

Abstract
Resolution of acute inflammation is an active process governed by specialized pro-resolving mediators (SPM) including resolvin D2 (RvD2) that activates a cell surface G protein–coupled receptor (GPCR), GPR18/DRV2. Here, we investigated RvD2-DRV2-dependent resolution mechanisms using DRV2-deficient mice (DRV2-KO). In polymicrobial sepsis initiated by cecal ligation and puncture (CLP), RvD2 (~2.7 nmol/mouse) significantly increased survival (>50%) of wild-type (WT), reduced hypothermia and bacterial titers compared to vehicle-treated CLP mice that succumbed at 48h. Protection by RvD2 was abolished in DRV2-KO mice. Mass spectrometry-based lipid mediator metabololipidomics demonstrated that DRV2-KO infectious exudates gave higher pro-inflammatory leukotriene (LT) B4 and pro-coagulating thromboxane (TX) B2, as well as lower SPM, including RvD1 and RvD3, compared to WT. RvD2-DRV2-initiated intracellular signals were investigated using mass cytometry (CyTOF) which demonstrated that RvD2 enhanced phosphorylation of CREB, ERK1/2 and STAT3 that were absent in DRV2-KO macrophages. Monitored by real-time imaging, RvD2-DRV2 interaction significantly enhanced phagocytosis of live E. coli, an action dependent on PKA and STAT3 in macrophages. Taken together, we identified an RvD2-DRV2 axis that activates intracellular signaling pathways that increase phagocytosis-mediated bacterial clearance, survival and organ protection. Moreover, these results provide evidence for RvD2-DRV2 and their downstream pathways in pathophysiology of infectious inflammation.
Keywords: eicosanoids, phagocytes, innate response, resolution of infection, pro-resolving mediators
Introduction
Inflammation is a protective response to defend the host against infection and injury (1). Ungoverned and excessive inflammation is an underlying pathology of many prevalent diseases, including cardiovascular diseases, diabetes, arthritis and sepsis (2-4). Complete resolution of acute inflammatory responses was thought to be a passive process with dissipation or dilution of local chemoattractants and pro-inflammatory mediators, allowing tissues to return to homeostasis (1). In recent years, we obtained first evidence that resolution of self-limited inflammation is not merely a passive termination, but rather an actively orchestrated programmed response that is rapidly turned on during acute inflammatory challenges, permitting inflamed and injured tissues to return via catabasis to function (5, 6). This event is driven in part by temporal lipid mediator class switching from generation of pro-inflammatory mediators (e.g. leukotriene B4) to the biosynthesis of lipoxins (7) and Specialized Pro-resolving Mediators (SPM) (8-11). SPMs are evolutionally conserved chemical structures derived from polyunsaturated fatty acids including eicosapentaenoic acid (EPA)-derived E-Series resolvins and docosahexaenoic acid (DHA)-derived D series resolvins, protectins and maresins. Complete stereochemistries of these SPM are established and total organic synthesis achieved that also confirmed their potent pro-resolving actions. SPM are potent mediators governing not only innate immune responses in host defense, but also pain, organ protection and tissue remodeling (recently reviewed in (8)).
SPM derived from DHA including resolvinD2 (RvD2) were first identified and isolated from murine self-resolving exudates during the resolution phase of self-limited acute inflammation in vivo (12). The biosynthesis of RvD2 involves 17-lipoxygenation of DHA to 17S-hydroperoxy-DHA (17S-HpDHA) that is further transformed enzymatically to a 7(8)epoxide-containing intermediate in leukocytes via 5-lipoxygenase (LOX), followed by enzymatic hydrolysis to form RvD2. Endogenous RvD2 production is documented in human serum, plasma (13), adipose tissue (14), placenta (15), lung (16), breast milk (17), and sepsis patients (18). With isolated human polymorphonuclear neutrophil (PMN), RvD2 increases intracellular phagosomal reactive oxygen species (ROS) generation for microbial killing (19). In whole blood at a single cell level using microfluidic chambers, RvD2 limits PMN chemotaxis and direct travel as well as increases random movement towards an IL-8 chemotactic gradient (20). RvD2 also decreases monocyte adhesion to adipocytes as well as their trans-adipose migration (21). RvD2 is a potent immunoresolvent that stereoselectively reduces excessive PMN trafficking in peritonitis and improves survival in sepsis (19). RvD2’s potent nanogram actions are also protective in disease models where RvD2 prevents inflammatory bowel disease such as colitis (22), alleviates inflammatory and fibromyalgia-induced pain (23, 24), increases survival following burn wound and reduces kidney and liver injuries in mice (25, 26), periodontitis (27) as well as nerve injuries as seen in Parkinson’s disease (28).
Resolution at cellular level consists of cessation of PMN entry into the tissue and elevated efferocytosis (i.e. macrophage phagocytosis of apoptotic PMN) (1). We introduced a quantitative definition of resolution of self-limited sterile acute inflammation denoted resolution indices that permit assessment of the resolution properties of SPM and pinpoint their unique mechanisms of action (6). SPM each lower the magnitude of leukocyte infiltration (Ymax) and/or shorten the resolution interval (Ri), which is the interval from the time point of maximum PMN infiltration to the time point of 50% PMN reduction in peritoneal exudates (17, 29). We also assessed the actions of SPM in resolution of E. coli infection using these resolution indices together with cellular composition and functions (30). During E. coli infection, RvD2 given at the peak of PMN infiltration (12h) significantly shortens the resolution interval by half, enhances PMN apoptosis and macrophage efferocytosis in self-limited E. coli infection (31). Based on these potent pro-resolving actions of SPM, we proposed the “cardinal signs of resolution” including clearance of debris (expurgatio reliquiorum), clearance of infective agents (expurgatio contagionem agentis), analgesia (doloris absentia) and gain of function (muneris lucrum) (32). With these cellular, molecular and quantitative definitions, we now appreciate the mechanisms controlling the active resolution programs of inflammation and have pinpointed the pro-resolving actions of SPM.
Polymicrobial sepsis is a complex scenario containing phases of both excessive inflammatory responses temporally associated with immunosuppressive states (33). Sepsis remains an unmet clinical challenge with high mortality rates and increasing incidence (34, 35). Recently, we identified a G protein-coupled receptor for RvD2 termed DRV2/GPR18, and demonstrated specific binding of RvD2 to human recombinant DRV2 (31). In human macrophages, RvD2 stimulates phagocytosis and efferocytosis in a DRV2-dependent manner. RvD2-DRV2 interaction stimulates phagocyte functions to accelerate resolution of bacterial infections. Here, we tested the hypothesis that the DRV2 receptor is pivotal in systemic infection and report that RvD2-DRV2 interactions stimulated bacterial clearance and reduced mortality in microbial sepsis. This ligand-receptor activation initiates specific intracellular signal transduction pathways (e.g. PKA, STAT3) that contribute to RvD2-DRV2 stimulated phagocytosis of live E. coli.
Materials and Methods
Cecal ligation and puncture (CLP)
Targeted deletion of mouse gpr18 (NM_182806) was constructed by Lexicon Pharmaceuticals in a 129/SvEv-C57B/6 mixed background (31). Male mice (10-12 weeks old) were used for all experiments and fed ad libitum Laboratory Rodent Diet 20-5058 (Lab Diet, Purina Mills). All experimental procedures used were approved by the Standing Committee on Animals of Harvard Medical School (protocol #02570) and complied with institutional and US National Institutes of Health (NIH) guidelines. CLP was carried out essentially as in (19). Mice were anesthetized with a mixture of oxygen and isoflurane 5% for anesthesia induction and 1.5% isoflurane for maintenance during surgery. After shaved, basal temperature was monitored. A cut (0.5cm) mid-line incision to mouse’s left side was performed; cecum was pulled out and ligated below the ileocecal valve with a 4-0 silk suture. Two punctures were performed with a 20-gauge needle, followed by a soft squeeze until stool was extruded. Cecum was then put back to the peritoneal cavity and animals were sutured with a 6-0 silk suture. Mice were then received 1μg of RvD2 intraperitoneally or 500μl saline with 0.1% ethanol (vehicle), and survival monitored over 120 hours. In a second group of animals, after 12h of CLP, peritoneal exudates were collected by lavaging with 5ml of PBS−/−. Peritoneal bacterial titers were determined by plating lavages on LB agar plates at 100X dilution, and the colony forming units (CFU) counted 24 hours later. Exudate cytokine levels were determined by proteome profiler mouse cytokine array following manufacturer’s instruction.
Lipid mediator metabololipidomics
Liquid chromatography–mass spectrometry (LC-MS)-MS-based metabololipidomics were performed with infectious exudates. Prior to sample extraction, ice-cold methanol containing deuterium labeled d4-LTB4, d4-5S-HETE, d4-PGE2, and d5-RvD2 internal standards (500pg each) were added to facilitate quantification. All samples were kept at −20°C for 45 min to allow protein precipitation and then subjected to solid-phase extraction as described (13). Extracted samples were analyzed by a liquid chromatography-ultraviolet-tandem mass spectrometry system, QTrap 5500 (AB Sciex) equipped with a Shimadzu LC-20AD HPLC (Tokyo, Japan). A Proshell 120 EC-18 column (100mm×4.6mm×2.7μm; Agilent Technologies, Santa Clara, CA, USA) was kept in a column oven maintained at 50°C, and lipid mediators (LM) were eluted with a gradient of methanol/water/acetic acid from 55:45:0.01 (v/v/v) to 100:0:0.01 at 0.5mL/min flow rate. To monitor and quantify the levels of targeted LM, a multiple reaction monitoring (MRM) method was devised with signature ion fragments for each molecule. Identification was conducted using published criteria including retention times and at least six diagnostic ions. Calibration curves were obtained using synthetic and authentic LM mixtures, including d4-LTB4, d5-LXA4, d4-PGE2, d5-RvD2, RvD1, RvD2, RvD5, PD1, MaR1, RvE1, RvE2, LXA4, LXB4, PGE2, PGD2, PGF2α, TXB2, LTB4 at 1.56, 3.12, 6.25, 12.5, 25, 50, 100 pg. Linear calibration curves for each compound were obtained with r2 values of 0.98-0.99. Quantification was carried out based on peak areas of the MRM transitions.
Proteome Profiler array
Peritoneal lavages were collected as described at 12h post CLP. Cell-free supernatants were collected by centrifugation. A 1:10 dilution of the supernatant (150μl) were incubated with the pre-coated Proteome Profiler array membranes (ARY028, R&D Systems) and processed according to the manufacturer’s instructions. Densitometric analysis of dot blots was performed using Image J software (National Institute of Health, Bethesda, MD, USA).
Real-time imaging of phagocytosis
Mouse bone marrow cells were collected and differentiated to macrophages with mouse GM-CSF (10ng/ml) for 6 days. These macrophages were plated onto 8-well chamber slides (0.5 × 105 cells/well) for overnight before the experiments. Chamber slides were kept in a Stage Top Incubation system for microscopes equipped with a built-in digital gas mixer and temperature regulator (TOKAI HIT model INUF-K14). Cells were treated with RvD2 (10nM), PKA inhibitor H89 (3μM; Sigma-Aldrich), RvD2 plus H89, or vehicle control for 15minutes at 37°C, followed by addition of Baclight Green-labeled E. coli at a proportion of 50:1 (E. coli: Macrophage) to initiate phagocytosis. Fluorescent images were then recorded every 10 min for 100 min (37°C) with Keyence BZ-9000 (BIOREVO) inverted fluorescence phase-contrast microscope (20X objective) equipped with a monochrome/color switching camera using BZ-II Viewer software (Keyence). Three separate experiments were performed. In each experiment, three fields (20x) per condition (per well) were recorded. Green fluorescence intensity was quantified using BZ-II Analyzer.
Cyclic AMP levels
For cAMP measurements, resident peritoneal MΦ from naïve WT or DRV2-KO mice were collected with 5ml PBS−/−, and plated in a 12-well plate (0.5×106 cells/well) with RPMI supplemented with 10% FBS. The next day, media was replaced with PBS+/+ and cells incubated with RvD2 (0.1 – 100 nM), forskolin (10μM; Sigma-Aldrich) or vehicle control for 15min at 37°C. Lysis buffer (100μl) was added to stop incubation and cells homogenized. cAMP levels were measured by ELISA following manufacturer’s instruction (Elite cAMP ELISA Assay kit, eEnzyme, CA-C315).
Macrophage phagocytosis of E. coli
Resident peritoneal MΦ were collected from naïve WT and DRV2-KO mice and plated onto 96-well plates (0.5×105 cells/well). RvD2 (1 pM-10 nM) or vehicle controls was incubated with MΦ for 15 min at 37°C, followed by incubation with FITC-labeled serum-treated zymosan particles at 10:1 ratio (zymosan: MΦ) or Baclight Green-labeled E. coli at 50:1 ratio (E. coli : MΦ) for 60 min at 37 °C. Plates were gently washed, extracellular fluorescence quenched by trypan blue, and phagocytosis determined by measuring total fluorescence (Ex 493/Em535 nm) using a fluorescent plate reader (Molecular Probes). The following inhibitors were added together with RvD2 or vehicle control: PKA inhibitor H89 (3μM; Sigma-Aldrich), pERK1/2 inhibitor (50μM; Tocris) and STAT3 inhibitor (100μM; Tocris)
Mass cytometry (CyTOF)
Peritoneal macrophages from DRV2-KO mice and WT were collected as in (18). Peritoneal cells were incubated with RvD2 (10 nM) for 0, 1, 5, 15 and 30 minutes at 37°C, followed by 1.6% of paraformaldehyde for 10 minutes at room temperature. Cells were barcoded following manufacture’s protocol with Palladium Isotopes (Pd 102, 104, 105, 108, and 110) (Fluidigm, Science). Brief, cells were washed twice using barcoding permeabilization buffer. Diluted barcodes were transferred to cells and incubated for 30 minutes at room temperature. Barcoded cells were washed twice in CyTOF staining buffer (PBS with 0.5% BSA and 0.1% sodium azide), and then pooled for staining. Pooled barcoded cells were incubated for 10 minutes with Fc block (Biolegend, San Diego, CA USA) for Fc receptor mediated non-specific antibody binding. Cells were then stained for 30 minutes with metal-label surface antibodies at room temperature, then wash twice in CyTOF staining buffer. Cells were then permeabilized in 80% ice-cold methanol for 10 min at −20°C. After washing twice to remove the methanol, cells were stained with metal conjugated antibodies for intracellular markers at room temperature for 30 minutes. The antibodies used for CyTOF were listed in Supplemental Table 3. Cells were then washed twice and stained in 500μl of 1:1000 Iridium intercalator (DVS Science, Toronto) diluted in PBS overnight at 4°C. Cells were then washed twice in CyTOF staining buffer and twice in MilliQ-filtered deionized water (36). Cells were reconstituted at a concentration of 5×106 cells/ml containing EQ calibration beads (EQ four elements Calibration Beads, Fluidigm, Science) according to manufacture’s protocol. Barcoded and cells were analyzed on Helios CyTOF (Fluidigm, Science) at event rate of 400-500 cells per second. The data were normalized using v6.3.119 Helios Software (Fluidigm) at LMA CyTOF facility at Dana Farber Cancer Institute (Boston, MA, USA). Files were debarcoded using Fluidigm Debarcoder application. Gating was performed in Cytobank Platform (Cytobank, Mountain View, CA, USA). Sequential gating strategy was used to analyze signaling events in resident peritoneal macrophages (CD11b+F4/80+). Phosphorylation levels of signaling molecules at 0, 1, 5, 15 and 30 minutes after exposure of RvD2 (10nM) in WT and DRV2-KO mice. Phosphorylation levels were calculated as the difference between the inverse hyperbolic sine (arcsinh) of the median signal intensity of at indicated time points and the arcsinh of the median signal intensity in unstimulated (0 min) signal (37).
Statistics
Statistical analysis was performed using Student’s t test (two group comparisons) and one-way ANOVA (multiple group comparisons). Kaplan-Meier survival curves were analyzed using a one-tailed log-rank (Mantel-Cox) test. In all cases a P<0.05 was considered significant.
Results
RvD2 protects mice from sepsis in a GPR18/DRV2 receptor-dependent manner
Since RvD2 displays potent actions and RvD2 receptor, namely GPR18/DRV2 was identified (18), we set out to investigate whether RvD2-DRV2 receptor axis is protective in polymicrobial sepsis. Using mice deficient in DRV2, we carried out cecal ligation and puncture (CLP), a polymicrobial systemic sepsis model that closely resembles human pathology (38). In wild-type (WT) animals, RvD2 significantly increased survival (>50%) in CLP compared to vehicle-treated mice that all perished before 48h (Figure 1A, left panel). In contrast, in DRV2 deficient mice (DRV2-KO), there were not significant differences in survival between RvD2-and vehicle-treated mice (Figure 1A, right panel). It was noted with vehicle-treated CLP mice, DRV2-KO succumbed earlier (12h) than WT (24h). Both WT and KO reached ~50% mortality between 24-30h. At 48h the remaining WT mice perished while KO had 20% survival. Log-rank test comparing the survival distributions of these two groups (WT CLP+veh vs. KO CLP+veh) did not reach statistical significance (P=0.87). It is possible, however, that there was genetic and/or functional compensation in DRV2-KO, e.g. expressions of other pro-resolving receptors and/or pro-inflammatory receptors might be altered in DRV2-KO. These could contribute to the apparent different outcome than that with WT mice survival following CLP.
Figure 1. RvD2 protects polymicrobial sepsis in a DRV2-dependent manner.

Mice were administered with RvD2 (1μg) or vehicle i.p. following CLP. (A) % survival of (left) wild type or (right) DRV2-KO mice. **p<0.01 log-rank (Mantel-Cox) test. NS: Non significant. Results are from 10 mice in each group. (B) Changes in body temperatures 24h after CLP. Results are expressed as mean±SEM from 5-8 mice/group. *p<0.05, CLP + veh. vs. CLP + RvD2 in WT group #p<0.05, vs. naive mice using unpaired Student’s t test. (C) Twelve hours after CLP, exudates were collected and microbial counts were determined. Results are expressed as mean±SEM from 4-6 mice/group. *p<0.05, CLP + veh. vs. CLP + RvD2 in WT group using unpaired Student’s t test.
We monitored body temperature at 24h after CLP, which induced hypothermia in mice compared to naïve mice. RvD2 reduced hypothermia (35.5°C±0.8) compared to vehicle controls (31.7°C±1.2) in WT, but not in DRV2-KO (Figure 1B). Additionally, in WT RvD2 enhanced bacterial killing, significantly reducing bacterial titers (105.5±32.3 cfu/cm2) compared to the vehicle-treated group (234.8±82.3 cfu/cm2), a response diminished in DRV2-KO (Figure 1C and supplemental figure 1). Thus, RvD2-DRV2 interactions in vivo increased survival in CLP mice, protected hypothermia and enhanced bacterial clearance from infectious exudates.
Lipid mediator metabololipidomics and proteome profiling
We next questioned whether DRV2-KO give heightened inflammatory status in sepsis, and carried out mass spectrometry (MS)-based metabololipidomics focusing on local acting lipid mediators (LM). Each LM was profiled using multiple reaction monitoring and identified by direct comparison with synthetic and authentic standards using matching criteria including retention time, characteristic fragmentation patterns, and at least six diagnostic ions (13). In infectious exudates obtained from CLP mice, we identified resolvins and other SPM, specifically eicosapentaenoic acid derived E-series Resolvins, docosahexaenoic acid derived D-series Resolvins, Protectins, and Maresin, as well as arachidonic acid derived leukotrienes, prostanoids and lipoxins (Figure 2A and supplemental table 1). A representative MS-MS spectrum of RvD1 used for identification is shown in Figure 2B. Principal Component Analysis (PCA) was used for exploring cross-covariance between WT and DRV2-KO. The 3D loading plot showed two distinct clusters, one with mostly pro-inflammatory and pro-coagulating LM (i.e. PG, LT and TX) that was associated with DRV2-KO. The other cluster contained pro-resolving mediators, e.g. RvE1-3, RvD1-5, PD1, MaR1, and LXA4, that were associated with WT animals (Figure 2C). We quantified each LM and found significant increases ~85% in pro-inflammatory and pro-coagulating LM e.g. prostaglandins plus thromboxane (Figure 2D), and leukotriene B4 plus 5-HETE (~95% increase; Figure 2E) in DRV2-KO compared to WT. In addition, DRV2-KO showed significant decreases ~60% in D-series resolvins including RvD1, AT-RvD1, RvD3, and AT-RvD3, compared to WT littermates (Figure 2F).
Figure 2. Lipid mediator metabololipidomics: DRV2-KO versus WT mice.

(A) MRM chromatographs (B) Representative MS-MS of RvD1. (C) principal component analysis of exudates lipid mediator 3D Loading Plot. (D) Exudate levels of prostanoids (PGs, TxB2), LTB4 plus 5-HETE, and D-series resolvins (RvDs) (pg/ml) in WT and DRV2-KO mice 12h after CLP. Results are expressed as mean±SEM from 4-5 mice/group *p<0.05 WT vs. DRV2-KO using unpaired Student’s t test.
Next, we carried out proteome profiling for CLP. Cell-free supernatants from infectious exudates showed significant up-regulation of a panel of cytokines compared to naïve mice at 12h with both WT and DRV2-KO (Figure 3A). RvD2 treatment significantly up-regulated a panel of proteins including matrix metalloproteinase (MMP)-2, MMP-3 and myeloperoxidase (MPO) in WT (Figure 3B and supplemental table 2). However, these cytokines were not significantly altered in DRV2-KO by RvD2 treatment. It was reported that loss of MMP-2 leads to increases in MCP-3 levels and exacerbates myocarditis in mice, pointing to a potential protective role of MMP-2 via its function in chemokine cleavage (39).
Figure 3. RvD2 regulates select proteins in DRV2-dependent manner.

Proteome Profiler cytokine array was carried out using 12h CLP infectious exudates. (A) Results are expressed as relative abundance to reference spots in a heat map with 111 proteins. (B) Levels of MMP2, MMP3 and MPO; mean±SEM from 3-6 mice/group. *p<0.05, CLP vs. CLP plus RvD2 in WT group; ##p<0.01 CLP plus RvD2 in WT vs. KO groups using unpaired Student’s t test. NS: Non significant. (Insets) Representative dot plot images.
In addition, we carried out statistical analysis between WT-RvD2 and KO-RvD2 groups and found statistically significant reduction in several proteins in KO mice compared to WT. These include IL-22, LIX (CXCL5), MMP-3, MMP-9, Pentraxin 2/SAP, Pentraxin 3/TSG-14, Reg3G, and Serpin F1 (Supplemental table 2). These proteins have been reported to have protective roles in regulating immune responses during infections. For example, IL-22 controls pathobionts via regulation of the complement system, promoting resistance after pathogen-induced intestinal damage (40). Reg3G restricts bacterial colonization of mucosal surfaces and reduces bacterial translocation (41). Pentraxin 3/TSG-14 improves survival in endotoxic shock and CLP (42). Therefore, reduction in these proteins in KO-RvD2 mice might contribute to their higher mortality rate and impaired bacterial clearance compared to WT-RvD2 group (Figure 1A&C).
Taken together, our results demonstrated that DRV2-KO showed dysregulated LM profiles, giving heightened pro-inflammatory LM and reduced SPM compared to WT in sepsis. Also, RvD2 regulates a select panel of inflammation-related proteins in a DRV2-dependent manner.
RvD2-DRV2-dependent intracellular signaling
Next, we investigated RvD2-DRV2-initiated intracellular signals using mass cytometry (CyTOF) with naïve mouse peritoneal macrophages. Macrophages collected from WT and DRV2-KO were incubated with RvD2 (10 nM) for 0-30 min and cells collected for staining with specific antibodies for cell surface markers, followed by permeabilization and staining of intracellular targets including a panel of phospho-proteins (see Methods). Macrophages were identified as CD11b+F4/80+ populations. See gating strategy in Supplemental Figure 1. RvD2 time-dependently increased phosphorylation of pAKT, pp38MAPK, pCREB, pS6, pERK1/2, pSTAT1, pSTAT3 and pSTAT5 each with different kinetics in macrophages. pAKT, pp38MAPK, pCREB, pS6 levels reached maximum at 1 min then gradually declined. pERK1/2 was detected at 1 min and peaked at 30 min after RvD2 stimulation (Figure 4A). In comparison, pSTAT1, pSTAT3 and pSTAT5 were also markedly increased as early as 1 min, and their levels remained elevated till 15-30 min. In macrophages from DRV2-KO, up-regulation of these phospho-proteins by RvD2 was abolished. In separate sets of experiments, pCREB levels were validated using flow cytometry (Figure 4B). Quantification of pERK1/2, pSTAT3 and pCREB using CyTOF and flow cytometry are shown in Figure 4C. These results indicate that RvD2 regulates phosphorylation of select kinases and transcription factors with different kinetics in macrophages in a DRV2-dependent manner.
Figure 4. RvD2-DRV2-dependent macrophage intracellular signaling.

(A) Heat maps of phosphorylated signaling molecules at 0, 1, 5, 15 and 30 minutes after exposure of RvD2 (10nM) in WT and DRV2-KO mice were obtained using CyTOF (see Methods). Phosphorylation levels were calculated as the difference between the inverse hyperbolic sine (arcsinh) of the median signal intensity in RvD2-treated peritoneal resident macrophages (at 0, 1, 5, 15, 30 min) and the arcsinh of the median signal intensity in vehicle-treated macrophages at 0 min. (bottom) gating strategy for macrophages (CD11b+F4/80+) and representative histograms of pCREB. (B) Flow cytometry for pCREB: gating strategy for macrophages (CD11b+F4/80+) and representative histograms. (C) Relative intensity of pERK1/2, pSTAT3, and pCREB. Results are mean±SEM from 3 independent experiments and samples were pooled from 5 mice in each group (WT vs. DRV2-KO) for each experiment. *p<0.05, **p<0.01 WT plus RvD2 vs DRV2-KO plus RvD2 using 2-tailed Student t-test.
STAT3 and PKA pathways mediates RvD2-DRV2-stimulated phagocytosis
Next, we investigated the role of select signaling components in RvD2-DRV2-stimulated phagocytosis identified using CyTOF, including PKA (that phosphorylates CREB), STAT3 (that is involved in phagosome maturation) and ERK. First, we monitored phagocytosis of live fluorescence-labeled E. coli by bone marrow-derived macrophages (BMDM) using real-time imaging. Fluorescence intensity increased with time (0-120 min) represents increased macrophage ingestion of E. coli (Figure 5A). RvD2 addition prior to E. coli (10 nM, 15 min) significantly enhanced phagocytosis (~95% at 100 min; Figure 5A). This RvD2 action was not observed in DRV2-KO BMDM, indicating the role of the RvD2-DRV2 axis in stimulating bacterial clearance via phagocytosis. Since cAMP activates PKA, which can phosphorylate pCREB (43), we determined cAMP levels with naïve peritoneal resident macrophages and found that RvD2 (10 - 100 nM) significantly increased cAMP levels in WT but not DRV2-KO (Figure 5B). A sub-optimal concentration of Forskolin (10 μM) was used as a control and taken as 100% (Figure 5B). This is consistent with earlier findings that RvD2 (10-100 nM) increases cAMP levels that are dependent on DRV2 in human macrophages (31).
Figure 5. PKA and STAT3 pathways mediates RvD2-stimulated phagocytosis.

(A) Differentiated mouse bone marrow macrophages collected from WT and DRV2-KO were plated onto 8-wells chamber slides (0.5 × 105 cells/well) and incubated with vehicle or RvD2 (10 nM) for 15 min at 37°C, followed by addition of BacLight Green-labeled E. coli to initiate phagocytosis. Fluorescent images were then recorded every 10 min. Three separate experiments were performed. In each experiment, three fields (20x) per condition (per well) were recorded. Results are mean fluorescent intensity (MFI); mean from 3 independent experiments with 3 mice/group in triplicates (3 fields/well). *p<0.05 using unpaired Student’s t test. NS: non-significant. (Insets) Representative fluorescent images at 100 min; scale bars, 50μm. (B) Mouse resident naïve macrophages were collected from WT and DRV2-KO, and incubated with 1-100 nM of RvD2 for 15 min and cAMP levels determined. Results are expressed as % increase of cAMP. cAMP levels in the presence of forskolin (10 μM) was taken as 100%; mean±SEM from n=3. *p<0.05 WT vs DRV2-KO. (C) Mouse resident naïve macrophages collected from WT were plated onto 8-wells chamber slides (1 × 105 cells/well) and incubated with vehicle or RvD2 (10 nM) in the presence or absence of a STAT3 inhibitor (NSC 74859; 100μM), PKA inhibitor (H89; 3μM) or ERK inhibitor (FR 180204; 10μM for 15 min at 37°C, followed by addition of BacLight Green-labeled E. coli to initiate phagocytosis. Fluorescent images were then recorded as in panel (A). Results are (left) mean fluorescent intensity (MFI) from a representative of n=3-4, (right) fold changes vs E. coli alone, mean±SEM from 3-4 independent experiments in triplicates or quadruplicates (3-4 fields/well). (D) Mouse resident naïve macrophages collected from WT were plated onto 96-well plates (0.5 × 105 cells/well) and incubated with vehicle or RvD2 (10 nM) in the presence or absence of a STAT3 inhibitor (NSC 74859; 100μM), PKA inhibitor (H89; 3μM) or ERK inhibitor (FR 180204; 10μM for 15 min at 37°C, followed by addition of FITC-labeled serum-treated zymosan (STZ) to initiate phagocytosis. Fluorescent images were then monitored using a plate reader. Results are mean±SEM from 3-4 independent experiments in triplicates. (C,D) *p<0.05, **p<0.01, ***p<0.001 using one-way ANOVA with post hoc multiple-comparison test (Newman-Keuls).
We next questioned whether PKA, STAT3 and ERK1/2 play a role in RvD2-stimulated phagocytosis. RvD2 incubation prior to E. coli addition (10 nM, 15 min) significantly enhanced phagocytosis. Incubation of macrophages with a STAT3 inhibitor (NSC 74859; 100 μM) or PKA inhibitor (H89; 3 μM) together with RvD2 significantly reduced RvD2-stimulated phagocytosis (Figure 5C). Similar results were obtained with BMDM, where H89 also reduced RvD2-enhanced phagocytosis of live E. coli. In comparison, a ERK inhibitor (FR 180204; 10 μM significantly change RvD2-stimulated phagocytosis (Figure 5C). Macrophage phagocytosis of serum-treated zymosan particles (STZ) was also carried out. STAT3 and PKA inhibition, but not ERK inhibition significantly blocked RvD2-stimulated phagocytosis (Figure 5D). Together, our results demonstrate that DRV2 contributes to RvD2’s pro-resolving actions in macrophage clearance of live E. coli and this action is dependent on cAMP-PKA and STAT3 signaling pathways (Figure 6).
Figure 6. Schematic representation of RvD2-DRV2-dependent signaling pathways involved in macrophage phagocytosis.
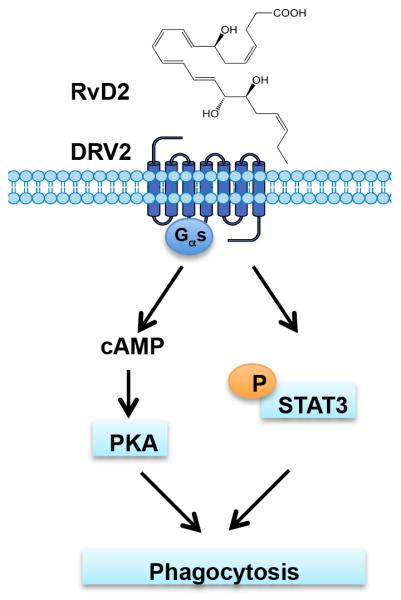
RvD2-DRV2 interactions initiate (i) Gαs protein coupling, leading to activation of cAMP-PKA signaling pathway and (ii) phosphorylation of STAT3, that contributed to macrophage phagocytosis.
Discussion
RvD2 is a potent immunoresolvent and controller of leukocyte traffic, and is protective in a wide range of disease models including airway and gastrointestinal inflammation (8, 19). In the present report, we demonstrated that RvD2-DRV2 interaction protected mice from sepsis, preventing hypothermia, enhancing phagocytosis-based bacterial clearance, and increasing survival. In infectious exudates collected from sepsis, DRV2-KO gave increased levels of PG, LT and TX, and reduced SPM (i.e. RvD1, RvD3, AT-RvD1, AT-RvD3) in infectious exudates (Figure 2). These results are consistent with those found in E. coli peritoneal infections where select SPM including AT-RvD1, RvD2, RvD5, PD1 and AT-PD1 were significantly reduced in DRV2-KO mice compared to WT. DRV2-KO also gave increased amounts of TX (31), indicating that DRV2-KO is associated with heightened inflammatory status during bacterial infection. Of interest, mice deficient in an RvD1 receptor, namely ALX, also gave heightened disease severity, including hypothermia and cardiac dysfunction during sepsis (44). Conversely, ALX transgenic mice (i.e. mice overexpressing human ALX) gave reduced inflammatory status with markedly decreased PMN infiltrates in peritonitis (45). Along these lines, mice overexpressing the human RvE1 receptor, namely ChemR23/ERV1, also showed lower PMN infiltration in peritonitis, and in addition, reduced ligature-induced alveolar bone loss (reviewed in ref (8)). Together, these results point to an endogenous role(s) of SPM receptors (e.g. ALX, ERV1 and DRV2) as checkpoint controllers of resolution during inflammation and bacterial infections.
We monitored a panel of phospho-proteins using CyTOF. These include transcription factors CREB, STAT1, STAT3 and STAT5, and kinases such as ERK1/2, p38MAPK and Akt (Figure 4A). We found that RvD2 significantly increased phosphorylation of CREB, the cAMP response element binding protein, in WT but not DRV2-KO macrophages (Figure 4A), suggesting that RvD2-DRV2 axis could regulate a panel of CRE-containing genes in a PKA-dependent manner (43). In mouse macrophages, RvD2 enhanced cAMP levels in a DRV2-dependent manner (Figure 5B). In addition, cAMP-activated protein kinase (PKA) mediated RvD2-stimulated phagocytosis (Figure 5C&D). Earlier we reported that RvD2 activates recombinant human DRV2 that is sensitive to cholera toxin, suggesting receptor coupling to a Gαs-like protein. Also, in human macrophages, RvD2 does-dependently increases cAMP, that was abolished when DRV2 was knocked down using specific shRNA (31). Thus, the present results together with earlier findings indicate that RvD2-DRV2 interactions initiate Gαs protein coupling, leading to activation of cAMP-PKA signaling pathway that enhanced macrophage phagocytosis (Figure 6). Activation of PKA by cAMP enhances Rac1 activity, leading to increased efferocytosis with peritoneal resident macrophages (46). Also, a cAMP analog activates EPAC (an GTP/GDP exchange factor), which in turn increases the levels of GTP-Rap1, leading to F-actin formation and enhanced phagocytosis of STZ in RAW264.7 murine macrophages (47). These components, namely Rac1, EPAC, Rap1, F-actin might also contribute to RvD2-stimulated phagocytosis of E. coli and STZ reported herein (Figure 5C&D). In this context, RvD1 rescues macrophages from oxidative stress-induced apoptosis during efferocytosis, suppressing activation of NADPH oxidase via cAMP-PKA signaling. This action is dependent on ALX (48). Thus, it is likely that Gαs-cAMP-PKA signaling cascade in macrophages is a shared pro-resolving mechanism for RvD1 and RvD2 receptors.
RvD2-DRV2 interactions also increased phosphorylation of STAT3, STAT5, ERK1/2, Akt and ribosomal protein S6 (Figure 4). S6 protein is a known downstream target of PI3K/Akt signaling pathway and the Raf/ERK pathway (49). In the case of RvE1, ERK/Akt/S6 pathway is involved in RvE1-stimulated phagocytosis in an ERV1-dependent manner (50). In addition, RvD1 and RvD2 increased phosphorylation of Akt and CREB in LPS-stimulate human monocytes (51). Inhibition of STAT3 phosphorylation decreases efferocytosis and M2 macrophage polarization in vitro (52). In addition, RAW264.7 murine macrophage phagocytosis of Staphylococcus aureus leads to up-regulation of genes involved in JAK-STAT pathway, including STAT3 and STAT5. Phosphorylation of STAT3 and STAT5 proteins is required for phagosome acidification and maturation (53). Thus, it is likely that STAT3 phosphorylation is also required for phagosome maturation during human macrophages phagocytosis. Disruption of this pathway with STAT3 inhibition diminished RvD2-enhanced phagocytosis of E. coli and STZ (Figure 5C&D). Along these lines, RvD1 up-regulates STAT3 in human monocytes (54). Together, these results indicate that each SPM stereoselectively activates its own specific receptor (e.g. RvD2-DRV2, RvD1-DRV1 and RvE1-ERV1). JAK-STAT and ERK/Akt/S6 are likely to be the common downstream pathways following receptor activation by SPM that are involved in SPM-initiated phagocytosis.
In summation, we provide evidence for an RvD2-DRV2 resolution axis that protects microbial sepsis, increasing bacterial clearance and improving survival. In addition, we identified DRV2 receptor-dependent signaling pathways in macrophages. Our present results suggest that these signaling components and pathways could represent “resolution signaling cassette/complex” in phagocytosis. Earlier reports established that other SPM activation of their receptors also mediate pro-resolving actions. For example, RvD1 regulates a panel of select micro-RNAs and their target genes involved in resolution of inflammation, including IκB kinase, IL-10, and 5-lipoxygenase in an ALX- and DRV1/GPR32-dependent manner (54). RvE1-ERV1 enhanced phagocytosis via Akt/S6 pathway (50). Resolvins and the other proresolution mediators from n-3 fatty acids are agonists of the resolution response and are conserved structures across the animal kingdom (8). Taken together, these findings provide new pro-resolution mechanisms of the host that may be of interest, providing potential therapeutic opportunities for the control of unwanted inflammation and infection that accompanies sepsis and infectious inflammatory diseases. Thus, SPM, their receptors and signaling pathways documented here provide examples of the potential for resolution pharmacology.
Supplementary Material
Acknowledgments
The authors thank Mary Small for assistance with manuscript preparation.
Grant numbers and sources of support: This work was supported in part by NIH grants R01GM38765 (C.N.S.) and R01GM38765-29S1 (S.L.).
References
- 1.Cotran RS. Inflammation: historical perspectives. In: Gallin JI, Snyderman R, Fearon DT, Haynes BF, Nathan C, editors. Inflammation: Basic Principles and Clinical Correlates. 3rd ed Lippincott Williams & Wilkins; Philadelphia: 1999. [Google Scholar]
- 2.Nathan C. Fresh approaches to anti-infective therapies. Sci Transl Med. 2012;4:140sr142. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126:23–31. doi: 10.1172/JCI82224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinarello CA, Joosten LA. Inflammation in rheumatology in 2015: New tools to tackle inflammatory arthritis. Nat Rev Rheumatol. 2016;12:78–80. doi: 10.1038/nrrheum.2015.180. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 7.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 8.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–567. doi: 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]
- 10.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12746–12751. doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307:C39–54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claria J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J Immunol. 2012;189:2597–2605. doi: 10.4049/jimmunol.1201272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keelan JA, Mas E, D'Vaz N, Dunstan JA, Li S, Barden AE, Mark PJ, Waddell BJ, Prescott SL, Mori TA. Effects of maternal n-3 fatty acid supplementation on placental cytokines, pro-resolving lipid mediators and their precursors. Reproduction. 2015;149:171–178. doi: 10.1530/REP-14-0549. [DOI] [PubMed] [Google Scholar]
- 16.Frediani JK, Jones DP, Tukvadze N, Uppal K, Sanikidze E, Kipiani M, Tran VT, Hebbar G, Walker DI, Kempker RR, Kurani SS, Colas RA, Dalli J, Tangpricha V, Serhan CN, Blumberg HM, Ziegler TR. Plasma metabolomics in human pulmonary tuberculosis disease: a pilot study. PLoS One. 2014;9:e108854. doi: 10.1371/journal.pone.0108854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnardottir H, Orr SK, Dalli J, Serhan CN. Human milk proresolving mediators stimulate resolution of acute inflammation. Mucosal Immunol. 2016;9:757–766. doi: 10.1038/mi.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalli J, Colas RA, Quintana C, Barragan-Bradford D, Hurwitz S, Levy BD, Choi AM, Serhan CN, Baron RM. Human sepsis eicosanoid and pro-resolving lipid mediator temporal profiles: correlations with survival and clinical outcomes. Crit. Care Med. 2016 doi: 10.1097/CCM.0000000000002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norling LV, Headland SE, Dalli J, Arnardottir HH, Haworth O, Jones HR, Irimia D, Serhan CN, Perretti M. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight. 2016;1:e85922. doi: 10.1172/jci.insight.85922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clària J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J. Immunol. 2012;189:2597–2605. doi: 10.4049/jimmunol.1201272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol. 2011;187:1957–1969. doi: 10.4049/jimmunol.1101305. [DOI] [PubMed] [Google Scholar]
- 23.Park CK, Xu ZZ, Liu T, Lu N, Serhan CN, Ji RR. Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J. Neurosci. 2011;31:18433–18438. doi: 10.1523/JNEUROSCI.4192-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein CP, Sperotto ND, Maciel IS, Leite CE, Souza AH, Campos MM. Effects of D-series resolvins on behavioral and neurochemical changes in a fibromyalgia-like model in mice. Neuropharmacology. 2014;86C:57–66. doi: 10.1016/j.neuropharm.2014.05.043. [DOI] [PubMed] [Google Scholar]
- 25.Bohr S, Patel SJ, Sarin D, Irimia D, Yarmush ML, Berthiaume F. Resolvin D2 prevents secondary thrombosis and necrosis in a mouse burn wound model. Wound Repair Regen. 2013;21:35–43. doi: 10.1111/j.1524-475X.2012.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue Y, Yu YM, Kurihara T, Vasilyev A, Ibrahim A, Oklu R, Zhao G, Nair AV, Brown D, Fischman AJ, Tompkins RG, Irimia D. Kidney and Liver Injuries After Major Burns in Rats Are Prevented by Resolvin D2. Crit Care Med. 2016;44:e241–252. doi: 10.1097/CCM.0000000000001397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maekawa T, Hosur K, Abe T, Kantarci A, Ziogas A, Wang B, Van Dyke TE, Chavakis T, Hajishengallis G. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3beta-C/EBPbeta pathway. Nat Commun. 2015;6:8272. doi: 10.1038/ncomms9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian Y, Zhang Y, Zhang R, Qiao S, Fan J. Resolvin D2 recovers neural injury by suppressing inflammatory mediators expression in lipopolysaccharide-induced Parkinson's disease rat model. Biochem Biophys Res Commun. 2015;460:799–805. doi: 10.1016/j.bbrc.2015.03.109. [DOI] [PubMed] [Google Scholar]
- 29.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery TW, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015;212:1203–1217. doi: 10.1084/jem.20150225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015;21:1071–1075. doi: 10.1038/nm.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 34.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 35.Nathan C. Making space for anti-infective drug discovery. Cell Host Microbe. 2011;9:343–348. doi: 10.1016/j.chom.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Gaudilliere B, Fragiadakis GK, Bruggner RV, Nicolau M, Finck R, Tingle M, Silva J, Ganio EA, Yeh CG, Maloney WJ, Huddleston JI, Goodman SB, Davis MM, Bendall SC, Fantl WJ, Angst MS, Nolan GP. Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med. 2014;6:255ra131. doi: 10.1126/scitranslmed.3009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mingueneau M, Krishnaswamy S, Spitzer MH, Bendall SC, Stone EL, Hedrick SM, Pe'er D, Mathis D, Nolan GP, Benoist C. Single-cell mass cytometry of TCR signaling: amplification of small initial differences results in low ERK activation in NOD mice. Proc. Natl. Acad. Sci. U. S. A. 2014;111:16466–16471. doi: 10.1073/pnas.1419337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 39.Westermann D, Savvatis K, Lindner D, Zietsch C, Becher PM, Hammer E, Heimesaat MM, Bereswill S, Volker U, Escher F, Riad A, Plendl J, Klingel K, Poller W, Schultheiss HP, Tschope C. Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation. 2011;124:2082–2093. doi: 10.1161/CIRCULATIONAHA.111.035964. [DOI] [PubMed] [Google Scholar]
- 40.Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz-Planillo R, Do N, Nunez G, Inohara N. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity. 2014;41:620–632. doi: 10.1016/j.immuni.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Fouts DE, Starkel P, Hartmann P, Chen P, Llorente C, DePew J, Moncera K, Ho SB, Brenner DA, Hooper LV, Schnabl B. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe. 2016;19:227–239. doi: 10.1016/j.chom.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dias AA, Goodman AR, Dos Santos JL, Gomes RN, Altmeyer A, Bozza PT, Horta MF, Vilcek J, Reis LF. TSG-14 transgenic mice have improved survival to endotoxemia and to CLP-induced sepsis. J. Leukoc. Biol. 2001;69:928–936. [PubMed] [Google Scholar]
- 43.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gobbetti T, Coldewey SM, Chen J, McArthur S, le Faouder P, Cenac N, Flower RJ, Thiemermann C, Perretti M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc. Natl. Acad. Sci. U. S. A. 2014;111:18685–18690. doi: 10.1073/pnas.1410938111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devchand PR, Arita M, Hong S, Bannenberg G, Moussignac RL, Gronert K, Serhan CN. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17:652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- 46.Frasch SC, Fernandez-Boyanapalli RF, Berry KZ, Leslie CC, Bonventre JV, Murphy RC, Henson PM, Bratton DL. Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of Rac activity. J. Biol. Chem. 2011;286:12108–12122. doi: 10.1074/jbc.M110.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim JG, Moon MY, Kim HJ, Li Y, Song DK, Kim JS, Lee JY, Kim J, Kim SC, Park JB. Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophages. J. Biol. Chem. 2012;287:5145–5155. doi: 10.1074/jbc.M111.257634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HN, Surh YJ. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 2013;86:759–769. doi: 10.1016/j.bcp.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513:1–13. doi: 10.1016/j.gene.2012.10.033. [DOI] [PubMed] [Google Scholar]
- 50.Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem. 2010;285:3451–3461. doi: 10.1074/jbc.M109.044131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu Z, Lamont GJ, Lamont RJ, Uriarte SM, Wang H, Scott DA. Resolvin D1, resolvin D2 and maresin 1 activate the GSK3beta anti-inflammatory axis in TLR4-engaged human monocytes. Innate Immun. 2016;22:186–195. doi: 10.1177/1753425916628618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soki FN, Koh AJ, Jones JD, Kim YW, Dai J, Keller ET, Pienta KJ, Atabai K, Roca H, McCauley LK. Polarization of prostate cancer-associated macrophages is induced by milk fat globule-EGF factor 8 (MFG-E8)-mediated efferocytosis. J Biol Chem. 2014;289:24560–24572. doi: 10.1074/jbc.M114.571620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu F, Zhou Y, Jiang C, Zhang X. Role of JAK-STAT signaling in maturation of phagosomes containing Staphylococcus aureus. Sci Rep. 2015;5:14854. doi: 10.1038/srep14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. 2011;25:544–560. doi: 10.1096/fj.10-169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.