

Freeze-frame synthetic proteins trap DNA reaction intermediates in single live cells, revealing origins of genome instability.
Keywords: DNA repair, DNA replication, homologous recombination, holliday junctions, RecQ family proteins, cancer, spontaneous DNA damage, Escherichia coli, RuvC, BLM
Abstract
DNA repair by homologous recombination (HR) underpins cell survival and fuels genome instability, cancer, and evolution. However, the main kinds and sources of DNA damage repaired by HR in somatic cells and the roles of important HR proteins remain elusive. We present engineered proteins that trap, map, and quantify Holliday junctions (HJs), a central DNA intermediate in HR, based on catalytically deficient mutant RuvC protein of Escherichia coli. We use RuvCDefGFP (RDG) to map genomic footprints of HR at defined DNA breaks in E. coli and demonstrate genome-scale directionality of double-strand break (DSB) repair along the chromosome. Unexpectedly, most spontaneous HR-HJ foci are instigated, not by DSBs, but rather by single-stranded DNA damage generated by replication. We show that RecQ, the E. coli ortholog of five human cancer proteins, nonredundantly promotes HR-HJ formation in single cells and, in a novel junction-guardian role, also prevents apparent non-HR–HJs promoted by RecA overproduction. We propose that one or more human RecQ orthologs may act similarly in human cancers overexpressing the RecA ortholog RAD51 and find that cancer genome expression data implicate the orthologs BLM and RECQL4 in conjunction with EME1 and GEN1 as probable HJ reducers in such cancers. Our results support RecA-overproducing E. coli as a model of the many human tumors with up-regulated RAD51 and provide the first glimpses of important, previously elusive reaction intermediates in DNA replication and repair in single living cells.
INTRODUCTION
Elucidating the enzymatic mechanisms of biological processes by in-cell genetic and in vitro biochemical analyses can be daunting because of the transient and elusive nature of reaction intermediates, which define the reaction mechanisms. Although genomics reveals the stable end products of reactions underlying genome instability and evolution—the new DNA sequences created—the intermediates themselves are seldom studied directly, especially in living cells. Here, we examine reactions of DNA repair by homologous recombination (HR), which can provoke genome instability/evolution. We report the engineering and use of synthetic proteins to detect, quantify, trap, and map a central DNA intermediate in HR, the Holliday junction (HJ): a four-way DNA junction [Fig. 1, A (iv) and B]. We use these engineered proteins to address fundamental questions in genome stability and replication in single living somatic cells: What is the primary use of/need for HR and how does a model RecQ family protein promote genome stability?
Fig. 1. RDG traps HJs, inhibiting recombination, repair, and protein action on HJs biochemically and in living cells.
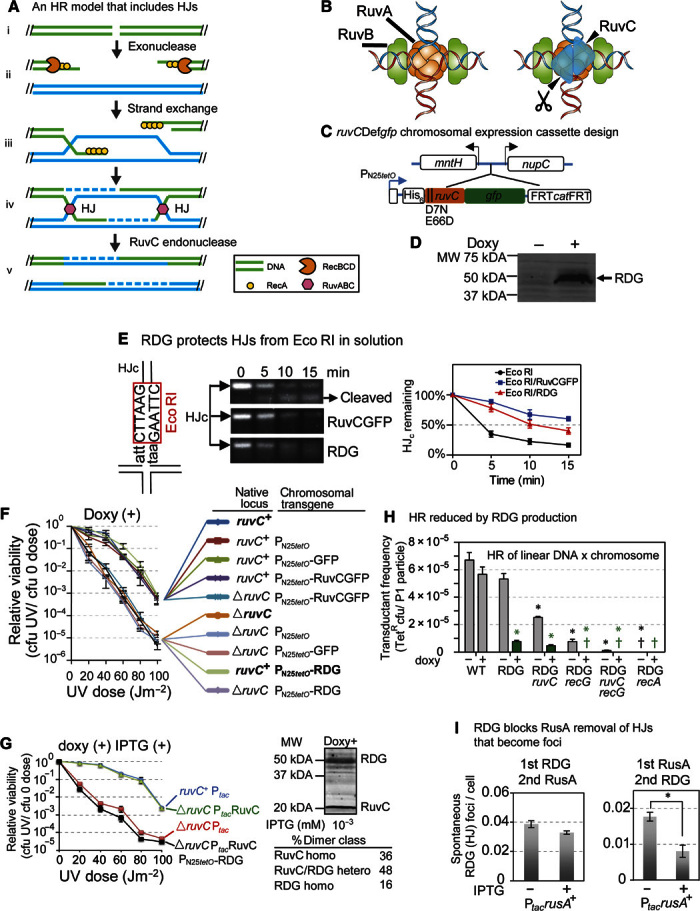
(A) Example of an HR model that includes HJs (with pink hexagons, RuvC, on them; see text S1 for HR and this model). Paired parallel lines, base-paired DNA strands; dashed lines, new DNA synthesis. (B) Illustration of RuvC (blue triangles) binding to HJ, adapted with permission from West (32). (C) Design of chromosomal regulatable ruvCDefgfp gene (see also fig. S1). PN25tetO, doxycycline-inducible promoter (39); arrows, directions of transcription. (D) RDG protein is induced with doxycycline (doxy; Western blot). (E) RDG protects HJ DNA (not linear duplex DNA; fig. S2D) from Eco RI cleavage in solution. Left: Eco RI site in synthetic immobile HJc DNA, 3 bp from the HJ center. Middle: Representative digestion of HJc by Eco RI inhibited by prebinding (fig. S2B) of RuvCGFP or RDG. Right: DNA band intensities normalized to that of HJc at time 0 of Eco RI treatment (means ± SEM, three experiments). (F) RDG production causes dominant-negative UV light sensitivity, implying failed DNA repair. The data imply that RuvC+ protein cannot act on HJs trapped by RDG. Native ruvC locus, either ruvC+ or deleted (ΔruvC) (left), and the protein produced from the chromosomal transgene (right): PN25tetO-RDG, PN25tetO-RuvCGFP, or PN25tetO promoter only. (G) In-cell titration of RDG/RuvC+ ratios shows that RDG remains dominant-negative, implying HJ trapping, when RDG levels are reduced to allow many RuvC+ homodimers. RuvC homodimers/RDG homodimers at 2.3 (black line) determined by Western blots. Right: percentages of RuvC homodimers and RuvC/RDG heterodimers expected at this ratio. RuvC and RDG levels controlled by IPTG-inducible Ptac and doxycycline-inducible PN25tetO, respectively. MW, molecular weight. (H) RDG inhibits HR in a phage P1 transduction assay in the presence (RDG) or absence of native RuvC or RecG HJ resolution proteins and in the absence of both (RDG ruvC, RDG recG, and RDG ruvC recG, respectively). RDG transcription induced (green) or repressed (gray). † or †, frequency < 1 × 10−7 colony-forming units (cfu) per particle; *P < 0.05 relative to the uninduced control. *P < 0.05 relative to the uninduced RDG control strain. (I) RDG protects HJs from RusA HJ endonuclease in living cells. RusA produced from an IPTG-inducible plasmid reduced RDG spontaneous HJ foci (foci described in Fig. 2) when produced before (right) but not after RDG (left). Left bar in each panel, no IPTG induction. *P < 0.05, two-tailed paired t test.
HR mends damaged DNA (text S1 and Fig. 1A) and in doing so also promotes genome instability that drives cancer (1) and evolution (2), adaptation of pathogens to our immune systems (3), and immune defenses (4). Most of the cancer-driving roles of HR occur in somatic cells repairing (mostly) endogenous DNA damage (5). Although HR commonly occurs between identical sister chromosomes in somatic cells (6), nonidentical homologous chromosomes sometimes recombine, which can cause “loss of heterozygosity” of alleles that promote cancer when mutant (7). Also in somatic cells, HR between repeated sequences promotes some cancer-driving chromosomal rearrangements, including large and small deletions and duplications (copy number alterations) and translocations (5, 8). In addition, human RAD51, the ortholog of Escherichia coli RecA—the conserved, ubiquitous, central HR catalyst—is overexpressed in diverse human tumors and associated with poor prognosis (9, 10), implicating HR in the maintenance or progression of cancer (11).
Here, we discover the primary uses of HR in single living somatic or vegetative (nonsexual) cells: the main DNA damage types that necessitate HR and the main cellular processes that cause them. We assess how frequent HR is in somatic cells. The importance of HR would be different whether used once per genome replication [estimated for E. coli and similarly per base pair for human DNA (12–14)] rather than once per hundred replications. We also demonstrate a genome-scale control and directionality of double-strand break (DSB) repair in the E. coli chromosome and address the functions of a model RecQ family protein in single living cells.
Five human orthologs of E. coli RecQ DNA helicase are genome-stabilizing cancer prevention proteins (15) important to human health, but their precise functions have been elusive in living cells. In biochemical experiments, purified RecQ both promotes RecA-mediated strand exchange, leading to HJs (16), and disentangles model double HJs (17). Although RecQ promotes the net accumulation of HJs in living cells (18) and allows the degradation of DNA at stalled replication forks (19, 20), whether it acts before or after HJs in cells is unknown (18), as are its main roles in cells. Of the five human orthologs, WRN (and yeast Sgs1) acts nonredundantly to reduce HJ levels in cells (21, 22). Sgs1 also works redundantly in DSB resection/HJ promotion (23–25), and RECQL4 is implicated in this role (26). BLM is implicated in HJ-level reduction (15). RECQL5 may act mainly on RNA (27). RECQL1, RECQL5, and RECQL4 also prevent genome instability and cancer in humans and mice (27–29), but whether via HR, and at what stage(s), is unknown.
We present engineered protein derivatives of E. coli RuvC four-way DNA junction (HJ)–specific endonuclease (Fig. 1B) (30–32) that trap HJs, inhibit their further chemistry with both purified proteins in solution, and in cells, label and quantify HJs as fluorescent foci in single living cells, and map sites of HJs in genomes via chromatin immunoprecipitation sequencing (ChIP-seq). HJs can form both as intermediates in HR, and HR independently when replication forks stall and “regress” (33, 34). We distinguish HR-generated HJs from non-HR–HJs in E. coli via the requirements of HR-HJ formation for specific HR proteins not required for fork regression in live E. coli (33), and as predicted by the biochemistry of the RecA and RecA-loader proteins (35). We discover the main sources and rates of formation of HR-HJs in vegetative E. coli and that the genomic footprints of HJs in DSB repair show chromosomal directionality. We also discover a novel “junction-guardian” role of RecQ, both promoting the formation of HR-HJs and preventing the formation of non-HR–HJs. By mining human cancer RNA data, we implicate the RecQ orthologs BLM and RECQL4 in similar roles in many human cancers.
RESULTS
A HJ trap is engineered from RuvC
We engineered endonuclease-defective, fluorescent protein fusions of four-way DNA junction–specific RuvC by substituting bases encoding catalytic amino acids (described in fig. S1 and text S1 for four-way junction specificity) (30–32). We also built an identical C-terminal green fluorescent protein (GFP) fusion to functional RuvC. RuvCDefGFP (RDG), RuvCDefmCherry (RDM), and RuvCGFP are encoded (separately) as doxycycline-inducible transgenes in a nongenic site between the mntH and nupC genes in the E. coli chromosome (Fig. 1, C and D), in cells that also have either the wild-type (WT) or deleted ruvC gene at the native locus, as indicated. We purified RDG and RuvCGFP proteins and confirmed that both bind model HJs in solution (fig. S2, A and B). RuvCGFP cleaves a model HJ, apparently uninhibited by the GFP tag (fig. S2C). RDG does not cleave the model HJ (fig. S2C), indicating that, as designed, RDG binds but does not cleave HJs in solution.
Purified RDG inhibits action of other proteins on HJs in solution
Two assays show that RDG inhibits the activities of other proteins at HJs, that is, “traps” HJs in solution. First, prebinding of either RDG or RuvCGFP to a model HJ with an Eco RI recognition sequence near the junction (fig. S2B) slowed cleavage by Eco RI endonuclease of HJ DNA (Fig. 1E) but not linear DNA (fig. S2D), indicating that both retard Eco RI specifically at a HJ. Second, we performed competition assays between RDG and Flp high-affinity site-specific recombinase/HJ resolvase (36), reported to have roughly similar HJ affinity to RuvC (32). More than half (55 ± 2%; mean ± SEM, three experiments) of the RDG bound to a model HJ containing the Flp recognition sequence resisted displacement by Flp (fig. S2, E to G). We conclude that RDG has HJ-trap activity in solution with affinity similar to Flp.
RDG inhibits action of other proteins on HJs in living cells
Three assays demonstrate that RDG inhibits HJ processing by other proteins in E. coli. First, RDG protects against RuvC, causing a “dominant-negative” sensitivity to ultraviolet (UV) light. ΔruvC cells are UV-sensitive (Fig. 1F, ΔruvC compared with ruvC+) (32) and became resistant with RuvCGFP production from the chromosomal transgene (Fig. 1, C and F, ΔruvC PN25tetO-RuvCGFP), showing that RuvCGFP substituted for RuvC. Thus, RuvCGFP was functional in cells as it was in solution (fig. S2C). By contrast, production of RDG caused a UV sensitivity similar to that of ΔruvC cells even in cells that also carry the native WT ruvC+ gene (Fig. 1F, ruvC+ PN25tetO-RDG; fig. S3, additional controls), implying that RDG blocked the activity of RuvC on HJs. Because RuvC functions as a dimer (Fig. 1B) (32), we constructed regulatable chromosomal cassettes for both RuvC and RDG to vary their ratios. We found that RDG prevents RuvC action, causing a dominant-negative UV sensitivity, even when the molar ratios were adjusted to produce a predicted 36% active-form RuvC homodimers (Fig. 1G, black line) and 16% RDG homodimers (48% heterodimers), assuming unbiased association of the RuvC and RDG monomer subunits. Because the Western blots measure denatured proteins (Fig. 1G and fig. S4B), this is predicted rather than measured directly. When RuvC homodimers were predicted to outnumber RDG at 50% RuvC to 8% RDG (41% heterodimers), cells became resistant to UV [fig. S4, A and B; 10−1 mM isopropyl-β-d-thiogalactopyranoside (IPTG)]. The data imply that RDG traps HJs in living E. coli, inhibiting RuvC action, even when RDG homodimers are only about half as numerous as functional RuvC homodimers (Fig. 1G), but not when RuvC homodimers exceed RDG homodimers by a factor of ≥6 (fig. S4, A and B). In experiments detailed below, we produced RDG homodimers in ≥50-fold excess of native RuvC homodimers (fig. S4C) to capture most or all HJs.
Second, RDG also inhibited recombination of linear DNA with the E. coli chromosome [transductional HR; per Magner et al. (18)], implying inhibition of the pro-HR activities of HJ-processing proteins, including RuvC and RecG. In transductional HR, RuvC and RecG partially substitute for each other such that cells deleted for ruvC or recG are somewhat HR-deficient, and ruvC recG double mutants are far more HR-deficient (Fig. 1H, gray bars; RDG not induced, compare RDG with its ΔruvC and ΔrecG derivatives and both with ΔruvC ΔrecG) (32). We found that induction of RDG (Fig. 1H, green symbols) reduced transductional HR by 6.6 ± 0.01 times in the presence of endogenous RuvC and RecG proteins (Fig. 1H, RDG green bar, induced, compared with RDG gray bar, not induced), regardless of native RuvC (Fig. 1H, RDG ruvC green bar), and somewhat more in ΔrecG or ΔruvC ΔrecG cells (Fig. 1H, green). The data imply that RDG blocks both RuvC and part of RecG HR-promoting action on DNA in living cells.
Last, RDG blocks the action of the RusA HJ and three-way junction endonuclease [reviewed by Mahdi et al. (37)] in living E. coli. In the following section, we show that RDG fluorescent foci correspond with HJs. Here, we used timed production experiments with regulatable RusA and RDG to show that the numbers of spontaneous foci of RDG are reduced if RusA is produced before RDG but are not reduced if RDG is produced first (Fig. 1I). The data imply that when RusA is produced first, it reduces cellular levels of spontaneous HJs (Fig. 1I, right), the remainder of which become RDG foci when RDG is produced. Further, when RDG was produced first, RusA had no significant effect on levels of spontaneous RDG foci (Fig. 1I, left). The data imply that RDG blocks the nuclease activity of RusA on HJs, seen as foci in living E. coli cells.
RDG binds and labels HJs in single living cells
We show that RDG forms fluorescent foci that are correlated with HJs from HR-DSB repair in E. coli cells (for example, Fig. 1A, HR repair model), as follows. We induced low levels of chromosomally encoded I–Sce I double-strand endonuclease (38) in proliferating E. coli with the I–Sce I cleavage site either near to or far from the replication origin (ori) (I-sites, red arrows, DSB; Fig. 2A) to create more or fewer reparable DSBs, respectively, in the more and fewer copies of those two chromosomal regions caused by replication (diagrammed in Fig. 2A). We validated the differential numbers of DSBs per cell as fluorescent foci of GamGFP, a DSB-specific trap protein (Fig. 2B), per Shee et al. (39).
Fig. 2. RDG foci represent HJs in living cells.
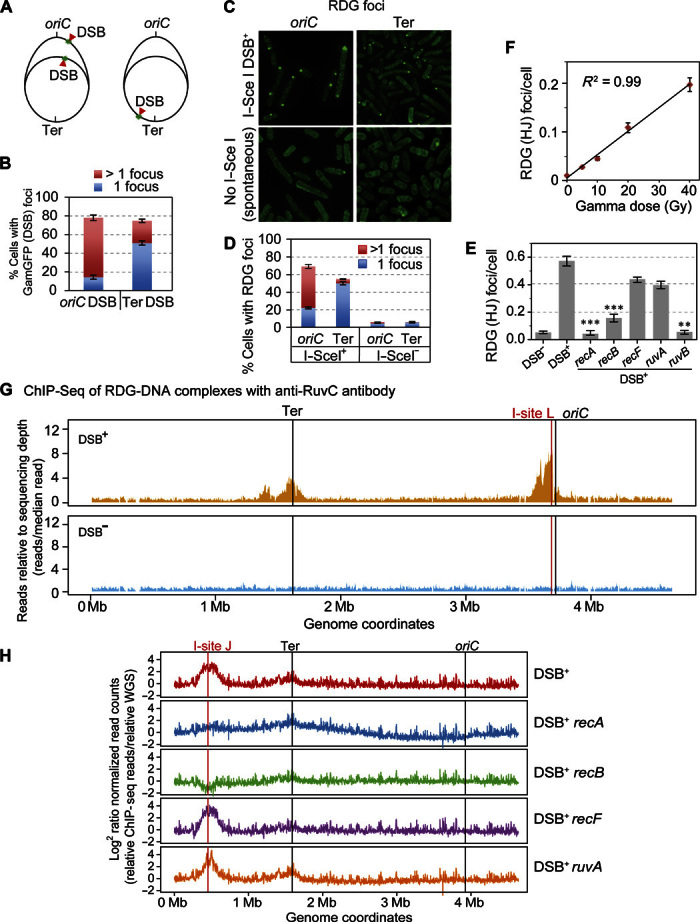
(A to E) Correlation of RDG foci with HR-DSB repair–generated HJs per living cell. (A) Strategy for E. coli chromosome cleavage with chromosomally encoded I–Sce I endonuclease at engineered cut sites (red arrows, DSB). Because of DNA replication, proliferating cells have more copies of oriC-proximal than oriC-distal DNA and thus will have more DSBs (and HR repair) per cell when cleaved by I–Sce I ori-proximally than ori-distally. (B) DSBs quantified as GamGFP focus, per Shee et al. (39), show that most of the cells with the oriC-proximal cut site have >1 GamGFP/DSB focus, and those with the ori-distal cut site have mostly 1 focus per cell, as previously described by Shee et al. (39). (C) Representative images of RDG foci after I–Sce I cleavage (top row) or spontaneous foci (bottom row). (D) I–Sce I–induced RDG foci are positively correlated with numbers of DSBs (and HJs) [quantification of images as in (B)], similarly to GamGFP foci in both oriC-proximal and oriC-distal sites. RDG foci increase with DSBs (P = 0.0001 for each locus, two-tailed unpaired t test), and more cells with >1 focus with ori-proximal than ori-distal cleavage (P = 0.0001, two-tailed unpaired t test). (E) HR protein dependence of I–Sce I/DSB–induced RDG foci. I–Sce I/DSB–induced RDG foci are reduced by a significant 26.1 ± 0.1 times in recA and 4.3 ± 0.1 (means ± SEM) times in recB null mutants indicating RecAB dependence, supporting their interpretation as HR-dependent foci formed during DSB repair. The dependence of RDG focus formation on RuvB supports their formation at four-way junctions. The independence of ruvA implies that RDG binds directly to DNA four-way junctions, as shown biochemically (44). **P < 0.01, ***P < 0.001, relative to DSB control, two-tailed unpaired t test. (F) RDG foci are positively correlated with dose of DSB-inducing γ radiation (R2 = 0.99; P < 0.001, Pearson’s correlation analysis). (G) RDG ChIP-seq shows that RDG localized DNA near a reparable I–Sce I–induced DSB (vertical red line) in the E. coli chromosome. I-site L, I–Sce I endonuclease site L. The large peaks disappear in DSB− cells (cells carrying the cut site but no I–Sce I enzyme). RDG ChIP-seq reads are normalized to the total reads in each sample and further normalized to the input genomic DNA. (H) RDG ChIP-seq shows enrichment at a different chromosomal I–Sce I cleavage site: I-site J, roughly halfway between the replication origin and terminus in the E. coli “right” replichore. The I–Sce I–induced RDG ChIP-seq peak is RecA- and RecB-dependent (indicating HR at a DSB), RecF-independent (indicating formation not at single-strand gaps), and RuvA-independent (supporting direct binding of RDG to four-way junctions, as for purified RuvC) (44). Figure S6B shows the DSB− (enzyme no cut site) controls. WGS, whole-genome sequencing.
We find that DSB repair induces RDG foci; we found 11 ± 3 and 9 ± 3 times more cells with RDG foci (means ± SEM) with I–Sce I cleavage than in uncleaved control cells (cut site, no enzyme; Fig. 2, C and D; values are for ori-proximal and ori-distal cleavage, respectively). Because of the extra DNA copies near the ori caused by replication (Fig. 2A), both more DSBs (Fig. 2B) and more opportunities for repair with a potential uncleaved sister chromosome are expected for ori-proximal than ori-distal DSBs (Fig. 2A). We found that ori-proximal cleavage of the chromosome produced 10 ± 3 times more cells with >1 RDG focus than did ori-distal cleavage (Fig. 2D). These data correlate the number of RDG foci with DSBs expected to be undergoing HR repair via HJs. The data also indicate that multiple events can be visualized as >1 focus per cell—the foci do not coalesce into a single spot.
Further, RecA and RecB proteins, which are required for HJ formation during HR-DSB repair (Fig. 1A) (40, 41), were required for I–Sce I–induced RDG focus formation (Fig. 2E). This supports the interpretation that DSB-induced RDG foci indicate HR-HJs. RecF loads RecA at non–DSB-instigated HR events (40–43) and is not required for I–Sce I–induced RDG foci, as expected (Fig. 2E).
RDG foci are also correlated with numbers of HR-reparable DSBs produced by gamma rays (Fig. 2F and text S2). In text S2, these and other data are used to estimate an efficiency of RDG detection of HJs of about 50%.
Four additional lines of evidence support the conclusion that the RDG foci represent HJs. (i) I–Sce I–induced (Fig. 2E) and spontaneous (Fig. 3A) RDG focus formation requires RuvB, which stabilizes purified RuvC binding to HJs in solution (32), and does not require RuvA, which is not required for RuvC HJ binding biochemically (44). Purified RuvC can recruit RuvB to model HJs in the absence of RuvA protein in solution (45). Because of the specificity of RuvC for HJs (text S1) (30) and the ability of RuvB to stabilize RuvC at HJs (45), our data imply that in living cells, RuvC/RDG can also bind HJs without RuvA and can recruit and be stabilized by RuvB, implying that RDG foci indicate HJs.
Fig. 3. Spontaneous HR repair HJs are replication-dependent and instigated by mostly non-DSB DNA damage.
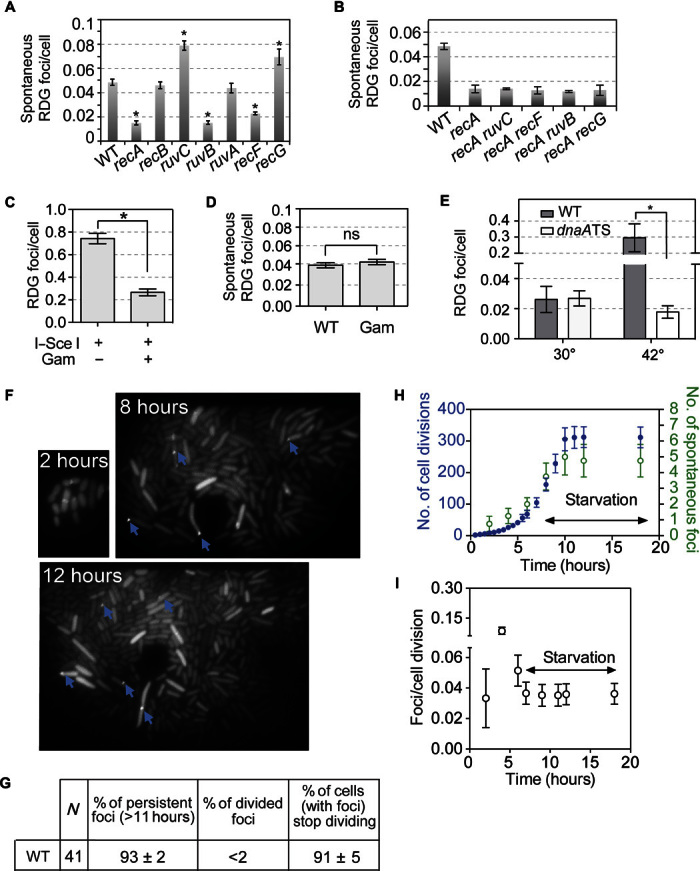
(A) Non-DSB damage provokes most spontaneous HJs in vegetative, growing E. coli: RecB independence indicates that most spontaneous DNA damage repaired is not DSBs. RecF dependence implies ssDNA gaps (42, 43). RuvB dependence: four-way junction–specific. recG mutant has increased spontaneous RDG foci, indicating that RecG reduces the steady-state level of spontaneous HJs, as predicted by Whitby et al. (91). (B) RecA dependence of the spontaneous HR-HJ foci in the various mutants. (C and D) DSB independence of most spontaneous HJ foci. Gam, DSB-specific DNA end–binding protein (92, 93) blocks I–Sce I–induced HJ foci (C), but does not block most spontaneous HJ/HR repair foci (D). n.s., not significant (control, fig. S13). (E) Replication dependence of most spontaneous RDG HJ foci. Spontaneous HR-HJ foci are reduced in dnaATS cells relative to WT at nonpermissive temperature (42°C), at which replication initiation is blocked. P = 0.03, two-tailed unpaired t test. (F to I) Microfluidic time-lapse imaging shows the birth, generation dependence, and persistence of spontaneous RDG HJ foci. (F) Representative images of microcolony with RDG foci (blue arrows) in WT strain. (G) Quantification of fates of spontaneous RDG HR-HJ foci and cells that acquire them; most spontaneous RDG foci persist over hours after formation; most cells with RDG foci cease to divide, indicating that the birth of each spontaneous focus reports on a new HJ event. (H) Quantification of generation dependence of the birth of spontaneous RDG foci. Rapid growth in glucose was followed by washing cells in the same medium lacking glucose to slow and halt cell divisions in the WT strain. Blue circles, number of cell divisions; green circles, cumulative number of spontaneous foci that appear in each microfluidic microcolony. (I) Rates of spontaneous RDG focus formation in rapid and stationary growth phases shown in (F).
Next, (ii) production of HJ endonuclease RusA before RDG reduced the number of spontaneous RDG foci, implying that the foci indicate HJs (Fig. 1I). (iii) The mCherry-tagged RDM forms spontaneous foci that colocalize with foci of a partial-function mutant RecA-GFP fusion protein (fig. S5) (46), but not with GFP alone, placing RDM foci in the cellular vicinity of DNA damage or repair (fig. S5). Foci of proteins bound to defined DNA sites are easily distinguishable at sites separated by 55 kb (39) [13 kb for Wang and Sherratt (47)], and are separated at ≥80 kb (39, 47), such that the colocalization here puts RDG roughly near RecA-bound DNA. (iv) Using ChIP-seq in the following section, we demonstrate that RDG binds sites of HR-DSB repair. We conclude that RDG foci indicate four-way DNA junctions in single living cells.
RDG ChIP-seq of HR-HJs at repairing DSBs shows genomic directionality of DSB repair
We mapped the genomic repair landscape of RDG at sites of DSB repair induced by I–Sce I cleavage by ChIP-seq using an antibody against RuvC. Figure 2G shows significant enrichment of DNA sequences near I-site L (red line), near the chromosomal replication origin, oriC (black line), and downstream of I-site L in the unidirectional replication path (fig. S6, additional controls). RDG enrichment extends 63 to 73 kb ori-proximally and 169 to 173 kb ori-distally from the cut site (changepoint analysis; two independent experiments). Surprisingly, a smaller enrichment occurred near the replication terminus (Fig. 2G), possibly from break-induced repair replication HJs that continue with the replication fork to the terminus (see Discussion). Both areas of enriched reads are DSB-dependent and are not observed in cells without I–Sce I cleavage (DSB−, Fig. 2G). Therefore, the enrichment reflects binding of RDG to DNA associated with DSB repair.
In Fig. 2H, with cleavage about halfway between the replication ori and terminus, reads were again enriched at the I-site and preferentially downstream in the replication path (143 to 145 kb ori-proximally and 207 to 207 kb ori-distally, changepoint analysis; two experiments). The RDG-DNA binding required the strand-exchange (HJ-producing) protein RecA and its DSB-specific loader (48) RecB (Fig. 2H), indicating HR-DSB repair specificity, and was independent of RecF [RecA loader at single-stranded DNA (ssDNA) gaps (42, 43)] and RuvA (Fig. 2H), which is not required for RuvC binding biochemically (44). We conclude that the ChIP-seq maps of RDG binding show the genomic locations of HJs formed by HR-DSB repair. These first glimpses of the genomic footprints of HJs during DSB repair demonstrate a directionality of DSB repair along the chromosome not observed previously, with more HJs ori-distally than ori-proximally (see Discussion).
Most spontaneous RDG foci result from HR DNA repair
With no DNA damage induced, RDG foci appeared spontaneously (figs. S7 and S8, A to C), most, we show, as a result of spontaneous DNA damage and HR repair. Appearance of most spontaneous RDG foci required the RAD51-orthologous strand-exchange HR repair protein RecA and its loader at ssDNA gaps (42, 43), RecF (Fig. 3, A and B, and fig. S8A), which is analogous with RAD52, the human RAD51 loading preparation protein (49). Purified RecA aids formation of regressed forks (RFs) in solution (50), but RecA is not required for the formation of, or RuvABC action on, RFs in living E. coli (33) and is required for HR repair (40, 41). About 75% of spontaneous RDG foci required RecA, RecF, and RuvB (Fig. 3A), supporting their origin as spontaneous HR repair events. The RuvB dependence supports focus occurrence at HJs. The different numbers of foci in these strains do not result from different growth rates/numbers of replication forks, as shown in fig. S9. Mutants that lack RecA and also RuvB or RecF show no further decrease in spontaneous RDG foci than recA single mutants, indicating that those proteins promote spontaneous RDG foci RecA dependently (Fig. 3B). The origin of the 25% RecA-independent spontaneous foci will be addressed in a separate study.
Replication-dependent non-DSB DNA damage underlies most HR-HJs in vegetative E. coli
Surprisingly, most HR protein–dependent RDG/HJ foci do not result from DSB repair, as shown in two ways. E. coli HR-DSB repair requires RecBC [RecA loader at DSBs (51)], analogous with human BRCA2, which loads RAD51 at DSBs (52–54), and not RecF, RecJ, or RecQ (40–43). In agreement, we see RecB dependence and RecF independence of I–Sce I–induced RDG foci (Fig. 2E, DSB+). By contrast, spontaneous RecA/HR-dependent RDG/HJ foci form independently of RecB (Fig. 3A), and were promoted by RecF (Fig. 3A), indicating repair of non-DSB DNA damage. Further, production of the phage Mu DSB end–binding protein Gam inhibits DSB repair (39) and I–Sce I–induced RDG foci (Fig. 3C) but did not affect spontaneous HR-dependent RDG foci (Fig. 3D and fig. S9, additional controls). We conclude that the main use of HJ-mediated HR in vegetative (nonsexual) E. coli is repair of DNA damage other than DSBs. The RecF dependence implicates ssDNA gaps, a substrate at which RecF loads RecA (42, 43).
The spontaneous DNA damage that necessitates HR-repair HJs is replication-dependent. We blocked replication initiation at the E. coli origin, oriC, using a dnaATS allele, which allows replication at 30°C but blocks oriC use at 42°C (55). We found 30.8 ± 0.2 times fewer spontaneous RDG foci per amount of DNA (fig. S8G) in dnaATS than the WT control strain at the 42°C restrictive temperature (Fig. 3E, normalized to amount of DNA per cell; fig. S8G), but no difference at permissive temperature (30°C). These data support DNA replication as the primary generator of the endogenous non-DSB DNA damage repaired by HJ-dependent HR normally during vegetative growth in E. coli. A model for HR repair of replication-generated ssDNA gaps is discussed below (see Discussion).
Microfluidics reveals spontaneous HR-HJ formation rates and their correlation with replication forks
We used time-lapse microfluidic imaging to see the birth and fates of spontaneous HJ foci in growing E. coli microcolonies. We observed that most RDG/HJ foci persisted for at least 11 hours after they appeared (93 ± 2%), and the cells in which they appeared ceased to divide (Fig. 3, F and G, 91 ± 5%). The HJs trapped by RDG might prevent chromosome segregation, which could block division, activate a checkpoint, or both.
We found that spontaneous HJ/RDG focus formation was correlated with cell divisions (Fig. 3, F and H), at rates from 0.033 ± 0.019 to 0.087 ± 0.018 foci per division, depending on cell growth rate (Fig. 3I). The generation dependence of most spontaneous HJs provides independent support for DNA replication as the driver of most spontaneous HR-HJ events.
Further, spontaneous HJs were correlated with replication forks. We determined chromosome numbers per cell using flow cytometry (fig. S8, D and E) (56), quantified spontaneous HJ/RDG foci via microscopy, and found a nearly constant spontaneous RDG focus frequency per replication fork: 5.0 × 10−3 (±0.3 × 10−3) and 4.2 × 10−3 (±0.6 × 10−3) in rich and minimal medium, respectively (fig. S8F). Most cells with RDG foci had one focus per cell (70.6 ± 0.4%), with the minority having more than one (fig. S8B). Because multiple HR repair events appear as multiple foci per cell (Fig. 2, A to D), we infer that most spontaneous repair HJs result from one or few DNA lesions per cell, rather than genome-wide catastrophe. The data support DNA replication as the origin of most spontaneous DNA damage repaired by HJ-associated HR.
RecQ and RecJ promote spontaneous HR-HJ formation
RecQ promotes both formation and dissolution of HJs biochemically (16, 17, 57) and promotes net accumulation of HR-HJs in cells (18); however, whether RecQ promoted HJ formation, aiding HR, or inhibited HJ resolution, reducing spontaneous HR in cells, was unknown (18). We show that both RecQ DNA helicase and its partner RecJ ssDNA-dependent exonuclease promote spontaneous HR-HJ formation and spontaneous HR in vegetative E. coli cells.
We found that spontaneous RDG foci were reduced by deletion of recJ or recQ, or recJ and recQ genes (Fig. 4A and figs. S9 and S10). We performed timed expression studies to trap HJs with RDG before the production of RecQ. HJ formation proteins are expected to increase HJ levels independently of whether RDG is present first to trap the HJs, whereas proteins that act after HJ formation (on HJs), such as RusA, are stopped by the production of RDG first (Fig. 1I). We found that RecQ increased RDG/HJ focus levels whether it was produced before or after RDG (Fig. 4B). These data support RecQ and RecJ as promoters of HJ formation.
Fig. 4. RecQ and RecJ promote formation of spontaneous HR repair HJs and prevent non-HR–HJs caused by RecA overproduction in a model of RAD51-overexpressing cancers.
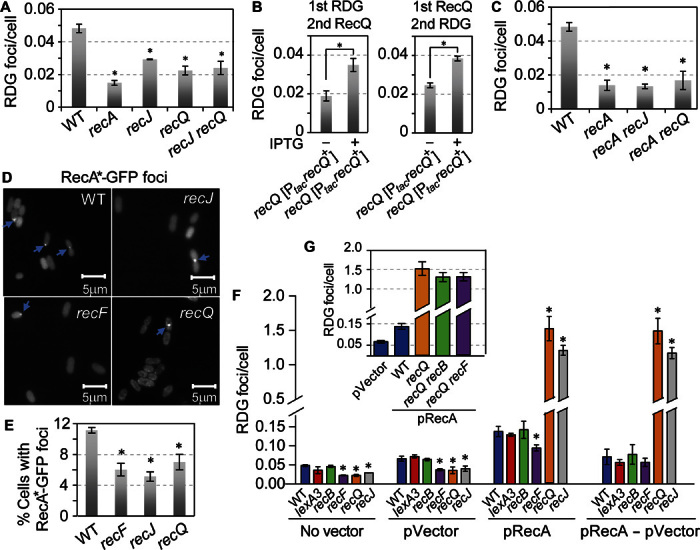
(A) RecQ and RecJ promote spontaneous HJ accumulation acting in the same pathway. P = 0.002, P = 0.002, and P = 0.006, two-tailed unpaired t test for recJ, recQ, and recJ recQ, respectively, compared with WT. (B) RecQ produced from an IPTG-inducible plasmid increased RDG spontaneous HJ foci when produced before (right) or after (left) RDG. Left bar in each panel, no IPTG induction. The data indicate that the RecQ promotion of RDG foci results from RecQ-promoted HJ formation, not RecQ-inhibiting HJ removal, which is blocked by RDG. (C) RecQ and RecJ promote spontaneous HJ/RDG foci via the RecA-dependent (HR repair) pathway, seen as no further reduction of HJ/RDG foci in recA recQ or recA recJ double mutants beyond that in the recA single mutant. (D and E) RecQ and RecJ promote RecA-GFP focus formation, suggesting that they act before RecA. (D) Representative images. Blue arrows indicate foci. (E) Quantification. Spontaneous RecA4155-GFP foci (WT) are reduced in recJ, recF, and recQ null mutant strains. P = 0.001, P = 0.004, and P = 0.02, two-tailed unpaired t test (means ± SEM of three experiments, >1000 total cells scored in each experiment). (F) RecA overproduction causes increased apparent non-HR–HJs, opposed by RecQ and RecJ. RecA overproduction increased RDG foci 2.09 ± 0.02 times (P = 0.01, two-tailed unpaired t test, pVector compared with pRecA; blue bars), independently of RecF or RecB, implying a non-HR origin. There is no RecF dependence when the spontaneous HR-HJs are subtracted out (pRecA − pVector). Moreover, RecA overproduction increased RDG foci by 11- and 9-fold in recQ- or recJ null mutant strains, respectively (P = 0.001 and P = 0.0001, two-tailed unpaired t test). (G) The great increase in RDG foci caused by RecA overproduction in ΔrecQ cells is both RecF- and RecB-independent, implying that RecQ prevents non-HR–HJs. We suggest that HJs are regressed replication forks caused by overproduced RecA (Fig. 6B).
Further supporting this conclusion, the following data suggest that RecQ and RecJ act before RecA. We observed that deletion of recQ or recJ from recA cells did not reduce spontaneous HJs further than in recA cells (Fig. 4, A and C), indicating their action in the same pathway as RecA. The observation that ΔrecQ ΔrecA and ΔrecJ ΔrecA double mutants are more similar to ΔrecA than to ΔrecQ or ΔrecJ single mutants (Fig. 4C) argues [per Avery and Wasserman (58)] that RecQ and RecJ act upstream of RecA in their pathway. We also saw that spontaneous foci of a mutant RecA-GFP fusion protein (46) were reduced in ΔrecJ or ΔrecQ strains (Fig. 4, D and E). These data imply that RecQ and RecJ act before RecA (strand exchange) in HR and may promote RecA loading onto ssDNA. RecF is the RecA loader at ssDNA gaps (42, 43), and 46 ± 11% of the spontaneous RecA-GFP foci were RecF-dependent (Fig. 4E), implying that these spontaneous RecA-GFP foci represent RecA on DNA at ssDNA gaps. We discuss a possible pre-RecA role of RecQ and RecJ in postreplication ssDNA gap repair in the Discussion.
We examined spontaneous HR events themselves to verify these conclusions. We found that most spontaneous HR that recombined close chromosomal direct repeat sequences required RecF, RecQ, and RecJ (in addition to RecA) (Fig. 5, A and B), showing a pro-HR role for RecQ, compatible with HJ formation and incompatible with inhibition of HJ resolution—the two ways that RecQ could have promoted net accumulation of HR-HJs in cells (18). The RecF dependence of most spontaneous RDG foci (Fig. 3, A and B) and HR events (Fig. 5B) supports recombinational ssDNA gap repair as the origin of most spontaneous HJ foci in vegetative, growing E. coli cells (model, Discussion). The data demonstrate that RecQ and RecJ promote HR-HJ formation in living cells, and imply that they act before RecA during replication-induced ssDNA gap repair, and that this is a primary role normally in growing cells.
Fig. 5. Dependence of spontaneous intrachromosomal HR on RecF, RecQ, and RecJ, and lack of increase in spontaneous HR with RecA overproduction.
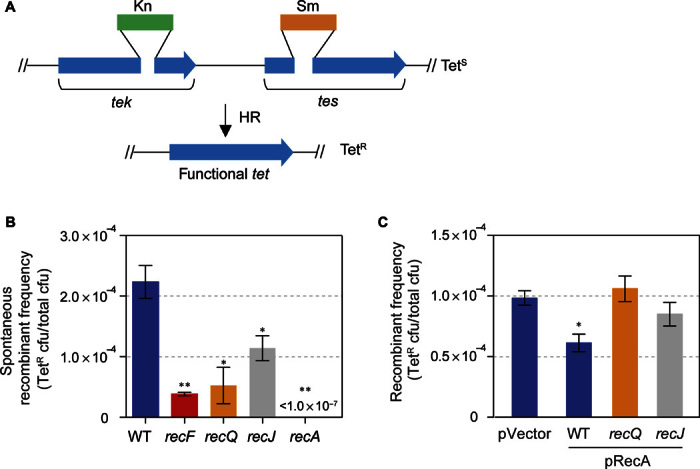
(A) Strategy of the intrachromosomal direct repeat recombination assay of Corre et al. (82). (B) In cells without artificial RecA overproduction, most spontaneous HR events require RecF, RecQ, and RecJ (and all of them require RecA) and do not require RecB (82). The data indicate that most of the spontaneous HR events are instigated by ssDNA gaps [per Morimatsu and Kowalczykowski (42) and Sakai and Cox (43)], not, for example, RecFQJ-independent, RecB-dependent DSB repair (for example, Fig. 2E) (40, 41). These data support the model shown in Fig. 8. (C) Spontaneous HR events are not increased by RecA overproduction. This finding suggests that the increased RDG/HJ foci seen with overproduced RecA (Fig. 4, F and G) are not increased HR-HJs, and supports the interpretation that they reflect regressed replication forks (illustrated in Fig. 6Biii). The small decrease in spontaneous HR with overproduced RecA compared with the pVector control (P = 0.02, unpaired t test) could be caused by titration of HJ resolution capacity in the cells via their increased non-HR–HJ levels (Fig. 4F). We hypothesize that the RecQ and RecJ independence of the spontaneous HR events in RecA-overproducing cells may indicate that DSB-instigated HR may dominate spontaneous events during RecA overproduction. This is expected because the presence of increased non-HR–HJs (probable RFs; Fig. 6Biii) is cleaved by RuvABC in living cells producing DSBs (33) and thus may cause DSB-instigated HR, which is RecQ- and RecJ-independent, to dominate.
RecQ and RecJ prevent non-HR–HJs in a cancer-state model
The human RecA ortholog RAD51 is overexpressed in a wide range of tumors with p53 defects (11), and is correlated with poor prognosis (9, 10); but how increased RAD51 supports the cancer state is unknown. We modeled RAD51 overexpression in cancers by overproducing RecA in E. coli and discovered that RecA overproduction increased HJ focus levels by a significant 2.09 ± 0.02 times [Fig. 4F, pVector versus pRecA (blue bars)].
The following data indicate that the increased RDG/HJ foci caused by RecA overproduction are not HR-HJs but rather are non-HR–HJs, such as regressed replication forks. Although RecA overproduction caused more HJ foci (Fig. 4F), it did not increase intrachromosomal HR events in close direct repeat sequences (Fig. 5C), showing a lack of correlation between HR and the extra HJ foci observed.
In addition, the increased RDG foci caused by RecA overproduction were formed independently of RecA loader proteins that promote HR repair of DSBs, RecB (51), and ssDNA gaps, RecF (Fig. 4F) (42, 43). Note that spontaneous RDG foci are RecF-dependent (Fig. 4F, no vector and pVector). If these are subtracted from the additional RDG foci added by RecA overproduction (Fig. 4F, pRecA − pVector), then we see that all of the RecA overproduction–induced RDG foci are RecF-independent (Fig. 4F, pRecA − pVector). RecB and RecF are analogous with human BRCA2 (51–54, 59–61) and RAD52 (49). Because the RecF-dependent component of spontaneous HR-HJ/RDG foci (Fig. 4F, pVector) remained present when RecA was overproduced (Fig. 4F, pRecA), and only the additional RDG/HJ foci are unaffected by RecF (Fig. 4F, pRecA − pVector), we can rule out the possibility that overproduction caused RecA to become RecF-independent for HR. Therefore, RecA overproduction did not cause HR to become independent of the HR RecA loader. Rather, the extra HJs during RecA overproduction appear to arise by a different, non-HR process. We infer that the increased HJs in this RAD51-overexpressing cancer model result from non-HR events (uncorrelated with HR as illustrated in Fig. 5C, and independent of RecA loaders as shown in Fig. 4, F and G): we suggest from replication fork stalling and regression, which occurs independently of RecF (35) and RecB (Fig. 6A) (33), and, biochemically, is promoted by excessive RecA independently of loader proteins (35).
Fig. 6. Model: Promotion of regressed replication forks by overproduced RecA and prevention of their associated HJs by RecQ and RecJ.
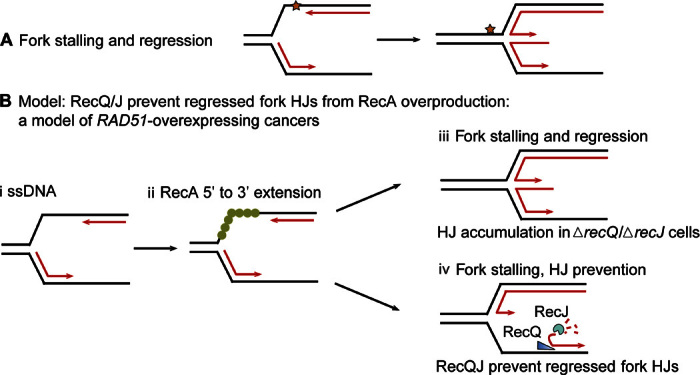
(A) Diagram of replication fork stalling at a replication-blocking DNA lesion (star) and regression to form a HJ. When forks stall, the potential energy of DNA supercoiling ahead of the fork drives the fork backward spontaneously (94), independently of RecA biochemically (94) and in cells (33). Lines, strands of DNA; parallel lines, base-paired strands; arrowheads, 3′ ends. (B) Model for fork regression caused by excess RecA and its prevention by RecQ/RecJ. We suggest that with RecA overproduction, forks regress spuriously, without a replication-blocking DNA lesion, because of RecA polymerization on ssDNA at the fork (i and ii) instigating strand exchange with the nascent sister duplex. (ii) RecA can be loaded on ssDNA at a fork and extended 3′ from the fork junction to the ssDNA-dsDNA (double-stranded DNA) junction at the 3′ leading strand end, a RecF-independent (35) [and RecB-independent (33)] independent substrate and reaction to promote (iii) fork regression. We suggest that after RecA loading onto the ssDNA, there can be two outcomes, which are influenced by the presence of RecQ or RecJ. (iii) In the absence of RecQ or RecJ, RF HJs will accumulate. (iv) In the presence of RecQ and RecJ, RecQ unwinding of the lagging strand at the fork (19, 69) and RecJ 5′-ssDNA–dependent exonuclease can prevent some RF HJs by making a ssDNA-end fork regression structure/substrate. This 3′-ssDNA end (arrowhead) would not be bound by RDG and may be degraded by ssDNA exonucleases, preventing HJs.
Surprisingly, we found that RecA overproduction resulted in RecQ and RecJ roles opposite to their roles in spontaneous HR (Fig. 5B) and HR-RDG focus formation (Fig. 4, A to E); RecQ and RecJ opposed accumulation of the extra non-HR–HJs caused by overproduced RecA (Fig. 4F). Cells that lack RecQ or RecJ (ΔrecQ or ΔrecJ) showed 11.1 ± 0.2 and 8.8 ± 0.1 times more RDG foci/HJs caused by RecA overproduction, respectively (Fig. 4F). The excessive RDG foci/HJs in ΔrecQ cells are also independent of the RecA loaders RecB and RecF (42, 43), which are used in HR repair (Fig. 4G). Moreover, we induced RDG, which traps HJs, before RecA overproduction (fig. S11). Therefore, the data indicate that RecJ and RecQ prevented the formation, rather than aided the postformation removal, of the non-HR–HJs caused by RecA overproduction. The RecQ/J role(s) in preventing non-HR–HJs (Fig. 4F) contrasts with the RecQ and RecJ roles in promoting HR-HJ formation that dominates spontaneous HJ events (Figs. 4, A to E, and 5B; see model in Discussion).
Thus, RecQ plays a novel junction-guardian role in E. coli: both promoting spontaneous HR (repair)–HJs and preventing apparent non-HR–HJs. RecQ/J may prevent the formation of non-HR–HJs caused by replication stalling and fork regression (model, Fig. 6B). We suggest that overproduced RecA promotes regressed-fork (FR), RF HJs abnormally by acting on undamaged (normal) forks (model, Fig. 6B), rather than just those with replication-blocking damage (star, Fig. 6A). We conclude that RecQ/J play a novel junction-guardian role in preventing non-HR–HJs that result from RecA overproduction (model, Fig. 6B) while simultaneously promoting HR repair HJs in ssDNA gap repair (model, Discussion).
Cancer transcriptome data show correlation of expression of human RecQ orthologs and HJ resolution proteins with RAD51 in the eight most common human cancers
We looked for RNA data correlations that might indicate human RecQ orthologs with potential similar junction-guardian roles in RAD51-overexpressing cancers. We hypothesized that if the RecQ junction-guardian function improves the fitness/growth of RecA-overproducing cells, then RAD51-overexpressing cancers might require proteins that play RecQ-like roles in preventing or reducing excess HJs caused by RAD51 overproduction. Thus, the genes encoding proteins with RecQ-like or HJ resolvase functions might be overexpressed in RAD51-overexpressing cancers. We found that the human RecQ orthologs BLM and RECQL4, as well as a HJ endonuclease gene, EME1, are frequently co-overexpressed with RAD51 in the eight most common cancers for BLM and in four of the eight most common cancers for RECQL4 (Spearman’s correlation analysis shown in Fig 7, A to E, and table S1).
Fig. 7. Increased BLM and RECLQ4 mRNA in RAD51-overexpressing human cancers of the eight, and four of the eight, most common cancer types, respectively.

Spearman’s rank correlation analyses of data from cBioPortal (88, 89), per Materials and Methods. Each data point represents the mRNA level in one patient sample relative to the reference population (Z score; Materials and Methods). Data from 129 to 1100 patient samples were analyzed per cancer type (table S1). (A) Increased BLM mRNA levels (y axis, Z scores) correlated with increased RAD51 mRNA (x axis, Z scores) in eight of eight of the most common cancer types (R2 > 0.25; P ≤ 6.23 × 10−21, Spearman’s rank correlation analysis). (B) Increased RECLQ4 mRNA levels (y axis, Z scores) correlated with increased RAD51 mRNA (x axis, Z scores) in four of eight of the most common cancer types (R2 > 0.25; P ≤ 2.07 × 10−8, Spearman’s rank correlation analysis). (C) Summary: correlation of increased RAD51 mRNA levels with increased levels of BLM and RECLQ4 RecQ orthologs, EME1 HJ resolution protein, and other RecQ orthologs and resolution proteins in patient tumor data of the eight most common cancers (numbers in parentheses, number of common cancers of the eight most common types with correlation). mRNA Spearman’s correlation coefficient calculated between these and other human RecQ orthologs and other HJ resolvases with RAD51 and with each other among the eight most common cancers is summarized in table S1 (R2 > 0.25 indicates moderate correlation; see table S1 for details). (D) Summary of Spearman’s R2 values for cancer RNAs of genes correlated with RAD51 overexpression (R2 > 0.25 for moderate correlation) and control genes poorly correlated or uncorrelated with RAD51 overexpression (for example, ACTB, which encodes a subunit of actin).
In both R2 values and numbers of cancer types involved, these very strong correlations (table S1) outscored current transcriptional correlation studies considered to be highly indicative to definitive. For example, CDH1 and ZEB1 (62), and KLF4 and KLF5 (63), showed R2 values of 0.15 and 0.08, both in only a single cancer type, compared with the R2 values for BLM with RAD51 (0.29 to 0.62) in the eight most common cancer types, and for RECQL4 with RAD51 (0.26 to 0.41) in four of the eight most common cancer types (Fig. 7, A to E). Thus, these are extremely robust correlations. Negative control and other genes did not show such correlations (for example, ACTB encoding a subunit of actin; Fig. 7D).
We hypothesize that the increased expression of the RecQ orthologs BLM and RECQL4 and the HJ resolvase EME1 may also prevent (or remove) excess HJs. The most coexpressed HJ resolvases and RecQ ortholog with BLM are EME1, GEN1, and RECQL4 (Spearman’s correlation analysis shown in fig. S12 and table S1). These correlations may indicate that EME1, GEN1, and RECQL4 may work together with BLM to reduce or prevent excess HJs caused by RAD51 overexpression, a common event in many and varied cancers. Further work is needed to test these hypotheses.
DISCUSSION
Engineered HJ-trap proteins
We showed that RuvC-derived, catalytically inactive fluorescent protein fusions (RDG, green; RDM, red) specifically bind, trap, and label four-way DNA junctions (HJs), allowing their quantification as fluorescent foci in single living cells (Figs. 1I, 2, A to F, and 3 and figs. S7 and S8) and their mapping in genomes via ChIP-seq (Fig. 2, G and H, and fig. S6). RDG binds and protects HJs from the action of other proteins, that is, traps HJs, when purified in solution (Fig. 1E and fig. S2) and in living cells (Figs. 1, F to I, and 2). The estimated efficiency of detection of HJs (or double HJs) as individual fluorescent foci is roughly 50% (text S2). Like the engineered DSB-trap proteins created previously (39), RDG and RDM are useful tools for studying DNA reaction intermediates in single living cells and in genomes.
A limitation of RuvC-based HJ traps is their dependence on E. coli RuvB (Figs. 2E and 3, A and B), probably via their specific protein-protein interactions (32). We are currently exploring whether this will limit the effectiveness of these tools in other organisms.
The main use of HJ-associated HR is repair of replication-instigated single-strand gaps
Although DSB repair mechanisms are the most studied HR reactions, we find that the main use of HJ-associated HR in vegetative (nonsexual) E. coli cells is not DSB repair. Surprisingly, spontaneous HJs are (i) 75% HR-based (Fig. 3, A and B), (ii) DSB-independent (Fig. 3, C and D), (iii) mostly RecFJQ-dependent (Figs. 3, A and B, and 4, A to C), (iv) replication-dependent (Fig. 3E), (v) correlated with replication fork numbers (Fig. 3, F to H, and fig. S8), and (vi) about 100 times less frequent than the once per E. coli genome replication estimated previously (Fig. 3, F to H; fig. S8; and text S2) (14, 40). Collectively, our data imply that repair of replication-induced ssDNA gaps (illustrated in Fig. 8) is the commonest use of HR and source of HJ intermediates in growing E. coli cells. Endogenous DNA damage is the primary instigator of repair-based genomic changes that drive cancer, genetic diseases, and microbial evolution (see Introduction). We suggest that single-strand lesions constitute the primary instigator of those changes. Other environmental conditions might produce other results.
Fig. 8. Model for RecQ and RecJ role in daughter-strand gap repair of spontaneous/endogenous DNA.
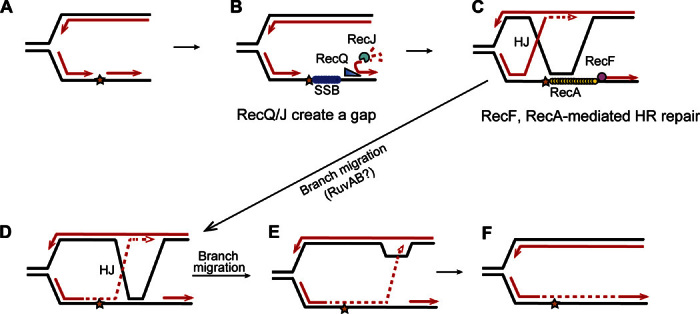
This model follows the ideas of Rupp and Howard-Flanders (95) and integrates the data (i) that most spontaneous RDG/HJ foci and intrachromosomal HR events are RecA- and RecF-dependent HR-HJs (Figs. 3, A and B, and 5B and fig. S10), reflecting RecA loading at single-strand gaps, and largely RecQ- and RecJ-dependent (Figs. 4, A to C, and 5B and fig. S10), with RecQ/J acting in the RecA-dependent pathway (Fig. 4C); (ii) that RecQ/J act before HJ formation, shown by their promotion of HJs even after RDG is produced (Fig. 4B); and (iii) that RecQ/J promote apparent RecA loading in assays of spontaneous RecA-GFP foci (Fig. 4, D and E) thought to reflect RecA-DNA nucleoprotein filaments (46). Model: During vegetative growth and DNA replication, (A) when a synthesis tract of lagging strand DNA (an Okazaki fragment, arrowheads, 3′ ends) encounters a replication-blocking lesion (star) in its template strand, (B) the adjacent Okazaki fragment can be unwound by RecQ DNA helicase, which translocates on DNA in the 3′ to 5′ direction as shown by Hishida et al. (69), exposing a 5′-ssDNA end, which is then degraded by RecJ 5′-ssDNA–dependent exonuclease. This creates a single-stranded gap, which can be coated by SSB [E. coli single-stranded DNA binding protein, like human replication protein A (96)]. (C) RecF is guided to a 5′ end of a ssDNA-dsDNA junction (43) and loads RecA onto SSB-coated ssDNA (42). The RecA-DNA nucleoprotein complex then promotes strand exchange (40, 41) so that the blocked 3′ Okazaki fragment end can displace the identical sequence (black line) in the sister chromosome, and prime continued DNA synthesis (dashed red line) using the new strand of the sister as a template. Lines, strands of DNA; red lines, new DNA strands; black lines, old strands; dashed red line, new DNA synthesis after strand exchange. (D) Branch migration of the HJ rightward, past the lesion, places newly synthesized DNA across from the lesion. Further branch rightward returns the newly synthesized strand to its original template (E) and returns the 3′ end to the original duplex (F), removing the HJ and D loop. The lesion is bypassed and, once it is part of dsDNA, can be repaired by excision repair pathways, which can act only when the damaged base or nucleotide is present in dsDNA [base excision repair and nucleotide excision repair; reviewed by Friedberg et al. (97)]. An alternative mode of resolution of the HJ is endonucleolytic cleavage, for example, by RuvC (not shown).
Genomic footprints of DSB repair HJs show genome-scale coordination and directionality of repair
When DSBs do occur, as when induced by I–Sce I endonuclease, their repair shows evidence of two modes of whole-genome control and directionality along the chromosome (Fig. 2, G and H, and fig. S6). First, I–Sce I–induced, RecA- and RecB-dependent HR-HJs occurred not only at the repairing DSB sites (between 60 and 200 kb on either side) but also at the chromosomal replication terminus region, megabases away (Fig. 2, G and H, and fig. S6). These data support a replicative model for HR-DSB repair (64), in which replication forks are primed by HR strand-exchange intermediates (replication restart at a DSB) that contain a HJ (Fig. 9C). The data suggest that the replication initiated by HR-DSB repair [break-induced replication (BIR)] continues from the DSB to the replication terminus (Fig. 9) (65), and imply that HR-initiated replication bubbles can sometimes drag their associated HJs from the DSB (65) all the way to the replication terminus (Fig. 9E), which is a new discovery. DSB repair in E. coli is largely dependent on the major replicative DNA polymerase, Pol III (65), and produces conservatively segregated new strands of DNA (65), supporting the model in Fig. 9, modified from the model suggested by Motamedi et al. (65). In yeast, BIR bubble migration was observed and thought to reflect D loops without HJs (similar to Fig. 1Aiii) (66), but we suggest that it might also, at least sometimes, include unresolved HJs, as we have observed here (Fig. 2, G and H, and fig. S6). These data support both the replicative nature of much of DSB repair and the frequent asymmetry (one-endedness) of the events, which allows extended genomic replication (for example, in contrast with two-ended mechanisms such as shown in Fig. 1A). Alternatively, the DSB repair–instigated HJs at the replication terminus might result from site-specific HJ-dependent resolution of chromosome dimers caused by crossing-over during HR-DSB repair, which is performed by the XerCD site-specific recombinase at the replication terminus dif site [reviewed by Lesterlin et al. (67)]. This seems less likely because the enrichment of terminus-proximal reads in ChIP-seq was far broader and larger than the 29–base pair (bp) dif site, to which XerCD site-specific HJs are confined.
Fig. 9. Model: One-ended DSB-induced repair (“BIR”) replisomes drag HJs to the E. coli replication terminus.
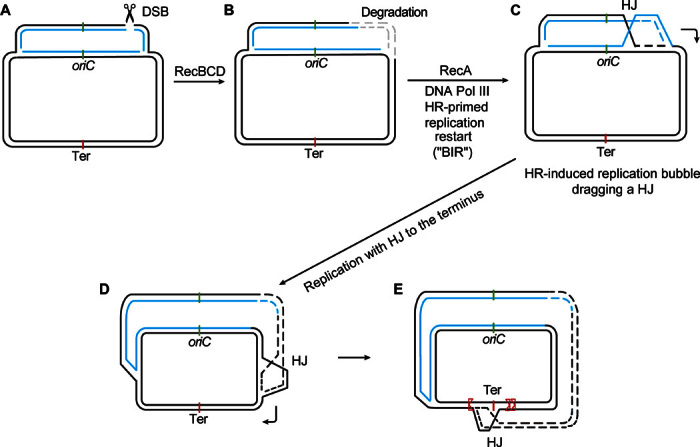
(A) to (C) incorporate ideas from Kuzminov (68), and (D) and (E) illustrate a model of Motamedi et al. (65). (A) Chi sites attenuate RecBCD double-strand exonuclease activity (40, 41) and are situated asymmetrically in the genome such that if a two-ended DSB were to occur, (B) the DNA on the replication terminus–proximal side would be likely to be degraded extensively (few active Chi’s in the ori-to-ter direction), whereas the DNA on the ori-proximal side would suffer less degradation (many active Chi’s in the ter-to-ori direction) (68). (C) Thus, the nondegraded DSB end would initiate HR repair by strand exchange that would prime a replication fork that would run in the chromosome’s natural ori-to-ter direction toward the terminus. (D and E) The model parts illustrated in (C) to (E) were offered previously by Motamedi et al. (65) in support of our observations that most DSB repair via HR requires the major replicative DNA polymerase (Pol III) and that the new strands are segregated conservatively (65), as observed subsequently in yeast BIR (98, 99). We suggested that the replication bubble proceeds toward the terminus, dragging an unresolved HJ behind it, which forces the new DNA strands out of the bubble, causing the observed conservative segregation of newly synthesized DNA strands in DSB repair replication (65). (E) With their trailing HJs, forks that begin on one side of the terminus will pause and accumulate at the terminus. Some that overshoot the midway point will be stopped at the unidirectional ter sites(s) (red, triangular sides stop oncoming forks) on the opposite side of the chromosome center point from which the bubble began. This pattern of HJ accumulation is seen in the ChIP-seq data shown in Fig. 2 (G and H).
Second, the data show skewed distributions of HJs around repairing two-ended DSBs in the E. coli chromosome with more HJs on the terminus-proximal side of the DSB than on the ori-proximal side of the DSB. That is, there are more HJs downstream than upstream of the DSB in the chromosomal replication paths (Fig. 2, G and H, and fig. S6) or “replichores.” For two different I–Sce I cleavage sites, there were more RecA- and RecB-dependent HJs terminus-proximally than ori-proximally (Fig. 2, G and H, and fig. S6). These data support the Kuzminov model (68) of asymmetrical DNA degradation and repair at DSBs in the E. coli chromosome (illustrated in Fig. 9, A to C) controlled by the asymmetrical distribution of chromosomal Chi sites. Chi sites attenuate RecBCD dsDNA exonuclease activity to allow HR end repair (40, 41) and fall asymmetrically in the E. coli genome with more active Chi sites upstream than downstream from any point in the replichores. Therefore, at any DSB in the genome, the ori-proximal DNA end is better protected from degradation by RecBCD exonuclease (more active Chi’s) and is more likely to be preserved, whereas the terminus-proximal DSB end is more likely to be degraded (few active Chi’s), shown in Fig. 9B (68). Preservation of the ori-proximal end and loss of the ter-proximal end (Fig. 9B) were proposed to cause one-ended DSB repair replication (BIR), with the direction of the repair replication preferentially from ori (the preserved end that initiates HR-mediated replication restart; Fig. 9, B and C) toward the terminus (Fig. 9, B to E). This model is compelling but untested. Our observation of more DSB-induced repair HJs terminus-proximally to DSBs (Fig. 2, G and H, and fig. S6) is predicted by the Kuzminov one-ended repair model if the HJs that accompany the strand exchange that primes BIR sometimes remain with the replication bubble, unresolved, and travel with the bubble toward the terminus (Fig. 9, C to E). The bias toward HJs ori-distally from DSBs might alternatively reflect two-ended DSB repair HJs (not shown in Fig. 9, but similar to that in Fig. 1A, iii and iv) following asymmetrical degradation (Fig. 9B). Other Chi-independent asymmetrical chromosomal features might underlie the HJ asymmetry observed (replication forks in progress, dominant directions of transcription). Additional work is needed to distinguish specific models. Regardless of the specific mechanism, the data demonstrate genome-scale control and directionality of DSB repair events along the E. coli genome.
A junction-guardian role of RecQ and its implication for BLM and RECQL4 human orthologs in RAD51-overexpressing cancers
We discovered a novel junction-guardian role for E. coli RecQ DNA helicase, ortholog of five human cancer prevention proteins (15), in promoting and preventing HJs. First, RecQ appears to act upstream of RecA loading in living cells (Fig. 4, D and E) and is required for the formation of most RecA-dependent spontaneous chromosomal HR-HJs (Fig. 4, A to C) and also for most spontaneous intrachromosomal HR events (Fig. 5B). Our data indicate that RecQ promotes HR-HJs during replication-instigated ssDNA gap repair (model illustrated in Fig. 8). Second, we also discovered that RecQ and RecJ prevent non-HR–HJs induced by RecA overproduction (Figs. 4, F and G, and 5C; model illustrated in Fig. 6B), an E. coli model of many p53-defective cancers with RAD51 overexpression (11). Our results are partly similar to previous observation of UV light–induced accumulation of plasmid DNA structures identified by bulk two-dimensional gel electrophoresis as either double-Y junctions or HJs (20), which, like our non-HR–HJ foci, were opposed by RecQ and RecJ but, unlike our results (Fig. 4, F and G), were dependent on RecF (20). Whereas those authors suggested a post-HJ role of RecQ and RecJ in removing regressed replication fork HJs, our data imply that RecQ and RecJ prevent the formation of (RecF-independent) RF HJs. That these non-HR–HJs are induced by overproduction of RecA models many p53-defective cancers with RAD51 overexpression (11).
RAD51 overexpression supports breast cancer metastases (9) and is correlated with decreased survival of lung cancer patients (10). We discovered that RecA overproduction significantly increased HR protein–independent HJs and that RecQ/J play a novel junction-guardian role in preventing these (Fig. 4, F and G; model, Fig. 6B). RecA overproduction did not cause RecF independence of HR (Fig. 4F), and HR was not correlated with increased RecA-induced HJs (Fig. 5C), such that most of the RecA overproduction–induced extra HJs (Fig. 4, F and G) are implicated to arise HR-independently. We suggest that these non-HR–HJs are regressed replication forks (Fig. 6Biii). We hypothesize that excess RecA/RAD51 causes fork regression spuriously on undamaged DNA (illustrated in Fig. 6B). RecQ and RecJ could prevent the HJ stage of fork regression by unwinding and digesting the lagging strand, as shown (Fig. 6Biv) and implicated by RecQ and RecJ biochemistry (69) and DNA degradation in cells (19, 70, 71). This dual, junction-guardian role (promoting HR-HJs, preventing RF-HJs) could be shared by one or more human RecQ orthologs. Supporting this hypothesis, we found that BLM and RECQL4 are co-overexpressed with RAD51 in the eight and in four of the eight most common cancer types, respectively (Fig. 7, fig. S12, and table S1). Moreover, the known human HJ resolution protein genes EME1 and GEN1 (72) are co–up-regulated with BLM and RAD51 (Fig. 7, fig. S12, and table S1). We suggest that at least one consequence of overexpressed RAD51 in tumors could be increased RFs, which are genome-destabilizing and cause genome evolution that could drive the cancer state (73), and that BLM and RECQL4 may prevent RF-HJs, whereas EME1 and GEN1 may cleave the RFs, creating DSB ends that cycle through repair and replication. Purified BLM can promote fork regression in solution (74), and RECQL4 promotes HR-HJs (26). Perhaps they also prevent RF-HJs in cells. Failure to remove RFs would block replication and chromosome segregation, so their removal would be expected to be selected in cancers. Further work is needed to test these hypotheses. We are working toward human cell–compatible HJ traps that may aid exploration of these and many other ideas concerning HJs and genome instability in human cancers.
MATERIALS AND METHODS
Strains, media, and growth
Strains used in this study are given in table S2. Bacteria were grown in Luria-Bertani-Herskowitz (LBH) rich medium or M9 minimal medium (75) supplemented with thiamine (10 μg/ml; vitamin B1) and 0.1% glucose or glycerol as a carbon source. Other additives were used at the following concentrations: ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), kanamycin (50 μg/ml), tetracycline (10 μg/ml), and sodium citrate (20 mM). P1 transductions were performed as described by Thomason et al. (76). Genotypes were verified by antibiotic resistance, polymerase chain reaction (PCR), and, when relevant, UV sensitivity and sequencing.
Cloning and chromosomal expression cassettes encoding RuvCGFP, RDG, and RDM
The ruvC gene (without stop codon) and gfpmut3 (77) (or mCherry) (39) gene were fused with a linking sequence (5′-GCTATCGACGAAAACAAACAGAAAGCGTTGGCGGCAGCA-3′) using the protocol described by Heckman and Pease (78) and ligated into the pET28a vector. The two mutations (D7N and E66D) of ruvCDef were generated by site-directed mutagenesis (Stratagene). The doxycycline-inducible PN25tetO promoter from pRF3 (39) was subcloned upstream of ruvC (or ruvCDefgfp or ruvCDefmCherry) to replace the T7 promoter of pET28a. A chloramphenicol resistance (cat) cassette flanked by FRT sites from pKD3 (79) was subcloned downstream from ruvC (or ruvCDefgfp or ruvCDefmCherry). To move the entire expression system into the chromosome using Red-mediated short-homology recombination (79), we inserted parts of mntH and nupC genes (~1 kb) upstream and downstream of PN25tetOruvC (or ruvCDefgfp or ruvCDefmCherry) as homologous sequences, respectively. A promoter PN25tetO-only construct was also moved into the chromosome as a control. The completed plasmid constructs are listed in table S3 (pLC1 to pLC4). Each entire expression cassette was moved separately into the chromosome of SMR19152 by Red-mediated recombineering using primers P1 and P2 (table S4) and then transduced into the strains listed in table S2. SMR19152 encodes constitutively produced TetR protein that represses the PN25tetO promoter in the absence of the inducer doxycycline (80). All constructs were verified by PCR, antibiotic resistance, and sequencing.
Protein purification
The ruvCgfp and ruvCDefgfp genes were subcloned into pET28a expression vectors and then transformed into the BL21 derivative strain (T7 Express lysY/Iq Competent E. coli, New England Biolabs). After induction with 0.1 mM IPTG, the proteins with an N-terminal His6-tag were produced at 20°C for 16 hours and pelleted by centrifugation at 7000 rpm for 30 min. The cell pellet was resuspended in lysis buffer [50 mM Hepes (pH 8.0), 300 mM NaCl, 5 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride] and disrupted by sonication (10 times for 30 s, on/off at output 5; XL-2000, Misonix). The total cell lysate was centrifuged at 30,000g for 40 min, and the soluble recombinant protein was purified by immobilized metal ion chromatography with a Ni-NTA column (Qiagen). After washing with wash buffer (lysis buffer with 50 mM imidazole added), the protein was eluted with elution buffer (lysis buffer with 500 mM imidazole) and further desalted against the storage buffer [20 mM Hepes (pH 8.0) and 20 mM NaCl]. The protein was concentrated to about 2 mg/ml and stored at −80°C. The Flp protein was purified to near homogeneity using DNA affinity enrichment as the final step (81).
Western blot
Proteins were separated by 10% SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membrane according to the manufacturer’s instructions (Amersham, GE Healthcare). The membranes were blocked with ECL Prime blocking agent (GE Healthcare) and probed with primary mouse anti-RuvC monoclonal immunoglobulin G (IgG) antibodies (Santa Cruz Biotechnology). The membrane was further probed with secondary polyclonal goat anti-IgG antibody (Bethyl Laboratories) and visualized by scanning in a multicolor imager Typhoon detection system (GE Healthcare).
UV sensitivity test
Saturated overnight, LBH cultures were diluted 100-fold in LBH medium and grown at 37°C for 1.5 hours, at which time doxycycline (100 ng/ml) was added to induce protein production. After 1 hour of induction, cells were plated on LBH solid medium containing doxycycline (10 ng/ml), and the plated cells were irradiated with different doses of UVC light using a Stratalinker 1800 UV crosslinker (Stratagene) and then incubated in the dark overnight at 37°C for colony quantification. Control cultures without doxycycline induction were treated identically.
Assembly of synthetic HJs
The oligonucleotides used for the assembly of HJs (HJa-HJc) that served as substrates in binding and cleavage assays (Fig. 1E and fig. S2, A to C) are listed in table S4. For HJa, assembled from oligonucleotides HJa1 to HJa4, the bases that form a 2-bp homology core in the central region of HJ for RuvC cleavage are shown in bold. For HJb, composed of oligonucleotides HJb1 to HJb4, the bases that form the recognition sequence for Flp are shown in bold. HJc, constructed from oligonucleotides HJc1 to HJc4, was the substrate for restriction enzyme digestion assays. The sequences that form the recognition sites for restriction enzymes in HJc are shown in bold. The four arms of HJb were equal in length, as were those of HJc, and surrounded an immobile branch point. To assemble a given junction, equimolar amounts of the four requisite oligonucleotides were combined in 50 mM tris-HCl (pH 8.0), 5 mM EDTA, and 1 mM dithiothreitol (DTT), and the mixture was placed for 5 min in a water bath maintained at 80°C. Heat was turned off, and the bath was allowed to cool slowly to room temperature on the bench top. Nearly quantitative assembly of the junction was verified by gel electrophoresis.
Biochemical HJ-binding assays
The binding and competition assays were performed in 50 mM tris-HCl (pH 8.0), 5 mM EDTA, 1 mM DTT, 5% glycerol, and bovine serum albumin (100 mg/ml), containing 2 pmol of the HJb per incubation mixture. RuvCGFP or RDG gave complete binding of the junction at a molar ratio of 1:10 (junction to protein). For the standard binding assays, incubations were carried out on ice for 15 min after protein addition. For the competition assays, the junction was preincubated with RDG on ice for 15 min (1:10, junction to protein) before addition of Flp (1:4 or 1:8, junction to protein) and incubated for an additional 15 min. The binding mixtures were fractionated by electrophoresis in a 3% agarose gel in 6.7 mM tris-HCl (pH 8.0), 3.3 mM sodium acetate, and 2 mM EDTA at 4°C for 75 min (10 V cm−1) with continuous recirculation of the buffer. The DNA bands were visualized under UV light after staining the gel with ethidium bromide. In some assays, the junction-bound proteins were electrotransferred to a PVDF membrane using a semidry transfer cell (Bio-Rad, 170-3940) and probed with antibodies to GFP (to detect RDG) or with antibodies to native Flp. The detection reaction used peroxidase-conjugated secondary antibodies (anti-mouse for GFP; anti-rabbit for Flp) in conjunction with an enhanced chemiluminescence-based substrate (Pierce). A mouse monoclonal antibody to GFP was purchased from Abcam. A polyclonal antibody to Flp was raised in rabbits against a synthetic Flp peptide. For protein detection, the GFP and Flp antibodies were diluted 1:1000 and 1:5000, respectively.
Biochemical HJ cleavage assays
The reaction mixtures (20 μl) containing 0.25 μM HJa and 1 μM purified protein in 20 mM Hepes (pH 8), 10 mM MgCl2, and 5% glycerol were incubated for 1 hour at 37°C before the addition of 5 μl of stopping buffer [100 mM EDTA (pH 8), 1% SDS, 20% glycerol, 0.1% bromophenol blue, and proteinase K (5 μg/μl)] followed by incubation at 55°C for 30 min. The samples were analyzed by 3% agarose gel electrophoresis at 100 V.
Restriction enzyme digestion in the presence or absence of RuvCGFP or RDG
The DNA substrate was a HJ containing the Eco RI recognition sequence in one of the arms. Digestion was performed on the free junction or after binding by RuvCGFP or RDG. Each reaction mixture contained 2 pmol of the junction and 2.5 U of Eco RI (New England Biolabs). Prebinding of the junction by RuvCGFP or RDG was carried out in the digestion buffer (supplied by New England Biolabs) for 10 min on ice before enzyme addition. The molar ratio of the junction to RuvCGFP or RDG was 1:10. Immediately following enzyme addition, the reaction mixtures were transferred to a 37°C water bath. Reactions were stopped by the addition of 0.2% SDS, and samples were analyzed by electrophoresis in 3% agarose gel at room temperature (10 V cm−1). DNA was visualized by ethidium bromide staining. Quantification in plots shows means ± SEM from three independent experiments.
Quantification of DNA band intensities
DNA bands from gels were analyzed using Quantity One software from Bio-Rad.
Quantitative P1 transduction and intrachromosomal direct repeat HR assays
Quantitative P1 transduction assays were as described by Magner et al. (18), with the modification that saturated overnight liquid LBH cultures were diluted 100 times in LBH medium with or without doxycycline (100 ng/ml) to induce RDG and grown at 37°C for 3 hours. Phage P1 grown on strain SMR6263 was used to transduce leu::Tn10 into recipient strains. The transductants were plated on the Tet plates with or without doxycycline to induce RDG. The frequencies of TetR transductants were determined by three P1 transductions with recipient cells in excess (multiplicities of infection of <0.01 phage per recipient cell). The direct repeat HR assay was as described by Corre et al. (82) with the modification that some of the isogenic strains additionally carry plasmid vector or isogenic RecA-overproducing plasmid and were or were not induced to overproduce RecA by adding 1 mM IPTG.
Microscopy and focus quantification
Images were visualized with an inverted DeltaVision Core Image Restoration Microscope (GE Healthcare) with a 100× UPlan S Apochromat (numerical aperture, 1.4) objective lens (Olympus) and a CoolSNAP HQ2 camera (Photometrics). Captured images were chosen randomly under the microscope. The images were taken with Z stacks (0.15-μm intervals) and then deconvoluted (DeltaVision SoftWoRx software) to see the whole cell for precise quantification of foci in each cell. For each independent experiment, >1000 cells (about 3000 cells for ΔrecA and ΔrecB strains) were counted using ImageJ software with visual inspection in each of three independent experiments.
Spontaneous and γ-radiation– and I–Sce I–induced foci
For the measurement of spontaneous HJ focus formation, saturated overnight cultures were diluted 1:100 in fresh LBH medium or 1:25 dilution in M9-glucose medium and incubated at 37°C for 1.5 hours before adding doxycycline (100 ng/ml). Cultures were incubated for another 4 hours before visualizing under the microscope. Growth curves of different strain backgrounds were similar (fig. S9). For I–Sce I–induced foci, saturated overnight cultures started in medium containing 0.1% glucose were diluted 1:100 or 1:25 in fresh LBH or M9-glycerol medium supplied with proline (50 μg/ml), respectively, and incubated at 37°C for 1.5 hours before adding doxycycline (100 ng/ml). After 1 hour, 0.005% arabinose was added and incubation was continued for another 3 hours. The procedure of γ-radiation–induced focus formation was performed as previously described by Shee et al. (39), except for using different doses of γ-radiation.
For experiments with temperature-inducible Gam production, cultures were inoculated into rich medium from saturated overnight cultures at 30°C for 1.5 hours before shifting to 37°C (to induce Gam production) and adding doxycycline (100 ng/ml; to induce RDG). Spontaneous foci were counted after another 4 hours of incubation. For Gam production under DSB-inducing conditions, the temperature was shifted and doxycycline was added for 1 hour before adding arabinose to induce I–Sce I to generate DSBs. Foci were counted after another 3 hours of incubation at 37°C. RecA was overproduced by adding 1 mM IPTG. For dnaATS temperature shift experiments, cultures were inoculated into rich medium from saturated overnight cultures at 30°C for 1.5 hours before shifting to 42°C (to inactivate DnaA) and adding doxycycline (100 ng/ml; to induce RDG). Spontaneous foci were counted after another 4 hours of incubation.
Microfluidics and time-lapse fluorescence microscopy
The procedure was as described by Shee et al. (39), except for a slight difference in growth condition. Saturated cultures of SMR19382 were diluted 100-fold in M9 glucose medium supplemented with vitamin B1, 0.5% casamino acid, and doxycycline (10 ng/ml) and grown at 37°C for 1 hour before loading cells into the microfluidic chamber (time 0). For the next 7 hours, cells were bathed with the same medium, but with only doxycycline (2 ng/ml) to allow division, then switched at 7 hours to the same medium without glucose, and bathed until 18 hours. The number of cell divisions and the appearance of RDG foci were captured using time-lapse microscopic photography throughout the experiment.
ChIP-seq library preparation and sequencing
Cell cultures were grown in LBH with 0.1% glucose to saturation overnight, then diluted 100-fold into 100 ml of LBH in 500-ml flasks, and incubated at 37°C for 2.5 hours before the addition of doxycycline (100 ng/ml) to induce RDG production. After 30 min, 0.005% arabinose was added to induce I–Sce I expression. After another 2.5 hours of incubation at 37°C, proteins and DNA were cross-linked by the addition of 1% formaldehyde for 30 min at room temperature. The cross-linking was quenched by the addition of 0.5 M glycine, and cells were harvested by centrifugation and washed once with tris-buffered saline. Cells were lysed as described by Bonocora and Wade (83) with lysis buffer containing lysozyme (4 mg/ml). Lysates were sonicated using the Bioruptor Pico (Diagenode) until most of the DNA fragments were between 300 and 500 bp. Ribonuclease A was added to eliminate RNA-related interactions. The immunoprecipitation and library preparation were performed at the same time as described by Bonocora and Wade (83). Sequencing was performed on an Illumina MiSeq.
Whole-genome sequencing
Cultures were grown as described in ChIP-seq library preparation, except that before that protocol’s cross-linking step, cells were collected by centrifugation and genomic DNA was extracted and purified using the DNeasy Blood & Tissue Kit (Qiagen). Libraries were prepared using Nextera XT kits (Illumina), and sequencing was performed on an Illumina MiSeq.
Sequencing data analyses and deposit
Before mapping, FASTX-Toolkit (v0.0.14) was used to preprocess the sequences: Adaptor sequences were removed, and reads were trimmed and filtered according to quality. The sequence alignment was performed by BWA-MEM (v0.7.12) with –M option to mark shorter split hits as secondary alignments and other parameters as default (84). Depending on the strain background, reads were mapped to either the W3110 genome [National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) Database accession: NC_007779.1] or the MG1655 genome (NCBI RefSeq accession: NC_000913.3). Reads that had multiple primary hits or low mapping quality were discarded. Potential PCR duplicates were removed by retaining only one pair of reads with the highest mapping quality when multiple read pairs were mapped to identical external coordinates (Picard Tool MarkDuplicates). BedGraph files that report the physical genomic coverage (taking into account the unsequenced part between read pairs) in each 2000-bp bin were generated from BAM files using deepTools (85). For ChIP-seq data, the read counts in the bedGraph files were normalized to the median coverage. For whole-genome sequencing data, the read counts in each bin were first normalized against total read counts, and then the log2 ratios of DSB and no-DSB samples were calculated. Plots were generated by R software. Genomic regions that contain ribosomal RNA gene clusters had very few uniquely mapped reads and were thus eliminated from the plots. All sequencing data are available in the European Nucleotide Archive (ENA) under study accession no. PRJEB14145.
Chromosome copy number determinations
Replication origins were counted to estimate the number of forks per cell by using rifampicin runoff analyses, as described by Joshi et al. (56) and Bremmer and Dennis (86). Saturated overnight cultures were diluted 1:1000 or 1:750 in LBH glucose or M9 glucose medium, respectively, containing doxycycline (100 ng/ml). After reaching OD600 (optical density at 600 nm) = 0.1, rifampicin and cephalexin were added to the culture, which was allowed to complete ongoing rounds of replication (runoff). Cells were fixed and stained with 4′,6-diamidino-2-phenylindole and then analyzed on a BD Biosciences LSRFortessa cytometer. Three independent experiments were performed and results were averaged.
Phage λred gam plaque assay for phage Mu Gam function
The phage λ plaque assay was described by Shee et al. (39), except for induction of Gam protein by shifting temperature. The expression of Gam was controlled by phage λ PR promoter, next to the λcIts857 gene, encoding a temperature-sensitive CI transcriptional repressor protein that represses λ PR at ≤33°C and allows transcription from λ PR at ≥37°C (39). Saturated tryptone broth (TB) cultures of E. coli grown at 30°C were diluted 1:10 in fresh TB with 0.2% maltose, 5 mM MgSO4, thymine (10 μg/ml), and vitamin B1 (10 μg/ml) and grown for half an hour at 30°C before shifting temperature to 37°C to induce production of Mu Gam (or were grown at 30°C continuously). After 2.5 hours of induction, an equal volume of 10 mM tris and 10 mM Mg (TM) buffer (pH 7.5) was added, and cells were mixed with an appropriate volume of λred gam Chi0 suspension, adsorbed without shaking for 10 min at room temperature, and then plated with the addition of 2.5 ml of molten soft BBL agar [1% BBL trypticase peptone, 0.1 M NaCl, 0.7% agar (pH 7.5)] onto BBL plates (same medium solidified with 1% agar). The plates were incubated overnight at 37°C or for 24 hours at 30°C before observing the size of plaques (fig. S13). The production of Mu Gam allows large plaque formation by λred gam (39) [reviewed by Smith (87)].
Cancer RNA statistical analyses
Cancer patient RNA sequencing correlation analyses were performed using Spearman’s rank correlation on data from cBioPortal (88, 89). RNA levels of specific genes in patient samples for the eight most common cancer types—breast, lung, acute myeloid leukemia, colon, kidney, thyroid, bladder, and prostate—were compared with those in the cBioPortal reference population to generate Z scores, and the Z scores were compared for each gene of interest against the similarly generated Z scores of another gene (for example, BLM mRNA versus RAD51 RNA; shown in Fig. 7A). The reference populations for each gene/cancer are all cancer samples that are diploid for the gene in question (by default for mRNA). The returned value indicates the number of SDs away from the mean of expression in the reference population (Z score). This measure is useful for determining whether a gene is up- or down-regulated relative to all other tumor samples. As detailed in table S1, the number of patient sample data sets per cancer type ranged from 129 to 1100 (table S1). Spearman’s correlation coefficient was computed to assess the relationship between mRNA levels among genes of interest.
Other statistical analyses
Wet-bench experiments: All were performed at least three times independently, and a two-tailed unpaired t test was used to determine significant differences unless otherwise specified. Error bars represent 1 SEM except where otherwise indicated. Pearson’s correlation coefficient was computed to assess the relationship between RDG foci/cell and different doses of γ-irradiation. Changepoint analyses were used to detect peaks in ChIP-seq data by detecting the inflection points of curves and were performed using the “changepoint” R package (90).
Supplementary Material
Acknowledgments
We thank J. D. Wang and S. J. Sandler for providing E. coli strains with a mutant recA-gfp fusion; C. Shee for helpful discussion; F. Gu for cytometry help; M. A. Mancini, F. Stossi, R. D. Danderkar for help with microscopic imaging; I. Golding and L. McLane for help with microfluidics; and J. Gibson, S. G. Kozmin, K. M. Miller, F. W. Stahl, and three anonymous reviewers for improving the manuscript. Funding: This work was initiated with support from NIH Director’s Pioneer Award DP1-CA174424 (to S.M.R.) and developed via a gift from the W.M. Keck Foundation (to S.M.R.). The work was also supported by the National Aeronautics and Space Administration (NASA) through the NASA Astrobiology Institute under Cooperative Agreement No. NNA13AA91A issued through the Science Mission Directorate (to P.J.H.); Cancer Prevention and Research Institute of Texas (CPRIT) grant RP140553 (to S.M.R.); CPRIT Baylor College of Medicine (BCM) Comprehensive Cancer Training Program Postdoctoral Fellowship grant RP160283 (to D.M.F.); NIH R01 grants CA190635 (to L.L.), GM102679 (to D.B.), GM106373 (to P.J.H.), and GM88653 (to C.H.); NSF grant MCB1049925 (to M.J.); Robert F. Welch Foundation Award F-1274 (to M.J.); the BCM Cytometry and Cell Sorting Core with funding from the NIH (P30-AI036211, P30-CA125123, and S10-RR024574); and the BCM Integrated Microscopy Core with funding from the NIH (HD007495, DK56338, and CA125123), CPRIT (RP150578), the Dan L Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. Author contributions: S.M.R. conceived the study. J.X., L.-T.C., Q.M., L.L., D.B., P.J.H., C.H., M.J., and S.M.R. designed the study, analyzed the data, and wrote the paper. L.-T.C., J.X., Q.M., C.-H.M., J.A.H., H.-Y.L., D.M., J.P.P., D.M.F., H.M.H., M.R., R.B.N., and X.S. performed the experiments. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Sequencing data are available in the ENA under study accession no. PRJEB14145. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601605/DC1
text S1. DNA repair by HR and RuvC specificity for four-way junctions.
text S2. About half of HJs are detected as RDG foci in living E. coli cells, and half of Gam-detectable DSBs result in HJ foci.
fig. S1. Design of a catalytically inactive RuvC for trapping HJs: RuvCDef.
fig. S2. Purified RDG binds, does not cleave, and inhibits action of other proteins on synthetic HJs in solution.
fig. S3. RDG inhibition of resistance to UV light requires induction of transcription of the ruvCDefgfp gene.
fig. S4. Titration of RuvC with RDG levels shows minimum RDG/RuvC ratio at which RDG outcompetes RuvC, preventing repair in living cells.
fig. S5. Spontaneous RDM foci colocalize with RecA-GFP strand exchange protein in E. coli cells.
fig. S6. RDG ChIP-seq localization requires DSBs and specific RuvC antibody.
fig. S7. Spontaneous RuvCGFP and RDG foci overlap with DNA stain.
fig. S8. Spontaneous RDG HR/HJ foci per cell correlate with varying chromosome and replication fork numbers.
fig. S9. Similar growth rates of various mutant strains used.
fig. S10. Reduced spontaneous RDG/HJ focus levels in recF, recQ, and recJ cells are restored by supplying RecF, RecQ, and RecJ from plasmids.
fig. S11. RecA overproduction is induced after RDG accumulation in cells.
fig. S12. Increased EME1 and GEN1 HJ resolvase mRNAs in BLM-overexpressing human cancers of the eight, and six of the eight, most common cancer types, respectively.
fig. S13. Validation of Mu Gam protein function.
table S1. Pairwise mRNA level correlation between human RAD51 with human HJ resolvases and RecQ orthologs in the eight most common cancer types.
table S2. E. coli K12 strains and plasmids used in this study.
table S3. Names and locations of new I-sites and alleles.
table S4. Oligonucleotides used in this study.
REFERENCES AND NOTES
- 1.Jeggo P. A., Löbrich M., How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 471, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Coghlan A., Eichler E. E., Oliver S. G., Paterson A. H., Stein L., Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends Genet. 21, 673–682 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Vink C., Rudenko G., Seifert H. S., Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol. Rev. 36, 917–948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenter A. L., Class-switch recombination: After the dawn of AID. Curr. Opin. Immunol. 15, 190–198 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai A. G., Lieber M. R., Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics 11 (suppl. 1), S1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz-Galván S., Jimeno S., Rothstein R., Aguilera A., Histone H3K56 acetylation, Rad52, and non-DNA repair factors control double-strand break repair choice with the sister chromatid. PLOS Genet. 9, e1003237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen M. F., Koufos A., Gallie B. L., Phillips R. A., Fodstad Ø., Brøgger A., Gedde-Dahl T., Cavenee W. K., Osteosarcoma and retinoblastoma: A shared chromosomal mechanism revealing recessive predisposition. Proc. Natl. Acad. Sci. U.S.A. 82, 6216–6220 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta A., Haber J. E., Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 6, a016428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiegmans A. P., Al-Ejeh F., Chee N., Yap P.-Y., Gorski J. J., Da Silva L., Bolderson E., Chenevix-Trench G., Anderson R., Simpson P. T., Lakhani S. R., Khanna K. K., Rad51 supports triple negative breast cancer metastasis. Oncotarget 5, 3261–3272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao G.-B., Wu Y.-L., Yang X.-N., Zhong W.-Z., Xie D., Guan X.-Y., Fischer D., Kolberg H.-C., Kruger S., Stuerzbecher H.-W., High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer 93, 137–143 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein H. L., The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 7, 686–693 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann A. R., Fuchs R. P., Gaps and forks in DNA replication: Rediscovering old models. DNA Repair 5, 1495–1498 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Sonoda E., Sasaki M. S., Buerstedde J.-M., Bezzubova O., Shinohara A., Ogawa H., Takata M., Yamaguchi-Iwai Y., Takeda S., Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 17, 598–608 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J., The importance of repairing stalled replication forks. Nature 404, 37–41 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Larsen N. B., Hickson I. D., RecQ helicases: Conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 767, 161–184 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Harmon F. G., Kowalczykowski S. C., RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 12, 1134–1144 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suski C., Marians K. J., Resolution of converging replication forks by RecQ and topoisomerase III. Mol. Cell 30, 779–789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magner D. B., Blankschien M. D., Lee J. A., Pennington J. M., Lupski J. R., Rosenberg S. M., RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol. Cell 26, 273–286 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Courcelle J., Hanawalt P. C., RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 262, 543–551 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Courcelle J., Donaldson J. R., Chow K.-H., Courcelle C. T., DNA damage-induced replication fork regression and processing in Escherichia coli. Science 299, 1064–1067 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Saintigny Y., Makienko K., Swanson C., Emond M. J., Monnat R. J. Jr, Homologous recombination resolution defect in Werner syndrome. Mol. Cell. Biol. 22, 6971–6978 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gangloff S., Soustelle C., Fabre F., Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 25, 192–194 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Zhu Z., Chung W.-H., Shim E. Y., Lee S. E., Ira G., Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mimitou E. P., Symington L. S., Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gravel S., Chapman J. R., Magill C., Jackson S. P., DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu H., Shamanna R. A., Keijzers G., Anand R., Rasmussen L. J., Cejka P., Croteau D. L., Bohr V. A., RECQL4 promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 16, 161–173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saponaro M., Kantidakis T., Mitter R., Kelly G. P., Heron M., Williams H., Söding J., Stewart A., Svejstrup J. Q., RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Futami K., Ogasawara S., Goto H., Yano H., Furuichi Y., RecQL1 DNA repair helicase: A potential tumor marker and therapeutic target against hepatocellular carcinoma. Int. J. Mol. Med. 25, 537–545 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Croteau D. L., Singh D. K., Hoh Ferrarelli L., Lu H., Bohr V. A., RECQL4 in genomic instability and aging. Trends Genet. 28, 624–631 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fogg J. M., Schofield M. J., White M. F., Lilley D. M. J., Sequence and functional-group specificity for cleavage of DNA junctions by RuvC of Escherichia coli. Biochemistry 38, 11349–11358 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Dickman M. J., Ingleston S. M., Sedelnikova S. E., Rafferty J. B., Lloyd R. G., Grasby J. A., Hornby D. P., The RuvABC resolvasome. Eur. J. Biochem. 269, 5492–5501 (2002). [DOI] [PubMed] [Google Scholar]
- 32.West S. C., Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 31, 213–244 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Seigneur M., Bidnenko V., Ehrlich S. D., Michel B., RuvAB acts at arrested replication forks. Cell 95, 419–430 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Schlacher K., Wu H., Jasin M., A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robu M. E., Inman R. B., Cox M. M., RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 8211–8218 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ringrose L., Lounnas V., Ehrlich L., Buchholz F., Wade R., Stewart A. F., Comparative kinetic analysis of FLP and cre recombinases: Mathematical models for DNA binding and recombination. J. Mol. Biol. 284, 363–384 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Mahdi A. A., Sharples G. J., Mandal T. N., Lloyd R. G., Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J. Mol. Biol. 257, 561–573 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Gumbiner-Russo L. M., Lombardo M.-J., Ponder R. G., Rosenberg S. M., The TGV transgenic vectors for single-copy gene expression from the Escherichia coli chromosome. Gene 273, 97–104 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Shee C., Cox B. D., Gu F., Luengas E. M., Joshi M. C., Chiu L.-Y., Magnan D., Halliday J. A., Frisch R. L., Gibson J. L., Nehring R. B., Do H. G., Hernandez M., Li L., Herman C., Hastings P. J., Bates D., Harris R. S., Miller K. M., Rosenberg S. M., Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife 2, e01222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuzminov A., Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63, 751–813 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dillingham M. S., Kowalczykowski S. C., RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 72, 642–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morimatsu K., Kowalczykowski S. C., RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: A universal step of recombinational repair. Mol. Cell 11, 1337–1347 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Sakai A., Cox M. M., RecFOR and RecOR as distinct RecA loading pathways. J. Biol. Chem. 284, 3264–3272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson F. E., West S. C., Substrate specificity of the Escherichia coli RuvC protein. Resolution of three- and four-stranded recombination intermediates. J. Biol. Chem. 269, 5195–5201 (1994). [PubMed] [Google Scholar]
- 45.van Gool A. J., Shah R., Mézard C., West S. C., Functional interactions between the Holliday junction resolvase and the branch migration motor of Escherichia coli. EMBO J. 17, 1838–1845 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renzette N., Gumlaw N., Nordman J. T., Krieger M., Yeh S.-P., Long E., Centore R., Boonsombat R., Sandler S. J., Localization of RecA in Escherichia coli K-12 using RecA–GFP. Mol. Microbiol. 57, 1074–1085 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Wang X., Sherratt D. J., Independent segregation of the two arms of the Escherichia coli ori region requires neither RNA synthesis nor MreB dynamics. J. Bacteriol. 192, 6143–6153 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson D. G., Kowalczykowski S. C., The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell 90, 77–86 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Kantake N., Madiraju M. V. V. M., Sugiyama T., Kowalczykowski S. C., Escherichia coli RecO protein anneals ssDNA complexed with its cognate ssDNA-binding protein: A common step in genetic recombination. Proc. Natl. Acad. Sci. U.S.A. 99, 15327–15332 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta S., Yeeles J. T. P., Marians K. J., Regression of replication forks stalled by leading-strand template damage: II. Regression by RecA is inhibited by SSB. J. Biol. Chem. 289, 28388–28398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Churchill J. J., Anderson D. G., Kowalczykowski S. C., The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of χ, resulting in constitutive recombination activation. Genes Dev. 13, 901–911 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Badie S., Escandell J. M., Bouwman P., Carlos A. R., Thanasoula M., Gallardo M. M., Suram A., Jaco I., Benitez J., Herbig U., Blasco M. A., Jonkers J., Tarsounas M., BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 17, 1461–1469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spies M., Kowalczykowski S. C., The RecA binding locus of RecBCD is a general domain for recruitment of DNA strand exchange proteins. Mol. Cell 21, 573–580 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Prakash R., Zhang Y., Feng W., Jasin M., Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 7, a016600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirota Y., Ryter A., Jacob F., Thermosensitive mutants of E. coli affected in the processes of DNA synthesis and cellular division. Cold Spring Harbor Symp. Quant. Biol. 33, 677–693 (1968). [DOI] [PubMed] [Google Scholar]
- 56.Joshi M. C., Magnan D., Montminy T. P., Lies M., Stepankiw N., Bates D., Regulation of sister chromosome cohesion by the replication fork tracking protein SeqA. PLOS Genet. 9, e1003673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Handa N., Morimatsu K., Lovett S. T., Kowalczykowski S. C., Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23, 1234–1245 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Avery L., Wasserman S., Ordering gene function: The interpretation of epistasis in regulatory hierarchies. Trends Genet. 8, 312–316 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jensen R. B., Carreira A., Kowalczykowski S. C., Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J., Doty T., Gibson B., Heyer W.-D., Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 17, 1260–1262 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carreira A., Kowalczykowski S. C., Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 108, 10448–10453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peña C., García J. M., García V., Silva J., Domínguez G., Rodríguez R., Maximiano C., García de Herreros A., Muñoz A., Bonilla F., The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 119, 2098–2104 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Farrugia M. K., Sharma S. B., Lin C.-C., McLaughlin S. L., Vanderbilt D. B., Ammer A. G., Salkeni M. A., Stoilov P., Agazie Y. M., Creighton C. J., Ruppert J. M., Regulation of anti-apoptotic signaling by Kruppel-like factors 4 and 5 mediates lapatinib resistance in breast cancer. Cell Death Dis. 6, e1699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosig G., Gewin J., Luder A., Colowick N., Vo D., Two recombination-dependent DNA replication pathways of bacteriophage T4, and their roles in mutagenesis and horizontal gene transfer. Proc. Natl. Acad. Sci. U.S.A. 98, 8306–8311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Motamedi M. R., Szigety S. K., Rosenberg S. M., Double-strand-break repair recombination in Escherichia coli: Physical evidence for a DNA replication mechanism in vivo. Genes Dev. 13, 2889–2903 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilson M. A., Kwon Y., Xu Y., Chung W.-H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P., Ira G., Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lesterlin C., Barre F.-X., Cornet F., Genetic recombination and the cell cycle: What we have learned from chromosome dimers. Mol. Microbiol. 54, 1151–1160 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Kuzminov A., Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol. 16, 373–384 (1995). [DOI] [PubMed] [Google Scholar]
- 69.Hishida T., Han Y.-W., Shibata T., Kubota Y., Ishino Y., Iwasaki H., Shinagawa H., Role of the Escherichia coli RecQ DNA helicase in SOS signaling and genome stabilization at stalled replication forks. Genes Dev. 18, 1886–1897 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sangurdekar D. P., Hamann B. L., Smirnov D., Srienc F., Hanawalt P. C., Khodursky A. B., Thymineless death is associated with loss of essential genetic information from the replication origin. Mol. Microbiol. 75, 1455–1467 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Fonville N. C., Bates D., Hastings P. J., Hanawalt P. C., Rosenberg S. M., Role of RecA and the SOS response in thymineless death in Escherichia coli. PLOS Genet. 6, e1000865 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wyatt H. D. M., Sarbajna S., Matos J., West S. C., Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Branzei D., Foiani M., Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Ralf C., Hickson I. D., Wu L., The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 281, 22839–22846 (2006). [DOI] [PubMed] [Google Scholar]
- 75.J. H. Miller, A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria (Cold Spring Harbor Laboratory Press, 1992). [Google Scholar]
- 76.Thomason L. C., Costantino N., Court D. L., E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. Chapter 1, Unit 1.17 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Cormack B. P., Valdivia R. H., Falkow S., FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 78.Heckman K. L., Pease L. R., Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nishihara K., Kanemori M., Kitagawa M., Yanagi H., Yura T., Chaperone coexpression plasmids: Differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 64, 1694–1699 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma C.-H., Rowley P. A., Macieszak A., Guga P., Jayaram M., Active site electrostatics protect genome integrity by blocking abortive hydrolysis during DNA recombination. EMBO J. 28, 1745–1756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corre J., Cornet F., Patte J., Louarn J. M., Unraveling a region-specific hyper-recombination phenomenon: Genetic control and modalities of terminal recombination in Escherichia coli. Genetics 147, 979–989 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonocora R. P., Wade J. T., ChIP-seq for genome-scale analysis of bacterial DNA-binding proteins. Methods Mol. Biol. 1276, 327–340 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Li H., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997, 1–3 (2013). [Google Scholar]
- 85.Ramírez F., Dündar F., Diehl S., Grüning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.H. Bremmer, P. P. Dennis, Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (American Society for Microbiology Press, 1987), pp. 1327–1342. [Google Scholar]
- 87.G. R. Smith, in General Recombination, R. W. Hendrix, J. W. Roberts, F. W. Stahl, R. A. Weisberg, Eds. (Cold Spring Harbor Laboratory Press, 1983) vol. 13, pp. 175–209. [Google Scholar]
- 88.Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N., Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N., The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Killick R., Eckley I. A., Changepoint: An R package for changepoint analysis. J. Stat. Softw. 58, 1–19 (2014). [Google Scholar]
- 91.Whitby M. C., Vincent S. D., Lloyd R. G., Branch migration of Holliday junctions: Identification of RecG protein as a junction specific DNA helicase. EMBO J. 13, 5220–5228 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Williams J. G., Radding C. M., Partial purification and properties of an exonuclease inhibitor induced by bacteriophage Mu-1. J. Virol. 39, 548–558 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abraham Z. H., Symonds N., Purification of overexpressed gam gene protein from bacteriophage Mu by denaturation-renaturation techniques and a study of its DNA-binding properties. Biochem. J. 269, 679–684 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Postow L., Ullsperger C., Keller R. W., Bustamante C., Vologodskii A. V., Cozzarelli N. R., Positive torsional strain causes the formation of a four-way junction at replication forks. J. Biol. Chem. 276, 2790–2796 (2001). [DOI] [PubMed] [Google Scholar]
- 95.Rupp W. D., Howard-Flanders P., Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 31, 291–304 (1968). [DOI] [PubMed] [Google Scholar]
- 96.Krejci L., Sung P., RPA not that different from SSB. Structure 10, 601–602 (2002). [DOI] [PubMed] [Google Scholar]
- 97.E. C. Friedberg, G. C. Walker, W. Siede, R. D. Wood, DNA Repair and Mutagenesis (ASM Press, 2005). [Google Scholar]
- 98.Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J. E., Lobachev K. S., Malkova A., Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Donnianni R. A., Symington L. S., Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 110, 13475–13480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blackwood J. K., Rzechorzek N. J., Bray S. M., Maman J. D., Pellegrini L., Robinson N. P., End-resection at DNA double-strand breaks in the three domains of life. Biochem. Soc. Trans. 41, 314–320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bennett R. J., Dunderdale H. J., West S. C., Resolution of Holliday junctions by RuvC resolvase: Cleavage specificity and DNA distortion. Cell 74, 1021–1031 (1993). [DOI] [PubMed] [Google Scholar]
- 102.Bonura T., Smith K. C., Sensitization of Escherichia coli C to gamma-radiation by 5-bromouracil incorporation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 32, 457–464 (1977). [DOI] [PubMed] [Google Scholar]
- 103.Kuzminov A., Homologous recombination—Experimental systems, analysis, and significance. EcoSal Plus 4, 10.1128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ariyoshi M., Vassylyev D. G., Iwasaki H., Nakamura H., Shinagawa H., Morikawa K., Atomic structure of the RuvC resolvase: A Holliday junction-specific endonuclease from E. coli. Cell 78, 1063–1072 (1994). [DOI] [PubMed] [Google Scholar]
- 105.Saito A., Iwasaki H., Ariyoshi M., Morikawa K., Shinagawa H., Identification of four acidic amino acids that constitute the catalytic center of the RuvC Holliday junction resolvase. Proc. Natl. Acad. Sci. U.S.A. 92, 7470–7474 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ghosh A. S., Young K. D., Helical disposition of proteins and lipopolysaccharide in the outer membrane of Escherichia coli. J. Bacteriol. 187, 1913–1922 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morigen O., Løbner-Olesen A., Skarstad K., Titration of the Escherichia coli DnaA protein to excess datA sites causes destabilization of replication forks, delayed replication initiation and delayed cell division. Mol. Microbiol. 50, 349–362 (2003). [DOI] [PubMed] [Google Scholar]
- 108.Saka K., Tadenuma M., Nakade S., Tanaka N., Sugawara H., Nishikawa K., Ichiyoshi N., Kitagawa M., Mori H., Ogasawara N., Nishimura A., A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res. 12, 63–68 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Cherepanov P. P., Wackernagel W., Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995). [DOI] [PubMed] [Google Scholar]
- 110.Gordon A. J. E., Satory D., Wang M., Halliday J. A., Golding I., Herman C., Removal of 8-oxo-GTP by MutT hydrolase is not a major contributor to transcriptional fidelity. Nucleic Acids Res. 42, 12015–12026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cairns J., Foster P. L., Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128, 695–701 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tehranchi A. K., Blankschien M. D., Zhang Y., Halliday J. A., Srivatsan A., Peng J., Herman C., Wang J. D., The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell 141, 595–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H., Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Damagnez V., Doutriaux M. P., Radman M., Saturation of mismatch repair in the mutD5 mutator strain of Escherichia coli. J. Bacteriol. 171, 4494–4497 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.R. S. Harris, “On a molecular mechanism of adaptive mutation in Escherichia coli,” thesis, University of Alberta (1997). [Google Scholar]
- 116.McKenzie G. J., Harris R. S., Lee P. L., Rosenberg S. M., The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. U.S.A. 97, 6646–6651 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Al Mamun A. A. M., Lombardo M.-J., Shee C., Lisewski A. M., Gonzalez C., Lin D., Nehring R. B., Saint-Ruf C., Gibson J. L., Frisch R. L., Lichtarge O., Hastings P. J., Rosenberg S. M., Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344–1348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McKenzie G. J., Magner D. B., Lee P. L., Rosenberg S. M., The dinB operon and spontaneous mutation in Escherichia coli. J. Bacteriol. 185, 3972–3977 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.J. M. Pennington, “On spontaneous DNA damage in single living cells,” thesis, Baylor College of Medicine (2006). [Google Scholar]
- 120.Shee C., Gibson J. L., Darrow M. C., Gonzalez C., Rosenberg S. M., Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 13659–13664 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fonville N. C., Blankschien M. D., Magner D. B., Rosenberg S. M., RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair 9, 403–413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shee C., Gibson J. L., Rosenberg S. M., Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli. Cell Rep. 2, 714–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601605/DC1
text S1. DNA repair by HR and RuvC specificity for four-way junctions.
text S2. About half of HJs are detected as RDG foci in living E. coli cells, and half of Gam-detectable DSBs result in HJ foci.
fig. S1. Design of a catalytically inactive RuvC for trapping HJs: RuvCDef.
fig. S2. Purified RDG binds, does not cleave, and inhibits action of other proteins on synthetic HJs in solution.
fig. S3. RDG inhibition of resistance to UV light requires induction of transcription of the ruvCDefgfp gene.
fig. S4. Titration of RuvC with RDG levels shows minimum RDG/RuvC ratio at which RDG outcompetes RuvC, preventing repair in living cells.
fig. S5. Spontaneous RDM foci colocalize with RecA-GFP strand exchange protein in E. coli cells.
fig. S6. RDG ChIP-seq localization requires DSBs and specific RuvC antibody.
fig. S7. Spontaneous RuvCGFP and RDG foci overlap with DNA stain.
fig. S8. Spontaneous RDG HR/HJ foci per cell correlate with varying chromosome and replication fork numbers.
fig. S9. Similar growth rates of various mutant strains used.
fig. S10. Reduced spontaneous RDG/HJ focus levels in recF, recQ, and recJ cells are restored by supplying RecF, RecQ, and RecJ from plasmids.
fig. S11. RecA overproduction is induced after RDG accumulation in cells.
fig. S12. Increased EME1 and GEN1 HJ resolvase mRNAs in BLM-overexpressing human cancers of the eight, and six of the eight, most common cancer types, respectively.
fig. S13. Validation of Mu Gam protein function.
table S1. Pairwise mRNA level correlation between human RAD51 with human HJ resolvases and RecQ orthologs in the eight most common cancer types.
table S2. E. coli K12 strains and plasmids used in this study.
table S3. Names and locations of new I-sites and alleles.
table S4. Oligonucleotides used in this study.