

Abstract
Synthesis of aza-rocaglates, nitrogen-containing analogues of the rocaglate natural products, is reported. The route features ESIPT-mediated (3+2) photocycloaddition of 1-alkyl-2-aryl-3-hydroxyquinolinones with the dipolarophile methyl cinnamate. A continuous photoflow reactor was utilized for photocycloadditions. An array of compounds bearing the hexahydrocyclopenta[b]indole core structure was synthesized and evaluated in translation inhibition assays.
Keywords: Rocaglate, Photocycloaddition, ESIPT, Flow Chemistry, Heterocycle
Graphical abstract
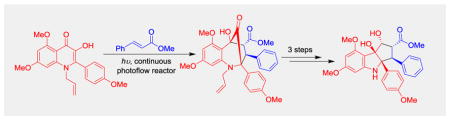
Aza-rocaglates were prepared from 3-hydroxyquinolinones and methyl cinnamate using ESIPT (3+2) photocycloaddition in a continuous photoflow reactor. A collection of compounds containing hexahydrocyclopenta[b]indole core was synthesized using this method and evaluated in translation inhibition assays.
Flavaglines are a family of natural products isolated from plants of the genus Aglaia which feature the cyclopenta[b]-benzofuran core structure.[1] After the first isolation of rocaglamide (1, Figure 1) by King and coworkers in 1982,[2] more than 100 natural flavaglines (rocaglates) have been isolated, characterized, and tested for insecticidal, anti-inflammatory, and anticancer activities.[3] Due to their intriguing structural complexity and biological activities, a number of synthetic chemists have undertaken syntheses of natural flavaglines as well as targeted derivatives with improved or novel biological properties.[4]
Figure 1.

Rocaglate and Related Natural Products, a Carbocyclic Analogue, and Aza-Rocaglate Structures.
However, with the exception of the carbocyclic analogue 4 for which biological data was not reported,[5] such studies have been limited to variations of the cyclopenta[b]benzofuran core.[4] Previously, we reported a biomimetic approach towards rocaglates using excited state intramolecular proton transfer (ESIPT)-mediated (3+2) photocycloaddition of 3-hydroxyflavones and dipolarophiles.[6] Later, we successfully extended the methodology to employ 1,2-dimethyl-3-hydroxyquinolinone (DMQ, 6) to access various cycloadducts.[7] For example, photocycloaddition of DMQ (6) with methyl cinnamate 7 provided bridged ketone cycloadduct 8 (Scheme 1).
Scheme 1.

Previous Studies on Photocycloadditions Using 3-Hydroxyquinolinones (3-HQ’s).
In the current study, we considered syntheses of nitrogen-containing rocaglate analogues employing ESIPT photocycloaddition of 2-aryl-3-hydroxyquinolinones such as 9 wherein it was thought that π-π stacking interactions between the 2-aryl substituent of substrate 9 and the phenyl group of methyl cinnamate 7 may favor the desired cycloaddition geometry (Scheme 2). Aza-aglain cycloadduct 10 may further undergo α-ketol (acyloin) rearrangement to 11 which may be followed by diastereoselective reduction to access aza-rocaglates such as 5 bearing the hexahydrocyclopenta[b]-indole scaffold. Herein, we outline synthetic and mechanistic studies regarding the aza-rocaglates as well as translation inhibition studies of the natural product variants.
Scheme 2.

Planned Synthesis of Aza-Rocaglates.
We first performed pilot studies with 2-aryl-3-hydroxyquinolinone substrate 12.[8b] Under photoirradiation of 12 with dipolarophile 7 (λ>330 nm), we did not observe satisfactory conversion to the corresponding cycloadduct 13 (Scheme 3). As our previous studies were performed using a N-methyl-3-hydroxyquinolinone substrate (cf. Scheme 1), 1-methyl-2-aryl-3-hydroxyquinolinone (N-Me-3-HQ, 15) was chosen as a modified substrate for ESIPT photocycloaddition. The synthesis of derivatives related to 15 has been previously reported.[8] However, using substrate 14 and polyphosphoric acid (PPA) as solvent,[8b] a lengthy workup and purification was required and only 34% of the desired product 15 was observed. In order to obtain practical amounts of 15, development of a new method for the synthesis of N-alkyl-3-hydroxyquinolinones was required. After evaluation of reaction conditions, we found that treatment of phenacyl anthranilate 14 with NaH (1.0 equiv.) in THF led to the production of 15 in 83% yield (Scheme 4A). A proposed mechanism (Scheme 4B) may proceed through formation of hemiaminal 14a followed by rearrangement to alkoxide 14b. 3,1-Benzoxazin-4-ones related to 14b have previously been reported from treatment of phenacyl anthranilates with acetic acid.[8c] Further reaction of 14b to iminium 14c followed by isomerization should generate the intramolecular hydrogen bond-stabilized enolate 14d (Scheme 4C).[9] A DFT model of 14d (Scheme 4C) shows a well-defined arrangement for further transformations.[10] Cyclization of 14d to benzoxazepine 14e followed by rearrangement to zwitterion 14f, elimination, and acidic workup provides 3-hydroxyquinolinone 15. Similar benzoxazepine structures have been reported previously in the literature by treatment of phenacyl anthranilates with phosphoryl chloride.[11] To the best of our knowledge, the synthesis of 1-alkyl-3-hydroxyquinolinones from phenacyl anthranilates under basic conditions has not been previously described.
Scheme 3.

Attempted (3+2) Photocycloaddition of 12.
Scheme 4.

A Synthesis of N-Me-3-HQ 15; B. Proposed Mechanism; C. DFT Model of Intermediate 14d (B3LYP_6–31G**++).
When substrate 15 was subjected to photoirradiation (λ>330 nm) in the presence of methyl cinnamate, cycloadducts were indeed generated. After condition optimization, we found that using trifluorotoluene and trifluoroethanol as co-solvents, aza-aglain derivatives 16 and 17 could be isolated in 40% yield (5:1 ratio). However, 48 h was needed to obtain the desired products in which case substantial decomposition was observed. Taking advantage of a recently developed continuous photoflow reactor,[12] we found that the reaction was complete in 9 h (46% yield, 89% yield b.r.s.m., 16:17= 5:1). In addition, significantly less decomposition was observed in comparison to batch reactions which facilitated product purification. Reduction of the minor isomer 17 using LiAlH4 provided an unstable diol which was acylated to afford the bis-para-bromobenzoate 18. The structure and relative stereochemistry of 18 was confirmed using HMBC and NOESY experiments.[10] Using sodium methoxide-mediated α-ketol rearrangement, followed by diastereoselective reduction, aza-rocaglate 20 could be obtained in 77% yield (2 steps) from cycloadduct 16. The structure of 20 was unambiguously confirmed by X-ray crystal structure analysis of the derived bromobenzoate 21 (Figure 2).[13] Interestingly, inspection of the X-ray structure reveals that the N-methyl moiety is coplanar to the adjacent aryl group indicating a non-pyramidalized nitrogen.[14] This is likely due to steric interaction of N-methyl moiety with the two nearby aryl substituents which appears to prevent nitrogen pyramidalization.
Figure 2.

Synthesis and X-ray Crystal Structure of Aza-Rocaglate 21.
In order to provide a plausible explanation for the differential reactivity of 3-hydroxyquinolinones 12 and 15, we measured their UV-Vis absorption spectra.[15] We found that in CH2Cl2:MeOH (2:1), both substrates possess an absorption band at approximately 370 nm which can be attributed to charge transfer excitation. More interestingly, we observed an additional absorption (310 nm) for substrate 12 (Figure 3). Therefore, we hypothesized that in solution, 12 may also be represented by an aromatic, dihydroxyquinoline tautomeric form which may diminish its photoreactivity.[16] In order to support this assertion, we synthesized 3,4-dimethoxyquinoline 22 by treatment of 12 with TMS diazomethane (Scheme 6) for evaluation of its UV absorption properties.[17] An absorption shoulder at 320 nm was also observed for compound 22 which provides information on the existence of tautomeric form 12a in solution.
Figure 3.

UV Absorption Spectra of 3-Hydroxyquinolinones 12 and 15 in Comparison to Dimethoxyquinoline 22.
Scheme 6.

Methylation of an N-H-3-hydroxyquinolinone.
We next targeted the synthesis of more highly functionalized aza-rocaglates. Starting with N-Me-Br-3-HQ 23 as substrate, obtained from commercially available materials in four steps,[10] photocycloaddition with methyl cinnamate 7 in the continuous flow reactor led to the production of cycloadduct 24 in 43% yield and excellent selectivity.[18] Ketol rearrangement, followed by diastereoselective reduction, provided aza-rocaglate 26 in good yield. (Scheme 7) Compound 26 was then further transformed to the corresponding hydroxamate 27[19] by saponification and coupling.
Scheme 7.

Synthesis of an Advanced N-methyl-aza-rocaglate 27.
As part of our study, we also targeted the synthesis of the N-H-containing aza-rocaglate 5. After an evaluation of nitrogen protecting groups for synthesis of 3-hydroxyquinolinone substrates, only alkyl protecting groups on the nitrogen could enable access to photocycloadducts (cf. Scheme 4). Therefore, an allyl protecting group was chosen to synthesize N-allyl-3-HQ 28 using the previously described method. Upon photoirradiation of substrate 28 in the continuous photoflow reactor, cycloadduct 29 could be obtained from N-allyl-3-HQ 28 in 43% yield (Scheme 8). Ketol rearrangement of 29 led to the production of 30 which was subjected to hydroxyl-directed reduction to afford N-allyl-aza-rocaglate 31. After a thorough evaluation of the allyl deprotection conditions, we found that using Pd (0) /1,4-bis(diphenylphosphino)-butane complex and Meldrum’s acid as nucleophile and proton source,[20] N-H-aza-rocaglate 5 could be obtained in 45% yield. Other N-deallylation conditions reported in the literature failed to provide the desired product.[21] In order to demonstrate the possibility for further functionalization of the N-allyl moiety, 31 was converted into acrylate 32 using olefin cross metathesis.[22]
Scheme 8.

Synthesis of N-H-aza-rocaglate 5.
Rocaglates have been shown to behave as potent inhibitors of translation by interfering with the activity of eukaryotic initiation factor (eIF) 4A, an RNA helicase necessary for cap-dependent protein synthesis.[23] We therefore assessed the biological activities of compounds 5, 26, 27, 31, and 32 in vitro in a translation assay programmed with the bicistronic mRNA, FF/HCV/Ren (Figure 4A). In this system firefly luciferase (FF) production is eIF4A-dependent whereas Renilla luciferase (Ren) is not.[24] Silvestrol (not shown) and rocaglates CR-1-31B[19a] and SDS-1-021-(−)[3e] potently inhibited FF production (5–10 fold) at 5 μM. The only aza-rocaglate to affect FF production was the N-methyl derivative 26, showing a ~30% reduction in FF production at 20 μM (Figure 4A). To assess activity towards cellular protein synthesis, HeLa cells were incubated with silvestrol, CR-1-31B, or 26 for 1 h and metabolic protein synthesis quantitated (Figure 4B). Whereas 100 nM silvestrol or CR-1-31B completely blocked protein synthesis, compound 26 showed modest activity at only at 50 μM (~20% inhibition).
Figure 4.

A. Effect of aza-rocaglates on in vitro translation of FF/HCV/Ren. B. Assessing translation inhibition activity of 26 on HeLa cells.
Consistent with these findings, we also found no evidence for translation inhibition over a 24 h period at concentrations up to 10 μM using a whole cell assay based on constitutive expression of the rapidly turned-over reporter protein firefly luciferase (Figure 5A). Likewise the compounds had minimal cytotoxic activity in a standard 3-day growth assay over the same concentration range (Figure 5B). Together, these results indicate that aza-rocaglates do not possess the same inhibitory potency towards protein synthesis in comparison to related rocaglates such as RHT.[19c]
Figure 5.

A. Translation inhibition (10 μM) in whole cells based on constitutive expression of firefly luciferase. B. 3-day growth assay (Human 293T cancer cells) over the same concentration.
In summary, we have employed ESIPT photocycloaddition methodology to synthesize aza-rocaglates. Our studies have uncovered differential photocycloaddition reactivities between N-H- and N-Me-substituted 2-aryl-3-hydroxyquinolinone substrates. A novel method to access 1-alkyl-2-aryl-3-hydroxyquinolinones was also developed for (3+2) photocycloaddition. Use of a continuous photoflow reactor facilitated synthesis of aza-rocaglates with both N-alkyl and N-H substitution. Initial protein synthesis and translation inhibition data indicates that aza-rocaglates do not possess activity in comparison to related rocaglates which provides further information on the SAR of the natural product scaffold.
Supplementary Material
Scheme 5.

(3+2) Photocycloaddition and Prototype Synthesis of an Aza-Rocaglate.
Acknowledgments
We thank the National Institutes of Health (GM-073855 to J.A.P, Jr. and R01CA175744 to L.W. and J.A.P, Jr.) and the Canadian Institutes for Health Research (MOP-115126 to J.P.) for research support. We thank Dr. Vincent Eschenbrenner-Lux for computational models, Prof. Aaron Beeler (BU) for helpful discussions, and Dr. Jeffrey Bacon (Boston University) and Matthew Benning (Bruker AXS) for X-ray crystal structure analysis. NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the NSF. Work at the BU-CMD is supported by R24GM111625.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Wenyu Wang, Department of Chemistry, Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States.
Dr. Regina Cencic, McGill University, Department of Biochemistry and The Rosalind and Morris Goodman Cancer Research Centre Room 810, 3655 Drummond St., Montreal, QC, H3G 1Y6 (Canada)
Dr. L. Luke Whitesell, Whitehead Institute for Biomedical Research (WIBR), Cambridge, Massachusetts 02142, United States
Prof. Dr. Jerry Pelletier, McGill University, Department of Biochemistry and The Rosalind and Morris Goodman Cancer Research Centre Room 810, 3655 Drummond St., Montreal, QC, H3G 1Y6 (Canada)
Prof. Dr. John A. Porco, Jr., Department of Chemistry, Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States
References
- 1.Ko FN, Wu TS, Liou MJ, Huang TF, Teng CM. Eur J Pharmacol. 1992;218:129. doi: 10.1016/0014-2999(92)90156-x. [DOI] [PubMed] [Google Scholar]
- 2.King ML, Chiang C, Ling H, Fujita E, Ochiai M, Andrew MT. J Chem Soc, Chem Commun. 1982;20:1150. [Google Scholar]
- 3.For recent reports on the chemistry and biology of flavaglines: Basmadjian C, Thuaud F, Ribeiro N, Désaubry L. Future Med Chem. 2013;5:2185. doi: 10.4155/fmc.13.177.Ribeiro N, Thuaud F, Nebigil C, Désaubry L. Bioorg Med Chem. 2012;20:1857. doi: 10.1016/j.bmc.2011.10.048.Ebada SS, Lajkiewicz NJ, Porco JA, Jr, Li-Weber M, Proksch P. Prog Chem Org Nat Prod. 2011;94:1. doi: 10.1007/978-3-7091-0748-5_1.Pan L, Woodard JL, Lucas DM, Fuchs JR, Kinghorn AD. Nat Prod Rep. 2014;31:924. doi: 10.1039/c4np00006d.Chu J, Cencic R, Wang W, Porco JA, Jr, Pelletier J. Mol Cancer Ther. 2016;15:136. doi: 10.1158/1535-7163.MCT-15-0409.Liu S, Wang W, Brown LE, Qiu C, Lajkiewicz NJ, Zhao T, Zhou J, Porco JA, Jr, Wang TT. EBioMedicine. 2015;11:1600. doi: 10.1016/j.ebiom.2015.09.018.
- 4.For recent studies on the synthesis of flavaglines, see: Thuaud F, Ribeiro N, Gaiddon C, Cresteil T, Désaubry L. J Med Chem. 2011;54:411. doi: 10.1021/jm101318b.Hawkins BC, Lindqvist LM, Nhu D, Sharp PP, Segal D, Powell AK, Campbell M, Ryan E, Chambers JM, White JM, Rizzacasa MA, Lessene G, Huang DCS, Burns CJ. Chem Med Chem. 2014;9:1556. doi: 10.1002/cmdc.201400024.Liu T, Nair SJ, Lescarbeau A, Belani J, Peluso S, Conley J, Tilloston B, O’Hearn P, Smith S, Slocum K, West K, Helble J, Douglas M, Bahadoor A, Ali J, McGovern K, Fritz C, Palombella VJ, Wylie A, Castro AC, Tremblay MR. J Med Chem. 2012;55:8859. doi: 10.1021/jm3011542.Lajkiewicz NJ, Cognetta AB, III, Niphakis MJ, Cravatt BF, Porco JA., Jr J Am Chem Soc. 2014;136:2659. doi: 10.1021/ja412431g.
- 5.Bruce I, Cooke NG, Diorazio LJ, Hall RG, Irving E. Tetrahedron Lett. 1999;40:4279. [Google Scholar]
- 6.a) Gerard B, Sangji S, O’Leary D, Porco JA., Jr J Am Chem Soc. 2006;128:7754. doi: 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]; b) Gerard B, Cencic R, Pelletier J, Porco JA., Jr Angew Chem. 2007;119:7977. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2007;46:7831. [Google Scholar]; c) Roche SP, Cencic R, Pelletier J, Porco JA., Jr Angew Chem. 2010;122:6683. doi: 10.1002/anie.201003212. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2010;49:6533. [Google Scholar]; d) Lajkiewicz NJ, Roche SP, Gerard B, Porco JA., Jr J Am Chem Soc. 2012;134:13108. doi: 10.1021/ja305342f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia B, Gerard B, Solano DM, Wan J, Jones G, II, Porco JA., Jr Org Lett. 2011;13:1346. doi: 10.1021/ol200032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For previous syntheses of 3-hydroxyquinolinones, see: Yushchenko DA, Bilokin MD, Duportail G, Mély Y, Pivovarenko VG. Tetrahedron Lett. 2006;47:905.Hradil P, Hlavác J, Lemr K. J Heterocyclic Chem. 1999;36:141.Hradil P, Grepl M, Hlavác J, Lycka A. Heterocycles. 2007;71:269.Hradil P, Kvapil L, Hlavác J, Weidlich T, Lycka A, Lemr K. J Heterocyclic Chem. 2000;37:831.
- 9.Zhu Y, Drueckhammer DG. J Org Chem. 2005;70:7755. doi: 10.1021/jo0513818. [DOI] [PubMed] [Google Scholar]
- 10.Please see the Supporting Information for details.
- 11.Gandhi SS, Bell KL, Gibson MS. Tetrahedron. 1995;51:13301. [Google Scholar]
- 12.For recent reviews on flow photochemistry, see: McQuade DT, Seeberger PH. J Org Chem. 2013;78:6384. doi: 10.1021/jo400583m.Su Y, Straathof NJW, Hessel V, Noël T. Chem Eur J. 2014;20:10562. doi: 10.1002/chem.201400283.Cambié D, Bottecchia C, Straathof NJW, Hessel V, Noël T. Chem Rev. 2016 doi: 10.1021/acs.chem-rev.5b00707.
- 13.CCDC 1457719 (21) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 14.For the X-ray structure of a N-methyl indoline with a typical pyrimidalized nitrogen, see: Chiaroni PA, Riche NLC. Acta Cryst. 1977;B33:3410.
- 15.For photophysical studies of 3-hydroxyquinolinones, see: Yushchenko DA, Shvadchak VV, Mély Y, Pivovarenko VG. New J Chem. 2006;30:774.Yushchenko DA, Shvadchak VV, Klymchenko AS, Mély Y. J Phys Chem A. 2007;111:8986. doi: 10.1021/jp071075t.
- 16.Venturella P, Bellino A, Piozzi F, Marino ML. Heterocycles. 1976;4:1089. [Google Scholar]
- 17.Spence TWM, Tennant G. J Chem Soc C. 1971:3712. [Google Scholar]
- 18.The relative stereochemistry of cycloadduct 24 was confirmed by reduction with NaBH4 to provide an alcohol derivative which was subjected to NOESY NMR studies. See Supporting Information for further details.
- 19.For the synthesis of rocaglate analogues and biological studies, see: Rodrigo CM, Cencic R, Roche SP, Pelletier J, Porco JA., Jr J Med Chem. 2012;55:558. doi: 10.1021/jm201263k.Wolfe AL, Singh K, Zhong Y, Drewe P, Rajsekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, Schatz JH, Rodrigo CM, Zhao C, Rondou P, de Stanchina E, Teruya-Feldstein J, Kelliher MA, Speleman F, Porco JA, Jr, Pelletier J, Rätsch G, Wendel HG. Nature. 2014;513:65. doi: 10.1038/nature13485.Stone SD, Lajkiewicz NJ, Whitesell L, Hilmy A, Porco JA., Jr J Am Chem Soc. 2015;137:525. doi: 10.1021/ja511728b.
- 20.a) Garro-Helion F, Merzouk A, Guibé F. J Org Chem. 1993;58:6109. [Google Scholar]; b) Wang H, Reisman SE. Angew Chem. 2014;126:6320. doi: 10.1002/anie.201402571. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2014;53:6206. [Google Scholar]
- 21.For N-deallylation methods, see: Alcaide B, Almendros P, Alonso JM, Aly MF. Org Lett. 2001;3:3781. doi: 10.1021/ol0167412.Escoubet S, Gastaldi S, Timokhin V, Bertrand M, Siri D. J Am Chem Soc. 2004;126:12343. doi: 10.1021/ja049859x.Kitov PI, Bundle DR. Org Lett. 2001;3:2835. doi: 10.1021/ol016278t.Chandrasekhar S, Reddy R, Rao RJ. Tetrahedron. 2001;57:3435.
- 22.Hutait S, Batra S. Tetrahedron Lett. 2010;51:5781. [Google Scholar]
- 23.Novac O, Guenier A, Pelletier J. Nucl Acids Res. 2004;32:902. doi: 10.1093/nar/gkh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Nat Rev Drug Discov. 2015;14:261. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.