 cells transiently expressing GFP-2xP4M were challenged with tetramethylrhodamine (TMR)-labeled IgG-SRBCs, the biosensor accumulated in the forming phagocytic cup relative to unengaged areas of the plasmalemma. On phagosome closure (∼2 min after initiation of internalization), the biosensor levels peaked (end of phase I in Figure 1E). Strikingly, seconds after this marked increase, GFP-2xP4M detached from the sealed phagosome and remained absent from the compartment for ∼8–9 min. Furthermore, the biosensor reappeared gradually in phagosomes, starting ∼10 min after the particle initially contacted the macrophage (Figure 1, F and G). The levels of GFP-2xP4M associated with the phagosome increased continuously for the next 15 min, surpassing the initial maximum observed upon closure and remaining high for at least 30 min after the initiation of phagocytosis (Figure 1, F and G). The reappearance of PtdIns4P was often accompanied by the formation of dynamic 2xP4M-positive tubules and vesicles that emanated from phagosomes toward the juxtanuclear region (Figure 1H). On the basis of similar observations made in >80 phagosomes from multiple cells and preparations, we defined a triphasic pattern of PtdIns4P dynamics: I) a transient increase at the phagocytic cup during phagosome formation, II) a virtually complete disappearance from the early sealed phagosome, and III) a gradual reappearance on maturing phagosomes that ultimately surpasses the density at the PM (Figure 1G).
The mechanisms underlying the changes in PtdIns4P were analyzed next. Conversion to other inositide species seemed likely to account for the observed changes. We therefore compared the dynamic changes of PtdIns4P with that of PtdIns(4,5)P2. As reported earlier (Botelho et al., 2000), when visualized using monomeric red fluorescent protein (mRFP)–PH-PLCδ, PtdIns(4,5)P2 underwent a transient accumulation soon after the macrophages engaged the target, followed by a precipitous decrease that was obvious even before phagosomal closure was completed (Figure 2, A and C). PtdIns(4,5)P2 remained undetectable thereafter, failing to reappear throughout the maturation period. Comparison of the changes in PtdIns4P and those of PtdIns(4,5)P2 in cells cotransfected with GFP-2xP4M and mRFP-PH-PLCδ revealed that 1) the initial increase in PtdIns(4,5)P2 does not incur depletion of PtdIns4P, which in fact also increases during the earliest stages of particle engagement; 2) the steep initial decrease in in PtdIns(4,5)P2 is not accompanied by significant changes in PtdIns4P, suggesting that other mechanisms, such as hydrolysis to diacylglycerol by phospholipase C, are involved; 3) the final stage of PtdIns(4,5)P2 disappearance coincides with a significant increase in PtdIns4P, consistent with hydrolysis by 5-phosphatases; and 4) the sharp decrease in PtdIns4P observed after the phagosome seals cannot be attributed to resynthesis of PtdIns(4,5)P2, which remains undetectable.
FIGURE 2:

Comparison of the changes in PtdIns4P content to those of PtdIns(4,5)P2 and PtdIns3P. (A) Time-lapse gallery of confocal micrographs of RAW264.7 cells coexpressing GFP-2xP4M and mRFP-PH-PLCδ, a PtdIns(4,5)P2 biosensor, during phagocytosis of IgG-SRBCs. (B) Time-lapse gallery of confocal micrographs of cells coexpressing GFP-2xP4M and mRFP-2xFYVE-EEA1, a PtdIns3P biosensor, during phagocytosis of IgG-SRBCs. Insets, magnifications of the area delimited by dotted white boxes; dashed circles show the location of the phagosome. Scale bars, 5 μm. (C) Time course of the changes in PtdIns4P and PtdIns(4,5)P2 during phagosome formation. The intensity of GFP-2xP4M fluorescence in the phagosome was measured and normalized to the plasmalemmal intensity; to enable comparison between cells and between experiments, the data are expressed relative to the maximum value (green line, white circles). A similar analysis was applied to mRFP-PH-PLCδ (red line, black circles), a PtdIns(4,5)P2 biosensor. (D) Time course of the changes in PtdIns4P and PtdIns3P during phagosome formation and maturation. PtdIns4P was monitored as before, using GFP-2xP4M. mCh-FYVE-EEA1 was used as a PtdIns3P probe; phagosomal intensity was measured after subtracting cytosolic fluorescence. Data are expressed relative to the maximum value. Values are means ± SEM of three and five independent experiments, respectively. Note the different time scales used in C vs. D. Initial contact between cells and IgG-SRBC was considered as time 0.
We next compared PtdIns4P and PtdIns(3,4,5)P3 in cells cotransfected with mCh-2xP4M and GFP-PH-Gab2 (Supplemental Figure S2A). As described earlier (Marshall et al., 2001), PtdIns(3,4,5)P3 is detectable in forming cups as targets are engaged, and its levels increase as the pseudopods extend. PtdIns(3,4,5)P3 persists on sealed phagosomes for ∼1 min and then disappears abruptly. Neither the initial accumulation of PtdIns4P nor its secondary disappearance appears to be caused by degradation of or conversion to PtdIns(3,4,5)P3.
We also tracked PtdIns4P simultaneously with PtdIns3P, visualized using tandem FYVE domains of EEA1. PtdIns3P appeared on the phagosomal membrane as PtdIns4P disappeared and was present throughout the period when PtdIns4P was absent (Figure 2, B and D). Remarkably, this divergent behavior was also observed at the later stages, when PtdIns4P reappeared as PtdIns3P disappeared. Although we are not aware of any mechanism capable of directly interconverting these lipids, the changes in PtdIns4P and PtdIns3P seem to be related, conceivably via signaling intermediates. Of note, on the basis of extensive work previously reported by us and others, we concluded that the phagocytic efficiency, the time course of phagocytosis, and the changes in PtdIns(4,5)P2, PtdIns(3,4,5)P3, or PtdIns3P were not noticeably affected by the expression of the 2xP4M probe, confirming that it is innocuous at the expression levels used in this study.
The reacquisition of PtdIns4P may represent a key—yet unappreciated—step in the transition from early to late phagosomes. We therefore endeavored to place the PtdIns4P changes in the context of defined stages of the maturation of the phagosome. To this end, we used different maturation markers. Early phagosomes were identified by the presence of Rab5. As shown in Figure 3A, mRFP-Rab5 was acquired by the nascent phagosomes as PtdIns4P disappeared and was lost before the reacquisition of PtdIns4P. This inverse relationship resembles the behavior described earlier for PtdIns3P, although this inositide persists longer on the early phagosomal membrane than does Rab5, which ceases to be visible after 3–5 min (Figure 3D). Together these data indicate that PtdIns4P is absent from the phagosome during the early phase of maturation.
FIGURE 3:

Late phagosomes and phagolysosomes contain PtdIns4P. (A) Time-lapse gallery of confocal micrographs of RAW264.7 cells transiently coexpressing GFP-2xP4M and mRFP-Rab5 during phagocytosis of IgG-SRBCs. (B) Confocal micrographs of RAW264.7 cells transiently coexpressing GFP-2xP4M and mRFP-Rab7 during the course of phagocytosis of IgG-SRBCs. (C) Confocal micrographs of cells expressing GFP-2xP4M and exposed to TMR-labeled 10-kDa dextran for 3 h and chased for 30 min to label lysosomes. Images are representative of the distribution noted during the indicated time intervals after the onset of phagocytosis. Insets, magnifications of the area delimited by dotted white boxes. Scale bars, 5 μm. (D) Relative timing of the acquisition by phagosomes of different phosphoinositides and canonical markers of phagosome maturation. Qualitative only, intended to illustrate the relative order of events.
The reappearance of PtdIns4P coincides roughly with the onset of the late stage of maturation, as defined by the acquisition of Rab7. Both GFP-2xP4M and mRFP-Rab7 are clearly visible on phagosomes between 10 and 30 min after sealing occurred (Figure 3, B and D). The formation of phagolysosomes follows soon thereafter. Fusion of phagosomes with lysosomes was assessed by preloading the latter with TMR-labeled dextran, using a well-established pulse-chase protocol (see Materials and Methods). Maturing phagosomes fused with dextran-loaded lysosomes slightly after PtdIns4P reacquisition (Figure 3C). A comprehensive perspective of the temporal relationship between the triphasic PtdIns4P changes relative to those of other phosphoinositides and the canonical stages of phagocytosis is provided in Figure 3D.
Disappearance of PtdIns4P from the phagosome
Because conversion to multiphosphorylated species did not appear to account for the disappearance of PtdIns4P from the nascent phagosome, we explored alternative mechanisms. Sac2 (INPP5F) was recently characterized as a PtdIns 4-phosphatase that functions in Rab5-positive early endosomes (Hsu et al., 2015; Nakatsu et al., 2015). Indeed, when coexpressed with Rab5 in RAW264.7 cells, GFP-Sac2 localized to endosomal structures (Supplemental Video S3). When analyzed under similar conditions, GFP-Sac2 associated with early phagosomes when they sealed (Figure 4A) at the same time as Rab5 did. Of importance, the association of Sac2 with phagosomes coincided with the disappearance of PtdIns4P, monitored using 2xP4M (Figure 4B).
FIGURE 4:

Sac2 recruitment and hydrolysis of PtdIns4P. (A) Confocal micrographs of RAW264.7 cells coexpressing mCherry-2xP4M, GFP-Sac2, and CFP-Rab5 during phagocytosis of IgG-SRBCs (the CFP channel is not shown). Insets, magnifications of the area delimited by dotted white boxes. Scale bar, 5 μm. (B) Time course of the changes in PtdIns4P and Sac2 during phagosome formation, from experiments like the one in A. PtdIns4P was monitored using mCherry-2xP4M and normalized to plasmalemmal mCherry-2xP4M intensity (red line, black squares); GFP-Sac2 fluorescence in the phagosome was calculated after subtraction of GFP-Sac2 cytosolic intensity (green line, white squares); data are expressed relative to the maximum value. Values are means ± SEM from three independent experiments. Pseudopod extension is considered as time 0. (C) Sac2 silencing efficiency of siRNA1 and siRNA2 in RAW264.7 cells measured by quantitative real-time PCR after reverse transcription; means ± SDs of three independent experiments and normalized to cells treated with nontargeting siRNA (control). (D) Time course of disappearance of PtdIns4P, assessed by kymography using GFP-2xP4M, during the early stages of phagocytosis. Top, region of the phagosome analyzed over time. Bottom, representative kymographs illustrating the disappearance of GFP-2xP4M from the base of the nascent phagosome over 120 s. The closure of the phagosome was considered as time 0. Images in A and D are representative of the distribution noted during the indicated time intervals after the onset of phagocytosis. (E) GFP-2xP4M phagosomal intensity was measured and normalized to GFP-2xP4M plasmalemmal intensity; data are expressed relative to the maximum value. Blue lines illustrate the kinetics of GFP-2xP4M disappearance in cells treated with nontargeting (control) siRNA; red lines illustrate cells treated with Sac2-targetted siRNAs. White and black squares and associated bars show the means ± SE of control and Sac2-knockdown (using siRNA1 and siRNA2) cells, respectively, from at least 10 phagosomes from three independent experiments. The intersections of the dotted lines indicate the t1/2 values of decay. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, and ns, not significantly different, relative to the nontargeting control.
We used gene silencing to assess whether this temporal coincidence reflected a causal relationship, that is, whether the acquisition of Sac2 was responsible for the loss of PtdIns4P. By independent electroporation of two different small interfering RNAs (siRNAs), the expression of Sac2 in RAW264.7 cells was depressed ∼65%, as determined by quantitative PCR (Figure 4C). This partial depletion of Sac2 resulted in a delayed disappearance of PtdIns4P from the formed phagosome (Figure 4, D and E) with both siRNA sequences. A fraction of the cells in which Sac2 had been silenced did not entirely lose 2xP4M fluorescence from phagosomes even 3 min postinternalization, contrasting with phagosomes from control cells (electroporated with nontargeting siRNA), which consistently lost PtdIns4P at ∼30–45 s after closure (Figure 4C). Although the partial inhibition observed may be attributed to incomplete gene silencing, other mechanisms could contribute to PtdIns4P reduction in the early phagosome.
We analyzed whether PLC activity contributes to the disappearance of PtdIns4P from phagosomes. Depletion of PtdIns4P could conceivably result from its direct hydrolysis by PLC (Zhang et al., 2013; Sicart et al., 2015) or may occur indirectly after its conversion to PtdIns(4,5)P2, the preferred substrate of the PLCs. Indeed, PLCγ1 and 2 are primarily responsible for the disappearance of PtdIns(4,5)P2 during phagosome formation (Azzoni et al., 1992; Liao et al., 1992; Scott et al., 2005). We therefore compared the rate of formation of diacylglycerol (DAG), the product of hydrolysis of phosphoinositides by PLC, with the disappearance of PtdIns4P. Because in macrophages DAG is rapidly converted to phosphatidic acid (Bohdanowicz et al., 2013), to better visualize its accumulation, we pretreated the cells with DAG kinase inhibitor II. In RAW264.7 cells cotransfected with mCh-C1-PKCδ—used as a probe for DAG (Shindo et al., 2003)—and GFP-2xP4M, DAG levels increased in extending pseudopods as the phagosome sealed, at a stage when PtdIns4P underwent only a modest increase (Figure 5, A and B). Maximal accumulation of DAG was observed as the PtdIns4P decreased sharply, consistent with the notion that PLC activity contributed directly or indirectly to the consumption of PtdIns4P. Accordingly, when recruited to the PM of otherwise resting cells using the rapamycin heterodimerization system, PLCβ3 (expressed as mRFP-FKBP-PLCβ3) caused a marked reduction in PtdIns4P (Figure 5D and Supplemental Video S4).
FIGURE 5:

Assessment of the role of PLC in PtdIns4P disappearance. (A) Confocal micrographs of RAW264.7 cells coexpressing GFP-2xP4M and mCh-C1-PKC∂ (a DAG biosensor) during phagocytosis of IgG-SRBCs; cells were pretreated with diacylglycerol kinase inhibitor II (30 μM) for 30 min to minimize the phosphorylation and rapid disappearance of DAG. (B) Time course of the changes in PtdIns4P and DAG during phagosome formation. PtdIns4P was monitored using GFP-2xP4M and normalized to plasmalemmal GFP-2xP4M intensity (green line, white squares); mCh-C1-PKCδ was used as DAG probe and normalized to mCh-C1-PKCδ cytosolic intensity (red line, black squares). Data are expressed relative to the maximum value. Values are means ± SEM from five independent experiments. Pseudopod extension was considered as time 0. (C) Recruitment of PLCβ3 to the PM through a rapamycin heterodimerization system. (D) RAW264.7 cells transiently coexpressing GFP-2xP4M, mRFP-FKBP-PLCβ3, and Lyn11-FRB were imaged before (left) and after (right) the addition of rapamycin (1 μM). (E) Recruitment of PLCβ3 to Rab7-positive compartments (late phagosomes) through a rapamycin heterodimerization system. (F) RAW264.7 cells transiently coexpressing GFP-2xP4M, mRFP-FKBP-PLCβ3, and iRFP-FRB-Rab7 imaged before (left) and after (right) the addition of rapamycin; PLCβ3 recruitment caused release of GFP-2xP4M from the PM and from late phagosomes within seconds; insets, inverted images of mRFP-FKBP-PLCβ3 fluorescence. Scale bars, 5 μm.
That PLC can directly hydrolyze PtdIns4P is suggested by the experiment illustrated in Figure 5F. In this instance, mRFP-FKBP-PLCβ3 was recruited to the membrane of late phagosomes using iRFP-FRB-Rab7 as the targeting determinant. As in the preceding case, addition of rapamycin resulted in depletion of PtdIns4P. It is important to note that, unlike the PM, the late phagosome contains PtdIns4P but no detectable PtdIns(4,5)P2. By analogy, it is conceivable that plasmalemmal PtdIns4P may have undergone direct hydrolysis by PLC.
Finally, we considered whether a fraction of the PtdIns4P may have been converted to PtdIns(3,4)P2 by class 2 phosphoinositide 3-kinases (PI3Ks; Supplemental Figure S2B). To investigate this possibility, we examined the dynamics of PtdIns(3,4)P2 during phagocytosis using a biosensor derived from the tandem-pleckstrin-homology-domain–containing protein (TAPP1) PH-TAPP1. We found a modest, discrete accumulation of PtdIns(3,4)P2 in extending pseudopods as the phagosome sealed (Supplemental Figure S2C), at a stage when PtdIns4P was undergoing accumulation. However, PtdIns(3,4)P2 was no longer detectable when PtdIns4P underwent the acute depletion that followed phagosome sealing. We also examined the disappearance of PtdIns4P from forming phagosomes in the presence of the PI3K inhibitor LY294002. PI3K inhibition severely impairs phagocytosis of large particles (>3–4 μm), but smaller ones are still ingested (Cox et al., 1999). We challenged cells pretreated with LY294002 with 1.6-μm latex beads and monitored PtdIns4P dynamics. As shown in Supplemental Figure S2D, inhibition of PI3Ks did not affect PtdIns4P disappearance from small phagosomes upon their scission from the PM. Thus we found no evidence to suggest that phosphorylation by LY294002-sensitive PI3Ks contributes measurably to the loss of PtdIns4P that follows phagosome sealing; we obtained similar results using wortmannin (unpublished data). Instead, hydrolysis by Sac2, possibly aided by PLC activity, is likely the major mechanism accounting for the early disappearance of PtdIns4P.
PtdIns4P reappearance in maturing phagosomes
We then investigated the mechanisms underlying PtdIns4P reappearance in late phagosomes. Mammalian cells express four different PtdIns4P kinases (PI4Ks): the class III enzymes PI4KIIIα/PI4KA and PI4KIIIβ/PI4KB and the class II enzymes PI4KIIα/PI4K2A and PI4KIIβ/PI4K2B (Balla, 2013; Boura and Nencka, 2015). PI4KA is responsible for maintaining the plasmalemmal pool of PtdIns4P (Balla et al., 2008; Nakatsu et al., 2012), whereas both PI4KB and PI4K2A function in the Golgi complex (Godi et al., 1999; Minogue et al., 2010). The class II enzymes are believed to synthesize endolysosomal PtdIns4P (Balla et al., 2002; Salazar et al., 2005; Jovic’ et al., 2012). To assess which of these enzymes is present on late phagosomes, we expressed fluorescently tagged chimeric constructs of each one of the PI4Ks in RAW264.7 cells and imaged them during the course of phagocytosis of IgG-SRBCs. Only PI4K2A accumulated noticeably in maturing phagosomes (Figure 6A). PI4K2A recruitment was first apparent ∼10 min after phagosome closure, and the kinase was still clearly detectable after 30 min. When GFP-PI4K2A was coexpressed with mCh-2xP4M, a correlation was observed between phagosomal acquisition of the kinase and reappearance of PtdIns4P (Figure 6Av). Quantitation of multiple experiments (Figure 6B) showed that acquisition of the kinase slightly preceded the reappearance of detectable amounts of the phosphoinositide in phagosomes. The recruitment of PI4K2A suggested by the heterologous expression experiments was validated by immunostaining. As illustrated in Figure 6Avi, association of the endogenous kinase with the late phagosome was also clearly observed using the PI4K2A-specific mouse monoclonal antibody 4C5G.
FIGURE 6:

PI4K2A recruitment and generation of PtdIns4P in maturing phagosomes. (A) Confocal sections of RAW264.7 cells expressing (i) GFP-PI4KA, (ii) GFP-PI4KB, (iii) GFP-PI4K2B, or (iv) GFP-PI4K2A or (v) coexpressing GFP-PI4K2A and mCh-2xP4M. (vi) Immunostaining of PI4K2A. Micrographs i–vi were acquired 20 min after initiation of phagocytosis. Insets, magnifications of the area delimited by dotted white boxes. (B) Time course of acquisition of PI4K2A and PtdIns4P during phagocytosis. PI4K2A was monitored measuring phagosomal GFP-PI4K2A and normalized to the mean fluorescence intensity of GFP-PI4K2A in the entire cell (green line, white circles). Data are expressed relative to the maximum value. PtdIns4P was monitored measuring phagosomal mCh-2xP4M and normalized to plasmalemmal mCh-2xP4M (red line, black circles). Data are expressed relative to the value upon phagosomal closure. Time 0 refers to the point when the PtdIns4P originally visible in the early phagosome disappeared entirely. (C, D) PI4K2A silencing efficiency in RAW264.7 (C) and COS-1-FcγRIIa (D) cells, measured by quantitative real-time PCR after reverse transcription; results were normalized to control siRNA cells and are shown as means ± SD of at least three independent experiments. (E) Confocal micrographs of COS-1-FcγRIIa cells expressing GFP-2xP4M treated with nontargeting (control) siRNA (left) and PI4K2A siRNA (right) after 40 min of phagocytosis. (F) Fluorescence of phagosome-associated GFP-2xP4M normalized to plasmalemmal GFP-2xP4M in cells treated with nontargeting (control) siRNA (open bar) and PI4K2A siRNAs (black bars). Data are means ± SEM of four independent experiments of at least 23 phagosomes each. Red arrows indicate phagosomes. *p < 0.05. Scale bars, 5 μm.
To assess the importance of PI4K2A-mediated synthesis in the reappearance of PtdIns4P in late phagosomes, we used siRNA to silence the expression of Pi4k2a. We used various combinations of six different oligonucleotides targeting the gene but attained a maximum of ∼40% reduction in gene expression (e.g., Figure 6C). Because gene silencing is notoriously difficult in myeloid cells, we used an alternative approach. Balla et al. (2005) reported considerable success in silencing PI4K2A in COS cells (a green monkey kidney cell line that is highly transfectable). Although COS cells are not inherently phagocytic, we and others showed that they acquire the ability to engulf IgG-opsonized targets when transfected with Fcγ receptors (Indik et al., 1991; Downey et al., 1999). Of note, the phagosomes formed by such cells undergo maturation and acidification and acquire bacteriostatic capacity (Downey et al., 1999). Indeed, COS-1-FcγRIIa cells (a line of COS-1 cells stably expressing FcγRIIa) transiently expressing GFP-2xP4M and allowed to ingest IgG-SRBCs recapitulated the triphasic changes in phagosomal PtdIns4P described earlier for macrophages (Supplemental Figure S3). We proceeded to silence PI4K2A using an oligonucleotide sequence (siRNA1) validated previously (Wang et al., 2003; Balla et al., 2005), as well as a newly designed sequence (siRNA2), to minimize the likelihood of off-target effects. Both sequences caused effective (siRNA1, ≥80%; siRNA2, ∼70%) gene knockdown (Figure 6D). Using this approach, we evaluated the role of PI4K2A in phagosomes. As illustrated in Figure 6E, although the presence of PtdIns4P in the plasmalemma and the initial phase of loss from the phagosome persisted, the late phase of reacquisition was markedly inhibited. The loss was somewhat heterogeneous (GFP-2xP4M was virtually absent from phagosomes in some cells, whereas in others, the intensity of the biosensor was only partially reduced or unaffected), an observation we attribute to the incomplete silencing of the PI4K2A gene. Nevertheless, the reduction of phagosomal PtdIns4P was highly significant (p < 0.0001; Figure 6F), implying that PI4K2A is at least partly responsible for the reformation of PtdIns4P in late phagosomes.
PtdIns4P is required for completion of phagosome maturation
The synchronous disappearance of PtdIns3P and resurgence of PtdIns4P coincide with the early-to-late transition of phagosomes. The phosphoinositide switch could be the cause or a consequence of the maturation process. This was analyzed by impairing the formation of PtdIns4P via silencing of PI4K2A and analyzing the effects of this inhibition on phagosome maturation. The early-to-late transition is characterized by a marked drop in luminal pH as the phagosomes acquire V-ATPases. We therefore compared the accumulation of an acidotropic fluorescent dye—an index of acidification—in phagosomes of cells treated with nontargeting or PI4K2A-targetting siRNA. We used cresyl violet, a fluorescent weak base that accumulates within acidic organelles (Figure 7A; Ostrowski et al., 2016). Its suitability as an indicator of acidic pH was confirmed by its accumulation in lysosomes, which were identified by preloading with labeled dextran using a well-established pulse-and-chase protocol (Figure 7B). Moreover, cresyl violet colocalized extensively with GFP-Rab7, whereas no significant colocalization was observed with GFP-Rab5 (Supplemental Figure S4), validating its use as a marker of late endocytic compartments. When analyzed 35–40 min after particle internalization, phagosomes of cells treated with nontargeting (control) siRNA regularly acquired cresyl violet (Figure 7C, left). Strikingly, cresyl violet was not detectable in most phagosomes of cells in which PtdIns4P reappearance was impaired using PI4K2A siRNA (Figure 7C, right). Of interest, cresyl violet–positive vesicles were often observed surrounding such phagosomes, suggesting that fusion, rather than the acidification of lysosomes, was impaired when PI4K2A was silenced.
FIGURE 7:
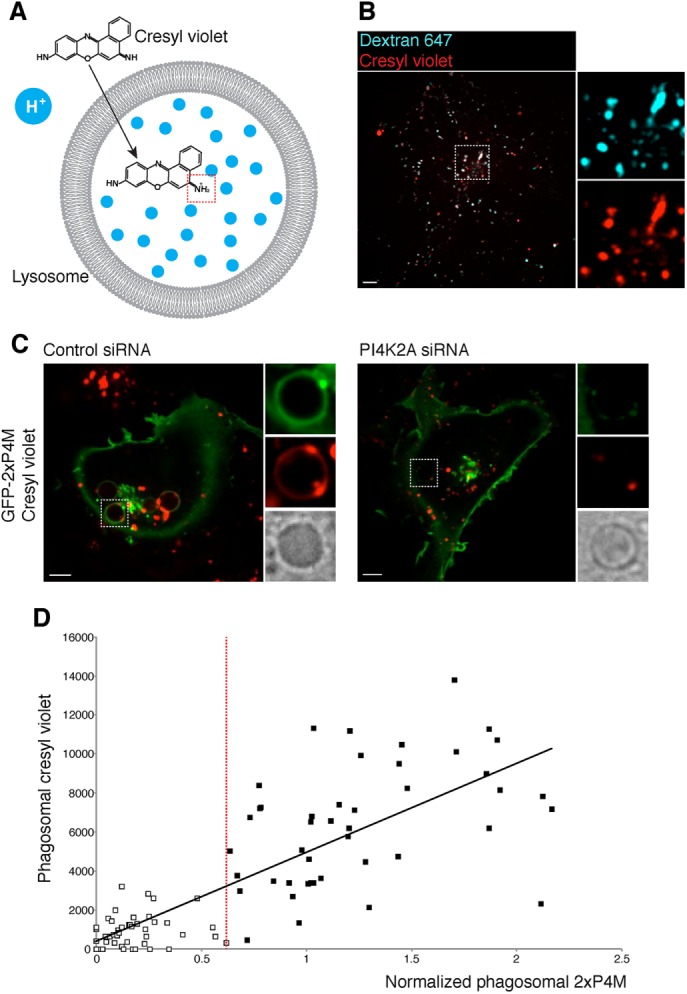
Phagosome acidification is impaired when PI4K2A is silenced. (A) Structure of cresyl violet and the proposed mechanism by which it accumulates in acidic compartments; note protonation of cresyl violet occurring in the dotted red box. (B) Single confocal section of COS-1-FcγRIIa cells where lysosomes were loaded with Alexa Fluor 647 10-kDa dextran (0.1 mg/ml, 3-h pulse, 30-min chase) followed by cresyl violet loading (1 μM, 2-min pulse); insets, magnifications of the area delimited by the dotted lines. C) Confocal micrographs of cells treated with nontargeting (control) siRNA (left) and PI4K2A siRNA (right) COS-1-FcγRIIa cells expressing GFP-2xP4M that were pulsed with cresyl violet after 40 min of phagocytosis of IgG-SRBC. Scale bars, 5 μm. (D) Plot relating cresyl violet acquisition with PtdIns4P levels in phagosomes, measured 40 min after phagocytosis; r2 = 0.64. The vertical red line represents an arbitrary threshold dividing two phagosomal populations based on phagosomal GFP-2xP4M intensity relative to that in the PM. White squares represent phagosomes with low PtdIns4P levels; black squares represent phagosomes with higher PtdIns4P levels.
Because not all cells/phagosomes were equally affected by the siRNA treatment, we quantified both the amount of PtdIns4P remaining on phagosomes (relative to that on the PM) and the extent to which they accumulated the acidotropic dye. Data obtained from 89 phagosomes in four separate experiments are collated in Figure 6D. There is a clear correlation (r2 = 0.64) between these parameters, strongly suggesting that accumulation of PtdIns4P by late phagosomes is essential for their full acidification.
Because prolonged and generalized absence of PI4K2A may have affected other cellular compartments, potentially causing indirect effects, we used an additional approach to assess the role of PtdIns4P in phagosome maturation. To circumvent such potentially nonspecific effects, we acutely depleted phagosomal PtdIns4P by recruiting a PtdIns4P-specific phosphatase, Sac1, using rapamycin-mediated heterodimerization. mCh-FKBP-Sac1 was recruited to Rab7-containing (late) phagosomes by coexpression with iRFP-FRB-Rab7 (Figure 8A). The cells were also cotransfected with GFP-2xP4M to monitor the effects of phosphatase recruitment on PtdIns4P. Addition of rapamycin caused depletion of PtdIns4P from phagosomes that had formed ≥35 min earlier (Figure 8B). Using this paradigm, we proceeded to study the association of the lysosomal-associated protein RILP (an effector of active Rab7) with phagosomes. We used a chimeric construct that encodes for the C-terminal domain RILP (Cantalupo et al., 2001); in control cells, GFP-RILP-C33 decorates the entire phagosomal membrane, consistent with extensive and sustained activation of Rab7 (Figure 8, C and D). Strikingly, in cells in which PtdIns4P was depleted by recruitment of Sac1, GFP-RILP-C33 acquisition by phagosomes was impaired compared with control cells (expressing mCh-FKBP not attached to Sac1; Figure 8, C and D). Note that in mCh-FKBP-Sac1–transfected cells, the construct was recruited normally to the phagosomes, indicating that the localization of iRFP-FRB-Rab7 was not affected. The targeting of iRFP-FRB-Rab7 to the membrane seemingly does not require activation of the GTPase. In addition, we observed that GFP-RILP-C33–positive vesicles often accumulated around PtdIns4P-depleted phagosomes, consistent with the earlier suggestion that fusion with late endosomes/lysosomes was impaired. Time-lapse video imaging supported the notion that incoming vesicles failed to fuse with phagosomes and showed no evidence that the observed vesicles were budding off the phagosomal membrane (Supplemental Video S5). Finally, when mRFP-Rab7 was transiently coexpressed with GFP-RILP-C33 in PI4K2A-silenced cells, a similar effect was observed. Rab7 accumulated in the phagosomal membrane, whereas RILP-C33 recruitment was generally impaired compared with control cells (Figure 8E).
FIGURE 8:

RILP acquisition is impaired in PtdIns4P-depleted phagosomes. (A) Recruitment of Sac1 to the PM through a rapamycin heterodimerization system. (B) COS-1-FcγRIIa cells transiently coexpressing GFP-2xP4M, mCh-FKBP-Sac1, and iRFP-FRB-Rab7 were challenged with IgG-SRBCs; phagocytosis was allowed to develop for 30 min before imaging. On PtdIns4P reappearance in phagosomes, cells were treated with rapamycin (1 μM) to induce recruitment of mCh-FKBP-Sac1, which resulted in PtdIns4P hydrolysis (release of GFP-2xP4M from phagosomes). Insets, mCh-FKBP-Sac1 fluorescence (black and white are inverted for clarity). (C, D) COS-1-FcγRIIa cells transiently coexpressing GFP-RILP-C33, mCh-FKBP-Sac1, and iRFP-FRB-Rab7; in control cells, mCh-FKBP-Sac1 was substituted with mCh-FKBP. The cells were challenged with IgG-SRBCs and treated with rapamycin (1 μM), and phagocytosis was allowed to proceed for 40 min. Cells were then washed, fixed, and imaged. *p < 0.05. (D) Representative confocal micrographs of mCh-FKBP- (left) and mCh-FKBP-Sac1-expressing cells (right); insets, magnifications of the dotted white boxes for each channel. Shown in C is GFP-RILP-C33 accumulation in phagosomes of cells expressing mCh-FKBP (open bars) or mCh-FKBP-Sac1 (black bars) phagosomes, showing a continuous pattern around the phagosome, and those with only punctate or no fluorescence were scored separately. (E) Confocal micrographs of COS-1-FcγRIIa cells coexpressing mRFP-Rab7 and GFP-RILP-C33 treated with nontargeting (control) siRNA (left) and PI4K2A siRNA (right) after 40 min of phagocytosis; insets, magnifications of the dotted white boxes. Scale bars, 5 μm.
DISCUSSION
We observed localized triphasic changes in the level of PtdIns4P during phagocytosis. These are summarized in schematic form in Supplemental Figure S5. Initially, PtdIns4P accumulated in the forming phagocytic cup. This coincided with an increase in PtdIns(4,5)P2 in extending pseudopods, which was reported earlier (Botelho et al., 2000). The accumulation of PtdIns4P in this setting may reflect localized synthesis required to satisfy the enhanced substrate demand for stimulated PtdIns(4,5)P2 generation. In this regard, we found a discrete accumulation of endogenous PI4KIIIα (PI4KA) in forming phagosomes (Supplemental Figure S2A). This raises the intriguing possibility that the PI4KIIIα complex (PI4KA-TTC7-EFR3) responsible for the plasmalemmal pool of PtdIns4P (Wu et al., 2014; Chung et al., 2015) may undergo stimulation at the cup.
On phagosome closure, PtdIns4P reaches a peak that coincides with the sudden disappearance of PtdIns(4,5)P2 from the vacuolar membrane. We believe that these events are linked in two ways. First, a previous study showed that the 5-phosphatases OCRL and INPP5B are recruited to nascent phagosomes (Bohdanowicz et al., 2012). These enzymes dephosphorylate PtdIns(4,5)P2, yielding PtdIns4P. In addition, cessation of PtdIns(4,5)P2 synthesis likely contributes to the accumulation of PtdIns4P. PIP5Ks that use PtdIns4P to synthesize PtdIns(4,5)P2 localize to the PM and are present at the phagocytic cup. However, these kinases are acutely released from the membrane of phagosomes as they undergo closure (Fairn et al., 2009). Detachment of the PIP5Ks while PtdIns4P production is ongoing would result in a localized burst of PtdIns4P. The observed transient accumulation of PtdIns4P likely results from the combination of the aforementioned phenomena.
The peak noted upon sealing is followed within 30–45 s by the virtual disappearance of PtdIns4P, which is undetectable in the membrane of early phagosomes for a period lasting ∼10 min (Figures 1, F and G, and 3D). Our data suggest that Sac2 mediates at least part of the hydrolysis of PtdIns4P, but PLC activity also seems to contribute either by depleting PtdIns(4,5)P2 and thereby accelerating PtdIns4P consumption or by directly hydrolyzing the latter (Figures 4 and 5). Exchange of PtdIns4P for other lipids (e.g., cholesterol) via contacts with the endoplasmic reticulum may have also occurred but was not investigated here. A period of ∼10 min when PtdIns4P is undetectable follows its abrupt disappearance after sealing. Strikingly, PtdIns4P then reappears. This secondary accumulation of PtdIns4P reaches its maximum ∼30 min after the onset of phagocytosis, attaining levels that clearly exceed those detected in the PM. The late reappearance of PtdIns4P coincides with and depends on the recruitment of PI4K2A.
Remarkably, the multiphasic changes in PtdIns4P are accompanied by opposite changes in the concentration of phagosomal PtdIns3P. The initial loss of PtdIns4P coincides with the emergence of PtdIns3P, and the converse is true once the early maturation stage is completed: PtdIns3P disappears abruptly as the phagosome regains PtdIns4P (Figure 3 and Supplemental Figure S6). A similar phospholipid conversion process was recently described to occur at the sorting-to-recycling endosome interphase (Ketel et al., 2016). Thus the identity and function of the early endosomal and phagosomal compartments may require not only acquisition of PtdIns3P, but also the simultaneous elimination of PtdIns4P. In this regard, it is noteworthy that the class III PI3K Vps34 that synthesizes PtdIns3P and the 4-phosphatase Sac2 coexist in early endosomes/phagosomes (Hsu et al., 2015; Nakatsu et al., 2015), whereas the PtdIns4P kinase PI4K2A and some myotubularins (3-phosphatases that break down PtdIns3P) localize to the late compartments (Tsujita et al., 2004; Lorenzo et al., 2006). These observations are compatible with the suggestion of Ketel et al. (2016) that activation of the myotubularins that eliminate PtdIns3P may be coupled to the acquisition of the kinases that foster PtdIns4P formation. We suggest that PtdIns4P, possibly in conjunction with PtdIns(3,5)P2, defines the identity and functional properties of the late phagosome/phagolysosome. This speculation is in accordance with recent reports showing that PtdIns4P plays a role in the fusion of autophagic vacuoles with lysosomes (Wang et al., 2015) and possibly also in phagolysosome biogenesis, as suggested from heterologous fusion experiments in cell-free systems (Jeschke et al., 2015).
How PtdIns4P regulates late maturation stages remains unclear. The only known PtdIns4P-specific effectors, such as the ceramide transfer protein CERT, the glucosylceramide transfer protein FAPP2. and the oxysterol binding protein OSBP1. function at the Golgi level (Graham and Burd, 2011). Our observations indicate that proper acidification requires the build-up of PtdIns4P in late phagosomes, an effect that may be mediated by RILP (Figures 7 and 8). Indeed, interference with RILP function was shown to impair phagolysosome formation (Harrison et al., 2003). RILP forms a complex with ORP1L that affixes dynein to Rab7-containing compartments (Johansson et al., 2007) and drives the centripetal motion of phagosomes toward the juxtanuclear region where lysosomes reside. It is of interest that ORP1L possesses a pleckstrin-homology domain known to bind phosphoinositides (Johansson et al., 2005). We speculate that PtdIns4P is required for the recruitment of ORP1L/RILP/dynein to maturing phagosomes.
In summary, we defined the dynamics of PtdIns4P during phagocytosis in live macrophages and began to elucidate the underlying mechanisms behind these changes and their functional implications. The phosphoinositide clearly undergoes pronounced changes and is likely to play additional, unappreciated roles in phagosome biology. The mechanism by which PI4K2A is recruited to maturing phagosomes and the possible role of PtdIns4P in phagosome resolution are of particular interest and should be the focus of future studies.
MATERIALS AND METHODS
Reagents
Sheep erythrocytes (10% suspension) were purchased from MP Biomedicals (Santa Ana, CA). Anti-sheep red blood cell antibodies were purchased from Cedarlane Laboratories (Burlington, Canada). Polystyrene microspheres (1.6 μm in diameter) functionalized with divinylbenzene were obtained from Bangs Laboratories (Fishers, IN). Rapamycin, human IgG, cresyl violet acetate, and wortmannin were from Sigma-Aldrich (Oakville, Canada). Fluorescent antibodies against mouse and rabbit were from Jackson ImmunoResearch Labs (West Grove, PA). Paraformaldehyde (16% [wt/vol]) was from Electron Microscopy Sciences (Hatfield, PA).
Cell culture
The murine macrophage cell line RAW264.7 and the fibroblastic COS-1 cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in medium RPMI 1640 (Wisent Bioproducts, Saint-Jean-Baptiste, Canada) supplemented with 10% heat-inactivated fetal bovine serum at 37°C under 5% CO2. Generation of COS-1 cells stably expressing FcγRIIa was previously described (Downey, 1999).
Plasmids
The following plasmids used in this study have been previously described: GFP-2xP4M-SidM, GFP-P4M-SidM, iRFP-FRB-Rab7, GFP-PI4KB, and GFP-PI4K (Hammond et al., 2014); GFP-PI4K2A and GFP-PI4K2B (Balla et al., 2002); mRFP-PH-PLCδ (Stauffer et al., 1998); mRFP-2xFYVE-EEA1 and GFP-PH-Gab2 (Bohdanowicz et al., 2010); mRFP-Rab5 and mRFP-Rab7 (Vieira et al., 2003); GFP-Sac2 (Nakatsu et al., 2015); mRFP-FKBP-Sac1 (Hammond et al., 2012); mRFP-FKBP (Szentpetery et al., 2010); GFP-RILP-C33 (Cantalupo et al., 2001); and Lyn11-FRB (Bohdanowicz et al., 2013). mCh-sialyltransferase was a kind gift from E. Rodriguez Boulan (Department of Cell Biology, Cornell University). GST-GFP-P4M was generated using GFP-P4M as a template and the following primers: forward, 5-gcggaattcatgtctaaaggtgaagaatt-3′; and reverse, 5′gcgctcgagttattttatcttaatggtttg-3′. The PCR product was digested with EcoRI and Xhol and cloned into a pGEX-6P1; mRFP-FKBP-PLCβ3 was generated using the constitutively active ΔXY-PLCβ3 template kindly provided by T. K. Harden and J. Sondek (University of North Carolina School of Medicine, Chapel Hill, NC) (Charpentier et al., 2014) and using the primers forward, 5′-atatcgatcggtccaggcgctgcagttgg-3′, and reverse, 5′-atattctagatcaaatgtagtccgaggcttcggtgtagatg-3′, for PCR amplification. The PCR product was digested with Pvu and XbaI and cloned into the mRFP-FKBP12-5-ptase domain construct obtained from plasmids amplified in Dam– bacterial strains and digested with the same restriction enzymes to replace the 5-ptase domain with the PLCβ3 construct; GFP in GFP-2xP4M was digested using NheI and BglIII and was replaced with mCherry to generate mCh-2xP4M-SidM.
Transient transfection
For transient transfections, ∼80% confluent monolayers of RAW264.7 cells were lifted by scraping and plated onto 1.8-cm glass coverslips at a density of 5 × 104 cells/coverslip. Macrophages were allowed to recover for 18–24 h and then transfected with FuGENE HD (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, 1 μg of plasmid DNA and 3 μl of the transfection reagent were mixed in 100 μl of serum-free RPMI and incubated for 15 min. The mix was then distributed equally into four wells of a 12-well plate containing the cells. Cells were typically used for experiments 12–18 h after transfection.
Protein purification
BL21-competent cells were transformed with GST-GFP-P4M. A single transformed colony was inoculated in 100 ml of Terrific Broth (Invitrogen) supplemented with 0.1% glycerol and incubated overnight at 37°C. The next day, 20 ml of the overnight culture was inoculated into 400 ml of Terrific Broth with glycerol and incubated for 2 h at 37°C. After the incubation, isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich) was added (final concentration 0.25 mM), and the culture was incubated at 30°C for 4 h. After the incubation, cells were lysed using FastBreak cell lysis reagent (Promega) according to the manufacturer’s instructions. The cell lysate was then centrifuged and the supernatant collected. The presence of GST-GFP-P4M was confirmed by Western blot using an α-GFP antibody (GFP(B-2); sc-9996; Santa Cruz Biotechnology, Dallas, TX). The supernatant was then incubated with Glutathione Sepharose 4B (GE Healthcare; previously washed three times with binding buffer, phosphate-buffered saline [PBS], pH 7.3) overnight at 4°C. The solution was then centrifuged and washed three times with binding buffer. Finally, the pellet was incubated in elution buffer (50 mM Tris-HCl plus 10 mM reduced glutathione, pH 8.0) for 30 min at room temperature, and the elution was collected. The presence of the purified GST-GFP-P4M was confirmed by immunoblotting with anti-GFP antibody (GFP(B-2).
Protein–lipid overlay assay
A PIP strip (Echelon Biosciences, Salt Lake City, UT) was blocked overnight at 4°C with PBS plus 0.1% Tween (PBS-T) plus 3% bovine serum albumin. The next day, the membrane was overlaid with purified GST-GFP-P4M (150 pmol/ml in blocking solution) and incubated overnight at 4°C with agitation. The membrane was next washed five times with PBS-T for 20 min. After the washes, the PIP strip was incubated with an anti-GFP antibody diluted in blocking buffer (1:500) for 1 h. The strip was then washed as described and incubated with anti-mouse horseradish peroxidase (HRP)–conjugated antibody (Jackson ImmunoResearch Labs) for 1 h and washed again. Finally, detection was performed using the Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Mississauga, Canada) according to the manufacturer’s instructions.
Gene silencing
The siRNAs (27mer Dicer-substrate) targeting mouse Sac2/Innp5F were from Origene (Rockville, MD). The targeting sequences were as follows: siRNA1, 5′-GGAAUGCGGUAUAAACGAAGAGGAG-3′; and siRNA2, 5′-ACAAGCCUGAGAAGAUUAUACCATC-3′. To silence PI4K2A, siRNA targeting mouse Pi4k2a was purchased from Dharmacon. The targeting sequences were 5′-UGAGGGAGCCUGUUAUCAA-3′, 5′-GGACACAGAUUGGGUGAUG-3′, 5′-GAAUCGGCCUGCCACCAAA-3′, 5′-GAGACGAGCCCGCUAGUGU-3′, 5′-CUACAAAGAUGCAGACUAUUU-3′, and 5′-CCAGAUGCCACCUGUGAUUUU-3′.
The previously used siRNA1 (5′-UGAAGCAGAACCUCUUCCUGAUU-3′; Jovic’ et al., 2012) and the siRNA2 (5′-CUACAAAGAUGCAGACUAUUU-3′) targeting PI4K2A in COS-1 cells were also from Dharmacon GE Healthcare (Mississauga, Canada). Oligonucleotides were delivered into the cells by electroporating 5 × 105 RAW264.7 cells with 200 pmol of the siRNA pool with the Neon transduction system (ThermoFisher Scientific, Burlington, Canada), using a single 20-ms pulse of 1750 V. Electroporated cells were allowed to recover for 24 h before being lifted once again for a second round of electroporation. Knockdown efficiency and phagocytosis were assessed 48 h after the initial electroporation. When transfection of plasmid DNA was required, cells were replated in 12-well plates after the second round of silencing and allowed to recover in fresh medium for 8 h. Then cells were transfected as described and used for experiments after an additional 18–24 h of incubation.
Oligonucleotides were delivered to COS-1 cells with HiPerFect transfection reagent (Qiagen, Toronto, Canada), following the manufacturer’s instructions. Cells were plated in six-well plates 24 h before silencing. Then cells were treated with a mix of the reagent and oligonucleotides for 18 h and allowed to recover in fresh medium for 6 h. A second round of silencing was performed for 18 h. Cells were allowed to recover for 30 h before experiments. When cells had to be transfected, they were replated in 12-well plates after the second round of silencing and allowed to recover in fresh medium for 8 h. Then cells were transfected as described and used for experiments 18–24 h after transfection.
Quantitative real-time PCR
Total RNA was isolated using the GeneJET RNA Purification kit (Thermo Fisher Scientific). Equal amounts of RNA were loaded as template for the generation of cDNA using the SuperScript VILO cDNA Synthesis kit (Thermo Fisher Scientific). Quantitative PCR was performed in a 96-well plate using the TaqMan System (Thermo Fisher Scientific) on a Step One Plus Real-Time PCR thermal cycler with Step One software (version 2.2.2; Applied Biosystems, Burlington, Canada). The Taqman gene expression assays for the reference gene and target gene were duplexed in triplicate. Target gene expression was determined by quantification relative to the reference gene and the control sample (ΔΔCt method). The Taqman gene expression assays used were as follows: Abt1 (reference gene), Mm00803824_m1; Pi4k2A, Mm01197215_m1; and Inpp5f (Sac2), Mm00724391_m1. In COS-1 cells, the reference gene was CDKN1A, Hs00355782_m1; and the target gene was PI4K2A, Hs00218300_m1.
Phagocytosis
For all phagocytosis assays, ∼5 × 104 cells were seeded onto 1.8-cm glass coverslips and allowed to grow for 18–24 h. For opsonization, 100 μl of a 20% SRBC suspension was washed with PBS and opsonized by incubation with 3 μl of rabbit IgG fraction against SRBCs at 37°C for 1 h. SRBCs were then washed three times with PBS. Alternatively, divinylbenzene-coated polystyrene beads were diluted 10-fold in PBS and opsonized by incubation with human IgG (final IgG concentration, 5 mg/ml) for 60 min at room temperature. Excess IgG was removed by washing the beads twice with PBS. When needed, SRBCs were prelabeled by incubation with succinimidyl esters for 20 min rotating at room temperature. Then the SRBCs were washed three times with PBS before opsonization as described. Phagocytosis was initiated by adding 75 μl of a 10-fold dilution of the SRBC or bead suspension to individual wells of a 12-well plate or directly into an imaging chamber containing a coverslip with cells. In all cases, phagocytosis was synchronized by sedimenting the particles by centrifugation (300 × g for 10 s).
Detection of acidification and lysosomal labeling
Cells were treated with cresyl violet acetate (1 μM final concentration) for 2 min and then washed twice with PBS and imaged. Lysosomes were loaded with 0.1 mg/ml dextran for 3 h at 37°C (5% CO2 balance air), followed by a chase period of at least 30 min.
Microscopy
For imaging, cells grown on glass coverslips were mounted in a Chamlide magnetic chamber (Seoul, South Korea), overlaid with 1 ml of Hanks’ balanced salt solution (Wisent Bioproducts) and maintained at 37ºC. Fluorescence microscopy was performed using spinning-disk confocal microscopes (Quorum Technologies, Guelph, Canada). Our systems are based on an Axiovert 200M microscope (Carl Zeiss) equipped with a 63× oil-immersion objective (numerical aperture 1.4) and a 1.5× magnifying lens. The microscopes carry a motorized XY stage (Applied Scientific Instrumentation, Eugene, OR), a Piezo Z-focus drive, and diode-pumped solid-state lasers emitting at 440, 491, 561, 638, and 655 nm (Spectral Applied Research, Richmond Hill, Canada). Images were recorded with back-thinned, cooled charge-coupled device cameras (Hamamatsu Photonics) under command of the Volocity software (version 6.2.1; PerkinElmer, Woodbridge, Canada). Selection of regions of interest, fluorescence intensity measurements, and brightness/contrast corrections were performed with ImageJ (version 1.48; National Institutes of Health, Bethesda, MD). Brightness and contrast parameters were adjusted across entire images and without altering the linearity of mapped pixel values. All images are representative of at least 15 determinations from three separate experiments.
Supplementary Material
Acknowledgments
R.L. is funded by the Connaught International Scholarship for Doctoral Students from the University of Toronto and by the National Council for Science and Technology/Consejo Nacional de Ciencia y Tecnologia of Mexico. This work was supported by Grant FDN-143202 from the Canadian Institutes of Health Research to S.G., Grant MOP-133656 from the Canadian Institutes of Health Research to G.D.F., funds from the Department of Cell Biology, University of Pittsburgh School of Medicine, to G.H., the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health to T.B., and Grant DK082700 from the National Institutes of Health to P.D.C.
Abbreviations used:
- Lyn11
N-terminal 11 amino acids of Lyn kinase
- PLC
phospholipase C
- PM
plasma membrane
- PtdIns(3,4,5)P3
phosphatidylinositol 3,4,5-trisphosphate
- PtdIns(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- PtdIns3P
phosphatidylinositol 3-phosphate
- PtdIns4P
phosphatidylinositol 4-phosphate.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0451) on November 9, 2016.
REFERENCES
- Azzoni L, Kamoun M, Salcedo TW, Kanakaraj P, Perussia B. Stimulation of Fc gamma RIIIA results in phospholipase C-gamma 1 tyrosine phosphorylation and p56lck activation. J Exp Med. 1992;176:1745–1750. doi: 10.1084/jem.176.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol Biol Cell. 2008;19:711–721. doi: 10.1091/mbc.E07-07-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Tuymetova G, Barshishat M, Geiszt M, Balla T. Characterization of type II phosphatidylinositol 4-kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem. 2002;277:20041–20050. doi: 10.1074/jbc.M111807200. [DOI] [PubMed] [Google Scholar]
- Balla A, Tuymetova G, Tsiomenko A, Várnai P, Balla T. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell. 2005;16:1282–1295. doi: 10.1091/mbc.E04-07-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohdanowicz M, Balkin DM, De Camilli P, Grinstein S. Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. Mol Biol Cell. 2012;23:176–187. doi: 10.1091/mbc.E11-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohdanowicz M, Cosío G, Backer JM, Grinstein S. Class I and class III phosphoinositide 3-kinases are required for actin polymerization that propels phagosomes. J Cell Biol. 2010;191:999–1012. doi: 10.1083/jcb.201004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohdanowicz M, Schlam D, Hermansson M, Rizzuti D, Fairn GD, Ueyama T, Somerharju P, Du G, Grinstein S. Phosphatidic acid is required for the constitutive ruffling and macropinocytosis of phagocytes. Mol Biol Cell. 2013;24:1700–1712, S1–S7. doi: 10.1091/mbc.E12-11-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura E, Nencka R. Phosphatidylinositol 4-kinases: function, structure, and inhibition. Exp Cell Res. 2015;337:136–145. doi: 10.1016/j.yexcr.2015.03.028. [DOI] [PubMed] [Google Scholar]
- Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, Hilbi H. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J Biol Chem. 2009;284:4846–4856. doi: 10.1074/jbc.M807505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier TH, Waldo GL, Barrett MO, Huang W, Zhang Q, Harden TK, Sondek J. Membrane-induced allosteric control of phospholipase C-β isozymes. J Biol Chem. 2014;289:29545–29557. doi: 10.1074/jbc.M114.586784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Nakatsu F, Baskin JM, De Camilli P. Plasticity of PI4KIIIα interactions at the plasma membrane. EMBO Rep. 2015;16:312–320. doi: 10.15252/embr.201439151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem. 1999;274:1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, Grinstein S. Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J Biol Chem. 1999;274:28436–28444. doi: 10.1074/jbc.274.40.28436. [DOI] [PubMed] [Google Scholar]
- Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 2012;33:397–405. doi: 10.1016/j.it.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Fairn GD, Ogata K, Botelho RJ, Stahl PD, Anderson RA, De Camilli P, Meyer T, Wodak S, Grinstein S. An electrostatic switch displaces phosphatidylinositol phosphate kinases from the membrane during phagocytosis. J Cell Biol. 2009;187:701–714. doi: 10.1083/jcb.200909025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- Graham TR, Burd CG. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011;21:113–121. doi: 10.1016/j.tcb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GRV, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, Irvine RF. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science. 2012;337:727–730. doi: 10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GRV, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol. 2014;205:113–126. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23:6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Hu F, Mao Y. Spatiotemporal control of phosphatidylinositol 4-phosphate by Sac2 regulates endocytic recycling. J Cell Biol. 2015;209:97–110. doi: 10.1083/jcb.201408027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indik Z, Kelly C, Chien P, Levinson AI, Schreiber AD. Human Fc gamma RII, in the absence of other Fc gamma receptors, mediates a phagocytic signal. J Clin Invest. 1991;88:1766–1771. doi: 10.1172/JCI115496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke A, Zehethofer N, Lindner B, Krupp J, Schwudke D, Haneburger I, Jovic M, Backer JM, Balla T, Hilbi H, Haas A. Phosphatidylinositol 4-phosphate and phosphatidylinositol 3-phosphate regulate phagolysosome biogenesis. Proc Natl Acad Sci USA. 2015;112:4636–4641. doi: 10.1073/pnas.1423456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Lehto M, Tanhuanpää K, Cover TL, Olkkonen VM. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell. 2005;16:5480–5492. doi: 10.1091/mbc.E05-03-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, Neefjes J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol. 2007;176:459–471. doi: 10.1083/jcb.200606077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovi’c M, Kean MJ, Szentpetery Z, Polevoy G, Gingras A-C, Brill JA, Balla T. Two phosphatidylinositol 4-kinases control lysosomal delivery of the Gaucher disease enzyme, β-glucocerebrosidase. Mol Biol Cell. 2012;23:1533–1545. doi: 10.1091/mbc.E11-06-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketel K, Krauss M, Nicot A-S, Puchkov D, Wieffer M, Müller R, Subramanian D, Schultz C, Laporte J, Haucke V. A phosphoinositide conversion mechanism for exit from endosomes. Nature. 2016;529:408–412. doi: 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- Levin R, Grinstein S, Canton J. The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev. 2016;273:156–179. doi: 10.1111/imr.12439. [DOI] [PubMed] [Google Scholar]
- Levin R, Grinstein S, Schlam D. Phosphoinositides in phagocytosis and macropinocytosis. Biochim Biophys Acta. 2015;1851:805–823. doi: 10.1016/j.bbalip.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Liao F, Shin HS, Rhee SG. Tyrosine phosphorylation of phospholipase C-gamma 1 induced by cross-linking of the high-affinity or low-affinity Fc receptor for IgG in U937 cells. Proc Natl Acad Sci USA. 1992;89:3659–3663. doi: 10.1073/pnas.89.8.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo O, Urbé S, Clague MJ. Systematic analysis of myotubularins: heteromeric interactions, subcellular localisation and endosome related functions. J Cell Sci. 2006;119:2953–2959. doi: 10.1242/jcs.03040. [DOI] [PubMed] [Google Scholar]
- Marshall JG, Booth JW, Stambolic V, Mak T, Balla T, Schreiber AD, Meyer T, Grinstein S. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J Cell Biol. 2001;153:1369–1380. doi: 10.1083/jcb.153.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue S, Chu KME, Westover EJ, Covey DF, Hsuan JJ, Waugh MG. Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIalpha at the trans-Golgi network. J Lipid Res. 2010;51:2314–2324. doi: 10.1194/jlr.M005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F, Baskin JM, Chung J, Tanner LB, Shui G, Lee SY, Pirruccello M, Hao M, Ingolia NT, Wenk MR, De Camilli P. PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J Cell Biol. 2012;199:1003–1016. doi: 10.1083/jcb.201206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F, Messa M, Nández R, Czapla H, Zou Y, Strittmatter SM, De Camilli P. Sac2/INPP5F is an inositol 4-phosphatase that functions in the endocytic pathway. J Cell Biol. 2015;209:85–95. doi: 10.1083/jcb.201409064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski PP, Fairn GD, Grinstein S, Johnson DE. Cresyl violet: a superior fluorescent lysosomal marker. Traffic. 2016;17:1313–1321. doi: 10.1111/tra.12447. [DOI] [PubMed] [Google Scholar]
- Salazar G, Craige B, Wainer BH, Guo J, De Camilli P, Faundez V. Phosphatidylinositol-4-kinase type II alpha is a component of adaptor protein-3-derived vesicles. Mol Biol Cell. 2005;16:3692–3704. doi: 10.1091/mbc.E05-01-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CC, Dobson W, Botelho RJ, Coady-Osberg N, Chavrier P, Knecht DA, Heath C, Stahl P, Grinstein S. Phosphatidylinositol-4,5-bisphosphate hydrolysis directs actin remodeling during phagocytosis. J Cell Biol. 2005;169:139–149. doi: 10.1083/jcb.200412162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo M, Irie K, Masuda A, Ohigashi H, Shirai Y, Miyasaka K, Saito N. Synthesis and phorbol ester binding of the cysteine-rich domains of diacylglycerol kinase (DGK) isozymes. DGKgamma and DGKbeta are new targets of tumor-promoting phorbol esters. J Biol Chem. 2003;278:18448–18454. doi: 10.1074/jbc.M300400200. [DOI] [PubMed] [Google Scholar]
- Sicart A, Katan M, Egea G, Sarri E. PLCγ1 participates in protein transport and diacylglycerol production triggered by cargo arrival at the Golgi. Traffic. 2015;16:250–266. doi: 10.1111/tra.12246. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Swanson JA. Phosphoinositides and engulfment. Cell Microbiol. 2014;16:1473–1483. doi: 10.1111/cmi.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentpetery Z, Várnai P, Balla T. Acute manipulation of Golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc Natl Acad Sci USA. 2010;107:8225–8230. doi: 10.1073/pnas.1000157107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujita K, Itoh T, Ijuin T, Yamamoto A, Shisheva A, Laporte J, Takenawa T. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817–13824. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, Schreiber AD, Stahl PD, Grinstein S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol. 2003;23:2501–2514. doi: 10.1128/MCB.23.7.2501-2514.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun H-Q, Zhu X, Zhang L, Albanesi J, Levine B, Yin H. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci USA. 2015;112:7015–7020. doi: 10.1073/pnas.1507263112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 2003;114:299–310. doi: 10.1016/s0092-8674(03)00603-2. [DOI] [PubMed] [Google Scholar]
- Wu X, Chi RJ, Baskin JM, Lucast L, Burd CG, De Camilli P, Reinisch KM. Structural insights into assembly and regulation of the plasma membrane phosphatidylinositol 4-kinase complex. Dev Cell. 2014;28:19–29. doi: 10.1016/j.devcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153:216–227. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.