

Abstract
A new dimeric macrolide xylopyranoside, cocosolide (1), was isolated from the marine cyanobacterium preliminarily identified as Symploca sp. from Guam. The structure was determined by a combination of NMR, HRMS, X-ray diffraction studies and Mosher’s analysis of the base hydrolysis product. Its carbon skeleton closely resembles that of clavosolides A–D isolated from the sponge Myriastra clavosa, for which no bioactivity is known. We performed the first total synthesis of cocosolide (1) along with its [α,α]-anomer (26) and macrocyclic core (28), thus leading to the confirmation of the structure of natural 1. The convergent synthesis featured Wadsworth–Emmons cyclopropanation, Sakurai annulation, Yamaguchi macrocyclization/dimerization reaction, α-selective glycosidation and β-selective glycosidation. Compounds 1 and 26 potently inhibited IL-2 production in both T-cell receptor dependent and independent manners. Full activity requires the presence of the sugar moiety as well as the intact dimeric structure. Cocosolide (1) also suppressed the proliferation of anti-CD3-stimulated T-cells in a dose-dependent manner.
Keywords: marine natural products, glycosides, macrolides, structure determination, total synthesis
Diving into symmetry
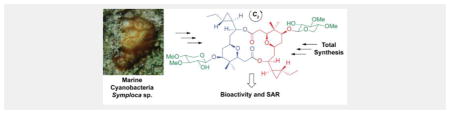
A new dimeric macrolide xylopyranoside, cocosolide, was isolated from marine cyanobacteria. Its structure was elucidated by a combination of spectroscopic methods and further confirmed by total synthesis. Along with its [α,α]-anomer and macrocyclic core, cocosolide was evaluated in various bioassays, which unveiled its role in immunosuppression. SAR study indicated the presence of the sugar moiety and the intact dimeric structure was required to achieve full activity.
Introduction
The chemical diversity of secondary metabolites reported to date has shown that marine cyanobacteria are an excellent resource for the discovery of new compounds.[1] Furthermore, marine cyanobacteria have been shown to be a source for secondary metabolites with interesting bioactivities and mechanisms of action.[2,3] In our continuing search for new metabolites from marine cyanobacteria, we have isolated a macrolide xylopyranoside trivially named cocosolide (1) from the lipophilic extracts of the soft, golden cyanobacterium preliminarily identified as Symploca sp. collected from Cocos Lagoon and Tanguisson reef flat, Guam. Cocosolide (1) is a symmetrical dimer, which structurally resembles the sponge metabolites clavosolides A-D isolated from a Philippine collection of the sponge Myriastra clavosa[4,5] and cyanolide A obtained from a Papua New Guinea collection of Lyngbya bouillonii (Figure 1).[6] Symmetrical dimeric secondary metabolites are rare in marine cyanobacteria. Two related symmetrical dimeric compounds tanikolide dimer[7] and malyngolide dimer[8] were reported from Lyngbya majuscula collected from Malagasy and Panama, respectively. To our knowledge, this is the second report of a dimeric macrolide glycoside isolated from cyanobacteria. An additional unusual structural feature is the presence of cyclopropyl groups, previously encountered in only a few cyanobacterial metabolites.[9] We report herein the details of isolation, structure elucidation, X-ray diffraction data of cocosolide (1), total synthesis and the biological activity studies of cocosolide (1), its anomer 26, monomeric analogues 2 and 3, and aglycon 28 (macrocyclic core).
Figure 1.

Structures of cocosolide (1) from Symploca sp., clavosolide A from the sponge Myriastra clavosa and cyanolide A from the cyanobacterium Lyngbya bouillonii.
Results and Discussion
Isolation and structure determination
The initial sample of the marine cyanobacterium Symploca sp., which appears as soft golden puffballs, was collected from a patch reef in Cocos Lagoon, Guam. The freeze-dried material was successively extracted with a mixture of CH2Cl2-MeOH (1:1) and aqueous MeOH (1:1). The combined extract was subsequently partitioned between EtOAc and H2O. The EtOAc-soluble portion was repeatedly fractionated by SiO2 chromatography followed by reversed-phase C18 HPLC to give a new compound, cocosolide (1, 8.0 mg, 0.08 % extract wt). Crystallization of cocosolide (1) in 10% EtOAc-hexanes furnished colorless crystals, and 1 indicated negative specific rotation similar to the previously described clavosolides.[4,5] The molecular formula of (−)-cocosolide (1) C46H76O16 was established from a high resolution ESIMS measurement of the [M + Na]+ peak at m/z 907.5009. Since the 13C spectrum (Table 1) showed only 23 signals, it was evident that 1 was a symmetrical dimer. The IR spectrum indicated the presence of alcohol, ester and ether functionalities by IR bands at 3462, 1741 and 1073 cm−1, respectively. Following the interpretation of DQF COSY and edited HSQC experiments, the 1H and 13C NMR signals were assignable to three partial structures C-2 to C-3, C-5 to C-14 and C-17 to C-21. The presence of a high-field methylene group H2-14 (δH 0.37, 0.23) that was coupled to two mutually coupled high-field methines H-10 (δH 0.68) and H-11 (δH 0.71) indicated the existence of a disubstituted cyclopropyl group in the C-5 to C-14 partial structure.
Table 1.
NMR Spectroscopic Data for Cocosolide (1) in CD3CN (600 MHz)
| position | δC mult. | δH (J in Hz) | COSYa | HMBC | NOESYb |
|---|---|---|---|---|---|
| 1, 1′ | 171.7, C | 2a, 2b, 3, 9 | |||
| 2a, 2a′ | 35.9, CH2 | 2.38, dd (17.2, 2.0) | 3 | 3 | 2b, 3, 15, 16 |
| 2b, 2b′ | 2.07, dd (17.2, 8.2) | 3 | 2a, 3, 15, 16 | ||
| 3, 3′ | 80.5, CH | 3.40, dd (8.2, 2.0) | 2a, 2b | 2a, 2b, 15, 16 | 2a, 2b, 15 |
| 4, 4′ | 39.2, C | 2a, 5, 6a, 6b 15, 16 |
|||
| 5, 5′ | 85.1, CH | 3.34, dd (11.4, 4.8) | 6a, 6b | 3, 6, 7, 15, 16 | 15, 17 |
| 6a, 6a′ | 37.4, CH2 | 1.80, m | 5, 6b, 7 | 8a | 5, 6b, 7 |
| 6b, 6b′ | 1.35, m | 5, 6a, 7 | 5, 6a | ||
| 7, 7′ | 76.3, CH | 3.38, m | 6a, 6b, 8a, 8b | 3, 5, 6a, 6b, 9 | 8a, 8b, 9 |
| 8a, 8a′ | 41.4, CH2 | 1.83, m | 7, 8b, 9 | 6b, 7, 8, 9 | 8b |
| 8b, 8b′ | 1.66, ddd (14.3, 4.1, 1.4) | 7, 8a, 9 | 8a | ||
| 9, 9′ | 77.6, CH | 4.30, dt (8.2, 1.4) | 8a, 8b, 10 | 11, 14a, 14b | 7, 10, 14a |
| 10, 10′ | 24.7, CH | 0.68, m | 9, 11, 14a, 14b | 9, 12a, 12b 14a, 14b |
14b |
| 11, 11′ | 20.6, CH | 0.71, m | 10, 12a, 12b 14a, 14b |
9, 12a, 12b, 13, 14b |
9, 12a, 12b |
| 12a, 12a′ | 27.2, CH2 | 1.37, m | 11, 12b, 13 | 10, 13, 14a, 14b | 11, 12b, 13 |
| 12b, 12b′ | 0.90, m | 11, 12a, 13 | 11, 12a, 13 | ||
| 13, 13′ | 13.8, CH3 | 0.85, t (6.9) | 12a, 12b | 11, 12 | 12a, 12b |
| 14a, 14a′ | 9.8, CH2 | 0.37, dt (8.3, 4.9) | 10, 11, 14b | 9, 12a, 12b, | 9, 11, 14b, |
| 14b, 14b′ | 0.23, dt (8.3, 4.8) | 10, 11, 14a | 10, 14a | ||
| 15, 15′ | 22.2, CH3 | 0.87, s | 3, 5, 16 | 2a, 2b, 3 | |
| 16, 16′ | 13.5, CH3 | 0.79, s | 3, 5, 15 | 2a, 2b | |
| 17, 17′ | 106.4, CH | 4.23, d (7.6) | 18 | 5, 18, 18-OH 19, 21a, 21b |
5, 21 |
| 18, 18′ | 74.2, CH | 3.18, ddd (8.3, 7.6, 4.8) | 17, 19 | 17, 18-OH, 19 | |
| 18, 18′-OH | 3.33, d (4.8) | ||||
| 19, 19′ | 85.4, CH | 3.02, t (8.3) | 18, 20 | 17, 18, 18-OH 20, 21a, 21b, 22 |
22 |
| 20, 20′ | 80.0, CH | 3.17, ddd (9.6, 8.3, 4.8) | 19, 21a, 21b | 18, 19, 21a 21b, 23 |
21a, 23 |
| 21a, 21a′ | 63.4, CH2 | 3.90, dd (11.6, 4.8) | 20, 21b | 17, 19, 20 | 20, 21b |
| 21b, 21b′ | 3.09, dd (11.9, 9.6) | 20, 21a | 21a | ||
| 22, 22′ | 60.4, CH3 | 3.49, s | 19 | 19 | |
| 23, 23′ | 58.5, CH3 | 3.38, s | 20 | 2 |
1H–1H COSY and proton(s).
NOESY correlations are from proton(s) stated to the indicated protons.
In addition, the 1H NMR spectrum indicated two singlets corresponding to two OMe groups (δ, 3.49, 3.38) and another two singlets corresponding to two methyl groups (δ, 0.87, 0.79). The large geminal coupling constant (J = 17.2 Hz) for the H2-2 methylene protons indicated that this methylene group is adjacent to the ester carbonyl.5 HMBC correlations (Table 1) from H2-2 (δH 2.38, 2.07) and H-3 (δH 3.40) to C-1 ester carbonyl (δC 171.7) and from H2-2a to C-4 quaternary carbon (δC 39.2) connected these two quaternary carbons to partial structure C-2 to C-3. The two methyl groups showed HMBC correlations to each other indicating the geminal arrangement of these methyl groups. Further HMBC correlations of these two methyls to C-3, C-4 and C-5 and HMBC correlation between H2-6 and C-4 linked C-4 to C-5 and thus established the C-1 to C-14 carbon skeleton. The partial structure C-17 to C-21 was identified as a pyranose sugar through HMBC correlation of the oxymethylene H2-21(δH 3.90, 3.09) to the C-17 (δC 106.4) anomeric carbon. The large diaxial coupling constant values (Table 1) observed for all oxymethines including the anomeric proton H-17 (J = 7.6 Hz) established a β-xylopyranose residue. HMBC correlations connected the two methoxy groups H3-22 (δ 3.49) and H3-23 (δ 3.38) to C-19 (δC 85.4) and C-20 (δC 80.0), respectively. The free OH group at δH 3.33 showed a COSY correlation to H-18 and HMBC correlation to C-17 and C-19, and therefore the position of the OH group was assigned to C-18. The oxymethine H-5 (δH 3.34) showed HMBC correlation to the anomeric carbon C-17 of the sugar moiety indicating the position of connectivity. Absence of other hydroxyl groups in the molecule and the HMBC correlation seen from H-3 to C-7 showed that C-3 through C-7 formed the tetrahydropyran ring. HMBC correlation between oxymethine H-9 and carbonyl carbon C-1 indicated the ester link at this position. These data established the symmetric 16-membered macrocyclic planar structure for cocosolide (1).
The relative stereostructure of cocosolide (1) was determined by single crystal X-ray diffraction. Cocosolide (1) is structurally related to the sponge metabolites clavosolides A-D. This unique structure of the clavosolides attracted many synthetic chemists. Willis and coworkers[10] in 2005 and Chakraborty and Reddy[11] in 2006 reported the total synthesis of (−)-clavosolide A. Both groups observed discrepancies in the 1H NMR spectrum in the cyclopropyl region between the natural product and their synthetic compound and reassigned the cyclopropyl carbinyl stereogenicity of the natural product. During the same period Smith and Simov[12] synthesized the revised structure of (−)-clavosolide A. The 1H and 13C NMR data of their synthetic (−)-clavosolide A proved to be in perfect agreement with the corresponding data reported for the natural (−)-clavosolide A.[5] The relative stereochemistry of the synthetic (−)-clavosolide A was confirmed by X-ray crystallography. The absolute stereochemistry of (−)-clavosolide A was assigned based on the absolute configuration of D-(+)-xylose, employed in the synthesis. The X-ray crystallography data of (−)-cocosolide (1) are in full agreement with the corresponding data reported for synthetic (−)-clavosolide A[12] for all stereogenic centers (Figure 2, Supporting Information Table S1). Therefore, we established the relative configuration for all stereogenic centers of the structure of (−)-cocosolide (1) based on the absolute configuration of synthetic (−)-clavosolide A.
Figure 2.

Computer-generated perspective drawing of the X-ray model of cocosolide (1).
Base hydrolysis of cocosolide (1) furnished the monomer 2 (Figure 3). Methylation of 2 with CH2N2 gave the methyl ester 3 (Figure 3). The structures of compounds 2 and 3 were determined by NMR studies and confirmed by HRMS studies. These two monomeric compounds 2 and 3 were prepared specifically for structure activity relationship studies, and subsequently the compound 3 was used to prepare the Mosher esters to establish the absolute stereochemistry of (−)-cocosolide (1). The two secondary hydroxy groups at C-9 and C-18 in compound 3 gave MTPA diesters. The MTPA ester of the sugar moiety (C-18) did not interfere with the stereochemical analysis given, and the Δδ values shown in Figure 3 indicated the absolute configuration at C-9 was S. Applying this stereochemical information in the X-ray crystallography data established the absolute stereochemistry for all stereogenic centers in (−)-cocosolide (1).
Figure 3.

Base hydrolysis product monomer 2 and corresponding monomer methyl ester 3 used for Mosher’s analysis [Δδ (δS–δR) values shown] and for biological studies.
Following its consistent isolation from subsequent Cocos Lagoon collections, we also encountered the same cocosolide- producing cyanobacterium at other collection sites, including the reef flat at Tanguisson, Piti, Fingers Reef, and north of Pago Bay. The cyanobacterium occurs in many shallow reef habitats around the island of Guam.
Total synthesis of cocosolide, its [α,α]anomer and macrocyclic core
Retrosynthetic analysis
Literature precedent for the synthesis of (−)-clavosolide A and (−)-cyanolide A indicated that the glycosylation of the symmetrical parent diol of a bis-macrolactone precursor led to a statistical mixture of [α,α]-, [β,β]-, and [α,β]-anomers, which reduced the overall yield of the total synthesis.[13–16] Thus, in designing the synthetic strategy to cocosolide we aimed to close the macrocycle via the dimerization of the permethylated-D-xylose-containing monomer 21, which could be obtained from glycosylation of alcohol 19. The Sakurai reaction of allylsilane 9 and aldehyde 12 was anticipated to stereoselectively provide a 3,3-disubstituted 2,6-cis-tetrahydropyran precursor 17, which was readily converted into 19 by oxidative cleavage of the double bond and subsequent reduction of the resulting ketone. The required allylsilane 9 could be derived from the known β-hydroxy ester 4.[17] The key intermediate 12 would be established through an asymmetric allylation of an appropriate aldehyde derived from the cyclopropane-containing acid 11 (Figure 4).
Figure 4.

Retrosynthesis analysis of cocosolide (1).
Synthesis of allylsilane 9
The synthesis of fragment 9 started with the protection of the known chiral ester 4[17] as its triethylsilyl (TES) ether, followed by treatment of the methyl ester with trimethylsilylmethyllithium to give methyl ketone 6 in 83% yield over two steps[18–23] (Scheme 1). Treatment of ketone 6 with KHMDS and N-phenyltrifluoromethanesulfonimide afforded enol triflate 7 in 91% yield. Kumada coupling of enol triflate 7 with (trimethylsilyl)methyl Grignard reagent and selective desilylation of TES ether in the presence of a catalytic amount of CSA gave allylsilane 9 in 61% overall yield.
Scheme 1.

Synthesis of allylsilane 9. a) TESOTf, 2,6-lutidine, DCM, 0 °C, 85%; b) LiCH2TMS, pentane, 0 °C, 98%; c) KHMDS, PhNTf2, THF, −78 °C, 91%; d) ClMgCH2TMS, Pd(PPh3)4, Et2O, 0 °C, 81%; e) CSA, MeOH, RT, 75%; DCM=dichloromethane; THF=tetrahydrofuran; TESOTf=triethylsilyl trifluoromethanesulfonate; KHMDS=potassium bis(trimethylsilyl)amide; PhNTf2=N-phenyl-bis(trifluoromethanesulfonimide); CSA=10-camphorsulfonic acid.
Synthesis of aldehyde 12
With the key intermediate 9 in hand, we next turned our attention to the synthesis of aldehyde 12, which contains a trans-disubstituted-cyclopropane moiety (Scheme 2). Thus, commercially available (S)-2-ethyloxirane 10 was subjected to optimized Wadsworth–Emmons cyclopropanation followed by an in situ saponification to furnish acid 11 in 82% yield.[24–28] This acid was then converted into homoallylic alcohol 11a via a three-step sequence involving a LiAlH4 reduction to furnish the corresponding alcohol, a TEMPO promoted oxidation to give rise to an aldehyde and Brown allylation to set the third stereocenter. Protection of the homoallylic alcohol of 11a as its benzyl ether, followed by oxidative cleavage of the terminal olefin with OsO4 and NaIO4 to produce the required aldehyde 12 in 26% overall yield from 11.
Scheme 2.

Synthesis of aldehyde 12. a) NaH, toluene, RT, then 10, 80 °C; b) NaOH (aq.), reflux, 82% (2 steps); c) LiAlH4, THF, 0°C; d) TEMPO, TCCA, DCM, 0 °C; e) (−)IPC2BOMe, AllylMgBr, Et2O, (1R,2R)-2-ethylcyclopropane-1-carbaldehyde, −82 °C to RT, then H2O2, NaOH(aq.), reflux; f) BnBr, NaH, THF, 0 °C to RT; g) NaIO4, OsO4, acetone/H2O, RT, 26% (5 steps); TEMPO=2,2,6,6-tetramethyl-1-piperidinyloxy; TCCA=trichloroisocyanuric acid; (−)IPC2BOMe=(−)-B-methoxydiisopinocampheylborane.
Synthesis of thioglycoside 16
Thioglycosyl donor 16 was accessed from the known ortho ester 14 as shown in Scheme 3. Treatment of ortho ester 14 with thiophenol in the presence of BF3·OEt2 afforded thioglycoside 15 in 36% yield.[29,30] Hydrolysis of the acetate group in 15 and reprotection of the resulting secondary alcohol with tert-butyldimethylsilyl triflate afforded the desired thioglycosyl donor 16 in 86% yield.
Scheme 3.

Synthesis of thioglycoside 13. a) PhSH, BF3·Et2O, DCM, RT, 36%; b) NaOMe, MeOH, RT; c) TBSOTf, 2,6-lutidine, DCM, 0 °C, 86% (2 steps); TBSOTf=tert-butyldimethylsilyl trifluoromethanesulfonate.
Assembly of subunits and completion of the synthesis of cocosolide (1)
With allylsilane 9, aldehyde 12 and thioglycoside 16 in hand, we were poised to complete the synthesis of cocosolide (1) (Scheme 4). The TMSOTf-promoted Sakurai annulation of allylsilane 9 with aldehyde 12, was found to occur rapidly at −78 °C to provide the 2,6-cis-tetrahydropyran (17) containing an exo-methylene in the 4-position. It is also worth noting that, most recently Millán et.al reported a concise methodology to construct the tetrahydropyran motif by using a three-component allylboration-Prins reaction sequence, which may provide us an opportunity to further optimize our synthetic scheme.[31] Dihydroxylation of 17 using the Upjohn method[32,33] and subsequent periodate cleavage afforded 18 in 85% yield. Reduction of the ketone with sodium borohydride gave rise to the secondary alcohol 19 in 92% yield as a single diastereomer.[12] Treatment of thioglycoside 16 with one equivalent of NBS at −25 °C in dry acetonitrile, followed by addition of aglycone (19), the glycosidation proceeded with 5:3 β-selectivity afforded the desired β-anomer 20 in 60% isolated yield.[34–37] Subsequent removal of the bis-benzyl ethers under hydrogenative conditions gave rise to diol 21. The primary alcohol in 21 was selectively converted into the corresponding acid with a one-step TEMPO-catalyzed oxidation protocol.[38] Macrodiolide formation under Yamaguchi’s conditions[39] provided macrocycle 22 in 43 % yield over two steps. Deprotection of 22 with TBAF in THF then furnished cocosolide (23), which was identical to the natural product in all respects.
Scheme 4.

Assembly of Subunits and Completion of the Synthesis of Cocosolide (1). a) TMSOTf, Et2O, 4Å MS, −78 °C, 75%; b) OsO4, NMO, NaIO4, acetone/H2O, RT, 85%; c) NaBH4, MeOH, −40 °C, 92%; d) NBS, MeCN, then 16, −25 °C to RT, 60%; e) H2, Pd/C, MeOH, RT, 83%; f) TEMPO, Bu4NCl, KBr, NaOCl, NaHCO3, DCM/H2O, 0 °C; g) 2,4,6-trichlorobenzoyl, DIPEA, RT, then DMAP, toluene, 80 °C, 43% (2 steps); h) TBAF, THF, RT, 82%; TMSOTf=trimethylsilyl trifluoromethanesulfonate; NMO=4-methylmorpholine N-oxide; NBS=N-bromosuccinimide; DIPEA=N,N-diisopropylethylamine; DMAP=4-(dimethylamino) pyridine; TBAF= tetra-n-butylammonium fluoride.
Synthesis of [α,α]-anomer of cocosolide (26)
With both 19 and 16 in hand, we prepared the [α,α]-anomer of cocosolide using the same strategy (Scheme 5),wherein, a highly α-selective glycosidation of aglycone 19 with thioglycoside 16 was achieved under the promotion of NIS, TfOH in CH2Cl2 to furnish the desired α-anomer 23 in 71% isolated yield.[40] Intermediate 23 was then elaborated to the [α,α]-anomer of cocosolide (26) in 19% yield by an identical strategy as described for 1, including removal of bis-benzyl ether, TEMPO-catalyzed oxidation, macrodiolide formation and desilylation. (Scheme 5).
Scheme 5.

Synthesis of [α,α]-anomer of cocosolide (26). a) NIS, TfOH, DCM, then 16, 71%; b) H2, Pd/C, MeOH, RT, 64%; c) TEMPO, Bu4NCl, KBr, NaOCl, NaHCO3, DCM/H2O, 0 °C; d) 2,4,6-trichlorobenzoyl, DIPEA, RT, then DMAP, toluene, 80°C, 40% (2 steps); e) TBAF, THF, RT, 75%; NIS= N-iodosuccinimide; TfOH= trifluoromethanesulfonic acid.
Synthesis of macrocyclic core of cocosolide (28)
The macrocyclic core of cocosolide (28) was prepared from 19 by a similar strategy as described for 1. Thus, protection of the secondary alcohol as its tert-butyldimethylsilyl (TBS) ether, followed by removal of the bis-benzyl ethers under hydrogenative conditions gave rise to the corresponding diol, which was then selectively converted into the corresponding acid with a one-step TEMPO-catalyzed oxidation protocol. Direct macrocyclization using Yamaguchi’s conditions afforded the C2 symmetric diolide 27 in 19% yield over four steps. Deprotection of 27 with TBAF in THF then furnished 28 in 71% yield.
Biological studies
To study the effect of cocosolide (1) on biological function, we treated an array of cell types (HCT116 colorectal cancer cells, RAW macrophage cells, Jurkat T-cell lymphoma cells; IC50 > 50 μM) with cocosolide (1) and found no modulation of cell viability. Structural considerations, including the dimeric nature coupled with the glycosylation feature, hinted at the possibility of dimeric surface targets for geometric and recognition consideration, respectively. We tested the effects in immortalized T-cells (Jurkat) as a model to evaluate immunomodulatory activity.[41] IL-2 production was induced via dual stimulation with phorbol 12-myristate 13-acetate (PMA, 80 nM) and phytohemagglutinin (PHA, 10 μg/mL), conditions for a T-cell receptor (TCR) dependent activation; or TCR-independent stimulants PMA (80 nM) and ionomycin (1 μM).[42] Cocosolide (1) and its [α,α]-anomer (26) equally and potently reduced IL-2 production without significantly affecting cell viability (Figures 5A and B). The susceptibility of the TCR-independent system was stronger, although both stimulations are abrogated in a dose-dependent manner. The macrocyclic core 28 and monomer ester 3 showed minimal effects in these assays compared with 1 and 26 (Figures 5C and D), indicating that the sugar and dimeric structure are important to the target recognition and engagement process.
Figure 5.

Biological effects of cocosolide (1), [α,α]-anomer 26, macrocyclic core 28 and monomer methyl ester 3 on T-cell systems. (A) Effect of 1 on IL-2 production by Jurkat cells and on cell viability in response to PMA/PHA and PMA/ionomycin stimulation. For comparison, cyclosporine A inhibited both activities at 1 μM by 90%. (B) Effect of cocosolide [α,α]-anomer 26 on IL-2 production by Jurkat cells and on cell viability in response to PMA/PHA and PMA/ionomycin stimulation. (C,D) Comparison of compounds 1 (natural product), 3 (monomer methyl ester), 26 ([α,α]-anomer) and 28 (aglycon) on their effect on IL-2 production in Jurkat cells (C) stimulated by PMA/PHA and (D) PMA/ionomycin. (E) Spleen cell proliferation assay. Anti-CD3 stimulated cells were cultured in triplicate wells in the presence of increasing concentrations of 1. Values shown represent the average spleen cell response from the mean triplicate values between two mice. * denotes p < 0.05 compared to DMSO.
To examine how cocosolide (1) may affect activated T cells, we stimulated spleen cells with CD3 and cultured the cells under increasing concentrations of cocosolide (1) for 72 h. As shown in Figure 5E, cocosolide (1) inhibited anti-CD3-mediated T-cell proliferation in a dose-dependent manner. Cell viability was not affected compared with DMSO-treated controls, suggesting that the observed suppression of T cell expansion by 1 is not attributed to cell death.
We also investigated possible modulation of Toll-like receptor 4 mediated pathways. Specifically, RAW264.7 macrophage cells were stimulated with lipopolysaccharides (LPS), and pretreatment with cocosolide (1) did not dampen the induction of NO production and also did not reduce the viability (up to 100 μM). Similarly, 1 did not exert antibacterial activity in our assays (Bacillus cereus, Pseudomonas aeruginosa, Mycobacterium tuberculosis). Taken together, cocosolide’s effects appear to be fairly cell- and pathway-type specific.
The non-cytotoxic effects are consistent with data reported for the structurally related clavosolides, for which a bioactivity remained to be established.[4,5] Clavosolides A and B were first reported from the sponge Myriastra clavosa but suggested to be of cyanobacterial origin because the structure did not relate to any known sponge metabolites and the presence of a high concentration of cyanobacterial cells was found in the sponge sample.[5] These dimeric metabolites along with two other analogues, clavosolides C and D were concurrently reported by a second group also from a cytotoxic extract of M. clavosa, displaying differential cytotoxicity in the NCI-60 cell panel.[4] Evaluation of the purified compounds revealed that these are non-cytotoxic. The structurally related macrolide cyanolide A was reported from the cyanobacterium L. bouillonii with potent molluscicidal activity against Biomphalaria glabrata, but non-cytotoxic effects against H-460 human lung adenocarcinoma and mouse neuroblastoma cells.6
Conclusions
This discovery of cocosolide (1) and the recent report of cyanolide A in a marine cyanobacterium verify the possible cyanobacterial origin for the related clavosolides. Rigorous biological studies necessitated an efficient total synthesis with a high degree of selectivity for the key steps and access to key analogues to probe structural requirements. We have achieved first total synthesis of cocosolide (1) along with its [α,α]-anomer (26) and macrocyclic core (28), thus leading to the confirmation of the structure of natural cocosolide. The convergent synthesis features Wadsworth–Emmons cyclopropanation, Sakurai annulation, Yamaguchi macrocyclization/dimerization reaction, α-selective glycosidation and β-selective glycosidation. The synthesis provided further quantities of the natural product and structural derivatives for biological studies. Initial biological studies suggested an immunosuppressive activity based on inhibition of IL-2 production and T-cell proliferation. Preliminary structure–activity relationship studies indicated the importance of the dimeric macrolide core and the glycosylation. Studies towards the mechanism of action and investigation of other biological effects are ongoing.
Experimental Section
Cocosolide (1)
Colorless crystals; mp 233–234 °C; [α]25D −65.3 (c 0.41, CH2Cl2); UV (MeOH) λmax (log ε) 210 (3.39) nm; IR (film) νmax 3462, 2957, 1741, 1464, 1252, 1165, 1096, 1073, 953 cm−1; 1H NMR, 13C NMR, DQF COSY, HMBC and NOESY data, see Table 1; HRESI/APCIMS m/z 907.5009 [M + Na]+ (calcd for C46H76O16Na, 907.5026). Comparison of 1H and 13C NMR spectra of cocosolide (1) from Cocos Lagoon and Tanguisson reef flat are shown in Supporting Information, Figures S1 and S2.
Biological reagents and general experimental procedures
Phorbol myristate acetate (PMA) was purchased from Promega (Cat# V1171; Madison, WI). Phytohemagglutinin (PHA) was from Sigma-Aldrich (Cat# L8902; St. Louis, MO) and ionomycin, calcium salt was purchased from EMD Chemicals, Inc (cat# 407952; Gibbstown, NJ). Human colon adenocarcinoma HCT116 cells and the human leukemic T-cell line Jurkat (Clone E6-1) were obtained from the American Type Culture Collection (Manassas, VA) and cultured either Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) for HCT116 cells and RPMI 1640 medium (Cellgro, Herndon, VA) for Jurkat cells, supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT). Cells were maintained at 37 °C humidified air in 5% CO2.
Cell viability assays
HCT116, RAW 264.7 and Jurkat cells were seeded in 96-well plates and 24 h later treated with up to 50 μM cocosolide (1) for 24 h (RAW 264.7) or 48 h (HCT116, Jurkat). Cell viability was measured using MTT reagent according to the manufacturer’s instructions (Promega, Madison, WI, USA).
Measurement of IL-2 production (Jurkat cells)
Jurkat cells (1 × 105 cells per well; 150 μL each well) were seeded in 96-well clear bottom plates and allowed to settle for one hour. Cells were then co-treated with compounds 1, 3, 26 and 28 (50 μM to 100 nM in EtOH) and TCR-dependent stimulants (80 nM PMA in DMSO and 10 μg/ml PHA in H2O) or TCR-independent stimulants (80 nM PMA in DMSO and 1 μM ionomycin in DMSO) along with EtOH/DMSO solvent controls. Cells were also treated with compounds alone to measure cell viability due to PMA/PHA and PMA/ionomycin toxicities. DMSO concentrations were maintained at 0.21% to minimize toxicity for Jurkat cells. After 24 h incubation, 50 μL of culture supernatant were removed from each well into a separate plate and used for measuring IL-2 production using an alphaLISA kit (PerkinElmer, Waltham, MA). Briefly, acceptor bead and anti-IL-2 antibody were incubated with 5 μL of supernatant for 60 min and donor beads added and incubated for further 30 min following which, IL-2 was quantified using Envision-Reader (PerkinElmer). The cells were used to measure viability after 48 h.
Proliferation assay
Red blood cells (RBC) were removed from freshly isolated whole spleen cells using ammonium chloride potassium (ACK) lysis buffer for 2 min at room temperature then washed free of lysis buffer using PBS. RBC-depleted spleen cells were then cultured in RPMI 1640 supplemented with 10% FCS (Invitrogen Life Sciences), 1x penicillin/streptomycin/neomycin (Gibco), and 10 mM HEPES buffer (Gibco) at a concentration of 106 cells/well in round-bottom 96-well plates at 37 °C. Anti-CD3e (0.05 μg/200 μL well) was added to stimulate cell proliferation in the presence of increasing concentrations of 1 (50 nM, 0.5 μM, 5 μM, and 50 μM). All treatment conditions were controlled to contain a final concentration of 1% DMSO solvent as used in drug preparation. At 72 h of culture, 1 μCu 3H-thymidine (Amersham Biosciences) in 50 μL of media was added per well and allowed to incorporate for 12–16 h. Cells were harvested and washed using an automated cell harvester (Perkin Elmer), and radioactivity was analyzed using a liquid scintillation counter. Cell proliferation is measured as counts per minute (cpm). Permission to use mice for this research project was obtained through the University of Florida IACUC board. Animals were cared for and euthanized according to procedures approved by the IACUC.
NO assay
RAW 264.7 cell assays to measure effects on LPS-induced NO production were performed as previously described.[43]
Antibacterial assays
Activity against Bacillus cereus, Pseudomonas aeruginosa, and Mycobacterium tuberculosis was assessed as we previously described.[44,45]
Supplementary Material
Scheme 6.

Synthesis of macrocyclic core of cocosolide (28). a) TBSOTf, 2,6-lutidine, DCM, 0 °C; b) H2, Pd/C, EtOAc, RT; c) TEMPO, Bu4NCl, KBr, NaOCl, NaHCO3, DCM/H2O, 0 °C; d) 2,4,6-trichlorobenzoyl, DIPEA, RT, then DMAP, toluene, 80°C, 19% (4 steps); e) TBAF, THF, RT, 71%.
Acknowledgments
This research was supported by the NIH, NCI grant R01CA172310, and by Shenzhen Peacock Plan (KQTD2015071714043444). We thank many students and staff of the UOGML for assisting VJP with collecting the sample in Guam over the years. We thank Rana Montaser and Yanxia Liu for initial biological testing. We also thank the Harbor Branch Oceanographic Institute at Florida Atlantic University spectroscopy facility for 600 MHz NMR spectrometer time, UV and melting point measurements, and the Florida Atlantic University, Jupiter Campus, for the use of their polarimeter and infrared spectrometer. This is contribution number 1027 from the Smithsonian Marine Station at Fort Pierce. The authors declare the following competing financial interest(s): H. Luesch is co-founder of Oceanyx Pharmaceuticals, Inc., which is negotiating licenses for patents and patent applications related to cyanobacterial metabolites.
Footnotes
Supporting information for this article is available on the WWW under ……
References
- 1.Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Nat Prod Rep. 2015;32:116–211. doi: 10.1039/c4np00144c. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Rein KS. Mar Drugs. 2010;8:1817–1837. doi: 10.3390/md8061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salvador-Reyes LA, Luesch H. Nat Prod Rep. 2015;32:478–503. doi: 10.1039/c4np00104d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson KL, Gustafson KR, Pannell LK, Beutler JA, Boyd MR. J Nat Prod. 2002;65:1303–1306. doi: 10.1021/np020193z. [DOI] [PubMed] [Google Scholar]
- 5.Rao MR, Faulkner DJ. J Nat Prod. 2002;65:386–388. doi: 10.1021/np010495l. [DOI] [PubMed] [Google Scholar]
- 6.Pereira AR, McCue CF, Gerwick WH. J Nat Prod. 2010;73:217–220. doi: 10.1021/np9008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutiérrez M, Andrianasolo EH, Shin WK, Goeger DE, Yokochi A, Schemies J, Jung M, France D, Cornell-Kennon S, Lee E, et al. J Org Chem. 2009;74:5267–5275. doi: 10.1021/jo900578j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutiérrez M, Tidgewell K, Capson TL, Engene N, Almanza A, Schemies J, Jung M, Gerwick WH. J Nat Prod. 2010;73:709–711. doi: 10.1021/np9005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwan JC, Meickle T, Ladwa D, Teplitski M, Paul V, Luesch H. Mol Biosyst. 2011;7:1205–1216. doi: 10.1039/c0mb00180e. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry CS, Bushby N, Charmant JPH, Elsworth JD, Harding JR, Willis CL. Chem Commun. 2005:5097–5099. doi: 10.1039/b509757f. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty TK, Reddy VR. Tetrahedron Lett. 2006;47:2099–2102. [Google Scholar]
- 12.Smith AB, III, Simov V. Org Lett. 2006;8:3315–3318. doi: 10.1021/ol0611752. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, Hong J. Org Lett. 2010;12:2880–2883. doi: 10.1021/ol101022z. [DOI] [PubMed] [Google Scholar]
- 14.Son JB, Kim SN, Kim NY, Lee DH. Org Lett. 2006;8:661–664. doi: 10.1021/ol052851n. [DOI] [PubMed] [Google Scholar]
- 15.Son JB, Kim SN, Kim NY, Lee DH. Org Lett. 2006;8:3411. doi: 10.1021/ol052851n. [DOI] [PubMed] [Google Scholar]
- 16.Barry CS, Elsworth JD, Seden PT, Bushby N, Harding JR, Alder RW, Willis CL. Org Lett. 2006;8:3319–3322. doi: 10.1021/ol0611705. [DOI] [PubMed] [Google Scholar]
- 17.Reiff EA, Nair SK, Henri JT, Greiner JF, Reddy BS, Chakrasali R, David SA, Chiu TL, Amin EA, Himes RH, et al. J Org Chem. 2010;75:86–94. doi: 10.1021/jo901752v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demuth M. Helv Chim Acta. 1978;61:3136–3138. [Google Scholar]
- 19.Mulzer J, Mantoulidis A, Oehler E. J Org Chem. 2000;65:7456–7467. doi: 10.1021/jo0007480. [DOI] [PubMed] [Google Scholar]
- 20.Durgnat JM, Warm A, Vogel P. Synth Commun. 1992;22:1883–1893. [Google Scholar]
- 21.Evans MA, Morken JP. J Am Chem Soc. 2002;124:9020–9021. doi: 10.1021/ja026703t. [DOI] [PubMed] [Google Scholar]
- 22.Storer RI, Takemoto T, Jackson PS, Ley SV. Angew Chem Int Ed. 2003;42:2521–2525. doi: 10.1002/anie.200351413. [DOI] [PubMed] [Google Scholar]
- 23.Gesinski MR, Rychnovsky SD. J Am Chem Soc. 2011;133:9727–9729. doi: 10.1021/ja204228q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armstrong A, Scutt JN. Org Lett. 2003;5:2331–2334. doi: 10.1021/ol0346887. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong A, Scutt JN. Chem Commun. 2004:510–511. doi: 10.1039/b316142k. [DOI] [PubMed] [Google Scholar]
- 26.Delhaye L, Merschaert A, Delbeke P, Briône W. Org Process Res Dev. 2007;11:689–692. [Google Scholar]
- 27.Bray CD, Minicone F. Chem Commun. 2010;46:5867–5869. doi: 10.1039/c0cc01333a. [DOI] [PubMed] [Google Scholar]
- 28.Kumar P, Dubey A, Harbindu A. Org Biomol Chem. 2012;10:6987–94. doi: 10.1039/c2ob25622c. [DOI] [PubMed] [Google Scholar]
- 29.Chayajarus K, Chambers DJ, Chughtai MJ, Fairbanks AJ. Org Lett. 2004;6:3797–3800. doi: 10.1021/ol048427o. [DOI] [PubMed] [Google Scholar]
- 30.Collot M, Savreux J, Mallet JM. Tetrahedron. 2008;64:1523–1535. [Google Scholar]
- 31.Millán A, Smith JR, Aggarwal VK. Angew Chemie Int Ed. 2016;55:2498–2502. doi: 10.1002/anie.201511140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider WP, McIntosh AV. 2,769,824. US Patent. 1956
- 33.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- 34.Nicolaou KC, Seitz SP, Papahatjis DP. J Am Chem Soc. 1983;105:2430–2434. [Google Scholar]
- 35.Paquette LA, Barriault L, Pissarnitski D, Johnston JN. J Am Chem Soc. 2000;122:619–631. [Google Scholar]
- 36.Kasai Y, Ito T, Sasaki M. Org Lett. 2012;14:3186–3189. doi: 10.1021/ol301278e. [DOI] [PubMed] [Google Scholar]
- 37.Seden PT, Charmant JPH, Willis CL. Org Lett. 2008;10:1637–1640. doi: 10.1021/ol800386d. [DOI] [PubMed] [Google Scholar]
- 38.Anelli PL, Biffi C, Montanari F, Quici S. J Org Chem. 1987;52:2559–2562. [Google Scholar]
- 39.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 40.Kusumi S, Tomono S, Okuzawa S, Kaneko E, Ueda T, Sasaki K, Takahashi D, Toshima K. J Am Chem Soc. 2013;135:15909–15912. doi: 10.1021/ja407827n. [DOI] [PubMed] [Google Scholar]
- 41.Schneider U, Schwenk HU, Bornkamm G. Int J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 42.Fischer BS, Qin D, Kim K, McDonald TV. J Pharmacol Exp Ther. 2001;299:238–246. [PubMed] [Google Scholar]
- 43.Ratnayake R, Liu Y, Paul VJ, Luesch H. Cancer Prev Res. 2013;6:989–999. doi: 10.1158/1940-6207.CAPR-13-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montaser R, Abboud KA, Paul VJ, Luesch H. J Nat Prod. 2011;74:109–112. doi: 10.1021/np1006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montaser R, Paul VJ, Luesch H. Phytochemistry. 2011;72:2068–2074. doi: 10.1016/j.phytochem.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.