

Abstract
Background
Colletotrichum graminicola and C. sublineola cause anthracnose leaf and stalk diseases of maize and sorghum, respectively. In spite of their close evolutionary relationship, the two species are completely host-specific. Host specificity is often attributed to pathogen virulence factors, including specialized secondary metabolites (SSM), and small-secreted protein (SSP) effectors. Genes relevant to these categories were manually annotated in two co-occurring, contemporaneous strains of C. graminicola and C. sublineola. A comparative genomic and phylogenetic analysis was performed to address the evolutionary relationships among these and other divergent gene families in the two strains.
Results
Inoculation of maize with C. sublineola, or of sorghum with C. graminicola, resulted in rapid plant cell death at, or just after, the point of penetration. The two fungal genomes were very similar. More than 50% of the assemblies could be directly aligned, and more than 80% of the gene models were syntenous. More than 90% of the predicted proteins had orthologs in both species. Genes lacking orthologs in the other species (non-conserved genes) included many predicted to encode SSM-associated proteins and SSPs. Other common groups of non-conserved proteins included transporters, transcription factors, and CAZymes. Only 32 SSP genes appeared to be specific to C. graminicola, and 21 to C. sublineola. None of the SSM-associated genes were lineage-specific. Two different strains of C. graminicola, and three strains of C. sublineola, differed in no more than 1% percent of gene sequences from one another.
Conclusions
Efficient non-host recognition of C. sublineola by maize, and of C. graminicola by sorghum, was observed in epidermal cells as a rapid deployment of visible resistance responses and plant cell death. Numerous non-conserved SSP and SSM-associated predicted proteins that could play a role in this non-host recognition were identified. Additional categories of genes that were also highly divergent suggested an important role for co-evolutionary adaptation to specific host environmental factors, in addition to aspects of initial recognition, in host specificity. This work provides a foundation for future functional studies aimed at clarifying the roles of these proteins, and the possibility of manipulating them to improve management of these two economically important diseases.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3457-9) contains supplementary material, which is available to authorized users.
Keywords: Fungal virulence, Maize anthracnose, Sorghum anthracnose, Fungal secondary metabolism, Fungal effectors, Hypersensitive response, Effector-triggered immunity, Plant disease
Background
Members of the fungal genus Colletotrichum cause anthracnose diseases on nearly every plant species grown for food or fiber worldwide [1, 2]. Colletotrichum graminicola (Ces.) Wils., and C. sublineola Henn., cause economically important anthracnose leaf blight and stalk rot diseases of maize (Zea mays L.), and sorghum (Sorghum bicolor [L.] Moench), respectively [3–6]. These two fungal sibling species are morphologically very similar, but reproductively isolated [5]. Results of molecular phylogenetic analyses suggest that they diverged from a common ancestor relatively recently, perhaps at the same time as the split between maize and sorghum (thought to be approximately 12 million years ago) [4, 5, 7–11]. There are no reports in the literature of C. graminicola infecting sorghum or of C. sublineola infecting maize in the field, and most studies agree that the two species are host-specific [6, 12–14]. We have found that C. sublineola can infect maize stalk epidermal cells, and maize leaf sheath cells that are dead or dying [15, 16]. This ability of C. sublineola to conditionally infect some maize tissues might explain two earlier papers that reported that maize was susceptible to isolates of Colletotrichum from sorghum [17, 18]. It also suggests that host range is determined by active recognition of and response to the non-pathogen by healthy tissues of the non-host, rather than structural barriers or the absence of some vital nutrient or other factor.
The determination of host range in plant pathogens is often attributed to the presence or absence of pathogen virulence factors, particularly specialized secondary metabolites (SSMs), and small-secreted protein (SSP) effectors [19–25].
The presence of particular SSMs has been associated with the determination of host range in some phytopathogenic fungi including Alternaria spp. [21] and Cochliobolus spp. [20]. The major classes of fungal SSMs include polyketides, peptides, terpenes, and indole alkaloids [26–28]. Each of these classes is associated with a specific family of proteins. These SSM-associated proteins are: polyketide synthases (PKS); nonribosomal peptide synthetases (NRPS); terpene synthases (TS); and dimethylallyl transferases (DMAT), respectively. Genes encoding these enzymes and other proteins involved in the production of the SSMs are often found physically associated in transcriptionally co-regulated gene clusters [29, 30].
Fungal effectors have been defined as SSPs that alter the structure or modulate the function of host cells to facilitate infection [31, 32]. Some effectors are translocated and operate in the host cytoplasm [33–36]. Others function in the plant cell apoplast [37]. Some effectors act as host specific toxins and induce apoptosis only in certain plant genotypes, conferring host specificity in several important necrotrophic pathogens [38, 39]. Examples of known effector categories include serine proteases, necrosis and ethylene-inducing protein 1-like proteins (NEP1-like proteins), and small cysteine-rich proteins [23, 40, 41].
Some plants have evolved an ability to recognize and respond to certain effectors by activating defense pathways via specific resistance (R) proteins, a phenomenon known as effector-triggered immunity (ETI). In these cases, the effectors act as avirulence (Avr) factors. Multiple rounds of mutation and selection of R and Avr genes during a co-evolutionary “arms-race” leads to the presence of multiple pathogenic races expressing different combinations of Avr genes within the pathogen population [42]. Recent evidence suggests that inducible non-host resistance in many agriculturally-important pathosystems, particularly involving closely related hosts, is due to ETI. In these cases all members of the non-host plant species contain the same R gene(s), whereas all members of the nonpathogenic microbial species contain the corresponding Avr gene(s) [43–52].
A number of recent comparative genomics studies have confirmed that genes encoding SSM-associated proteins and SSPs show evidence of rapid evolution in related pathogens with different host ranges [20, 25, 53–65]. Most of these studies have involved comparisons of relatively distantly related pathogens, and/or strains with diverse geographic origins. There have been comparatively few analyses of co-occurring, closely related sibling species. The goal of the present work was to identify, characterize, and compare candidate host specificity-related genes from two contemporaneous, co-occurring, host-specific strains of the sibling species C. graminicola and C. sublineola.
Results and discussion
The cytology of host specificity
Colletotrichum graminicola strain M1.001 was isolated from maize in Missouri in the late 1970s [66]. This strain caused typical, sporulating anthracnose lesions on maize leaves (cv. Mo17) within 3 days post inoculation (dpi), but on leaves of sorghum (cv. Sugar Drip) it produced only small reddish flecks, which failed to expand or sporulate even up to 7 dpi (Fig. 1a, d). Colletotrichum sublineola strain CgSl1 was isolated in the early 1980s from grain sorghum in Indiana [6]. This strain caused large, sporulating anthracnose lesions on sorghum, but not on maize leaves (Fig. 1b, c). Colletotrichum graminicola strain M1.001 readily infected and colonized multiple cells of detached leaf sheaths of maize by 48 h after inoculation (hpi) and C. sublineola strain CgSl1 did the same in sorghum sheaths by 72 hpi (Fig. 2a, b). In contrast, C. graminicola failed to infect leaf sheath cells of sorghum, and C. sublineola failed to infect maize leaf sheath cells, even up to 6 dpi (Fig. 2c, d). Sorghum responded within 48 hpi to C. graminicola appressoria by an accumulation of numerous vesicles containing red pigments, and maize responded to C. sublineola appressoria by the formation of iridescent papillae (Fig. 2c, d). Previous studies have determined that the red pigments consist of various anthocyanidin phytoalexins [67]. The maize papillae are composed primarily of callose [68]. Visible primary hyphae were always very small, and were produced in fewer than 1% of infection attempts in both non-host combinations. Unpenetrated cells beneath C. sublineola appressoria in maize leaf sheaths typically retained their ability to plasmolyze even up to 48 hpi, but cells containing rare penetration hyphae appeared granulated, and did not plasmolyze normally (Fig. 3a, b). Sorghum cells beneath C. graminicola appressoria usually plasmolyzed at 24 hpi, but by 48 hpi most of the cells had lost the ability to plasmolyze, whether they contained infection hyphae or not (Fig. 3c, d, Additional file 1: Figure S1). Most of the cells in the mock-inoculated maize and sorghum controls still plasmolyzed normally up to 72 hpi (Additional file 2: Figure S2). Colletotrichum sublineola and C. graminicola were able to colonize non-host leaf sheaths readily if the cells were killed first by a localized application of liquid nitrogen (Fig. 4a, b). These observations suggest that host specificity is based on active recognition of the non-pathogen by living non-host plant cells, followed by rapid deployment of defense responses targeting the infection sites, and ultimately plant cell death prior to, or coincident with, penetration. To identify potential candidates for factors that might trigger or facilitate this recognition, we compared the genomes of these two strains, with a particular focus on genes that were not conserved between them, and on genes encoding putative SSPs and SSM-associated proteins.
Fig. 1.

a maize leaf inoculated with C. graminicola, 7 dpi; b sorghum inoculated with C. sublineola, 7 dpi; c maize inoculated with C. sublineola, 7 dpi; d sorghum inoculated with C. graminicola, 7 dpi; e maize control, mock-inoculated with water, 7 dpi; f sorghum control, mock-inoculated with water, 7 dpi
Fig. 2.
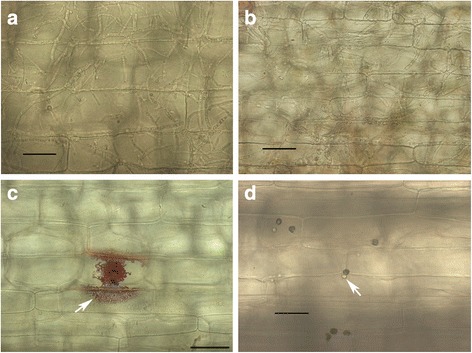
a C. graminicola hyphae in maize leaves, 48 hpi; b C. sublineola hyphae in sorghum leaves, 72 hpi; c C. graminicola on sorghum, 48 hpi, white arrow indicates red vesicles surrounding the appressorium; d C. sublineola on maize, 48 hpi, white arrow indicates an iridescent papillum beneath a melanized appressorium. Scale bars equal to 50 μm
Fig. 3.
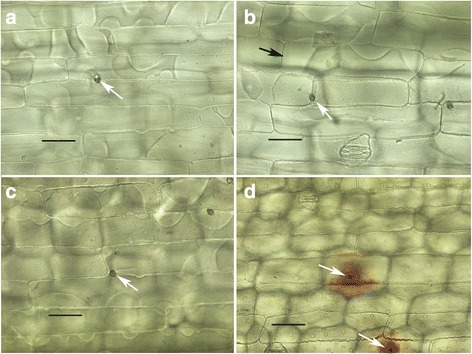
a CgSl1 on maize sheath, 48 hpi. Cell beneath appressorium (white arrow) plasmolyzes normally; b CgSl1 on maize sheath, small penetration hypha (white arrow) 48 hpi. Adjacent cell (black arrow) plasmolyzes normally. Cell containing penetration hyphae appears granulated, plasma membrane visible but appears abnormal; c M1.001 on sorghum sheath, 24 hpi, cells beneath appressoria (white arrow) still plasmolyze; d M1.001 on sorghum sheath, 48 hpi. No plasmolysis evident in any of the cells in the vicinity of the appressoria (white arrow). Scale bars equal to 50 μm
Fig. 4.

a CgSl1 growing in cells of maize sheaths killed by liquid nitrogen, 48 hpi; b M1.001 growing in cells of sorghum sheaths killed by liquid nitrogen, 48 hpi. Scale bars equal to 50 μm
The genomes of the C. graminicola and C. sublineola strains are very similar to one another, confirming their close evolutionary relationship
Colletotrichum graminicola and C. sublineola belong to a monophyletic clade of closely related Colletotrichum fungi that affect various graminaceous hosts [9, 10, 69]. We sequenced, assembled, and analyzed the genome of the CgSl1 strain of C. sublineola, and compared it with the previously published genome assembly and annotation of C. graminicola strain M1.001 [69]. The C. sublineola assembly was approximately 20% larger than the published M1.001 genome assembly (Table 1), although the amount of single-copy DNA was similar (Table 2). The C. sublineola genome was predicted to encode about 1300 more genes than the number previously published for C. graminicola [69] (Table 1, Additional file 3). Both genome annotations contained homologs for most or all of a set of 248 phylogenetically conserved genes, as identified by CEGMA, aka. the Core Eukaryotic Genes Mapping Approach [70], suggesting that both are relatively complete (Table 1).
Table 1.
Characteristics of the genome assemblies that were used in this study
| Genome features annotation | C. graminicola M1.001 BROAD | C. graminicola M5.001 MAKER | C. sublineolum CgSl1 MAKER |
|---|---|---|---|
| Source of Strain, Reference | Maize, Missouri, 1978 [66] | Maize, Brazil, 1988 [119] | Sorghum, Indiana, 1982 [6] |
| NCBI accession no. of assembly | ACOD01000001 | MRBI01000001 | MQVQ01000001 |
| Reference | [69] | This Manuscript | This Manuscript |
| Strain | M1.001 | M5.001 | CgSl1 |
| Total contig length (Mb) | 51.6 | 48.8 | 64.8 |
| Number of Contigs | 1,151 | 3,280 | 12,943 |
| Number of scaffolds | 653 | n/aa | 548 |
| N50 contig (kb) | 228.96 | 69.85 | 41.05 |
| N50 scaffold (Kb) | 579.19 | n/aa | 339.27 |
| GC-content (%) | 49.12 | 49.8 | 46.45 |
| Protein-coding genes | 12,006 | 15,052 | 13,311 |
| Conserved Genes [71] | 248 | 248 | 247 |
| Mean transcript length (bp) | 1399.1 | 1224.9 | 1389.9 |
| Number of exons | 32967 | 64326 | 35389 |
| Mean number of introns/gene | 1.745 | 3.2126 | 1.65 |
| Mean Intergenic distance (bp) | 2691 | 1599 | 2224.7 |
| Percentage coding | 32.81 | 36.89 | 29.5 |
| Percentage repetitive DNA in genome assembly | 3.12 | 2.83 | 3.14 |
a This assembly was not scaffolded
Table 2.
Results of a blastn analysis of genome similarity among three species of Colletotrichum
| C. graminicola (M1.001) | C. sublineola (CgSl1) | C. higginsianum | |
|---|---|---|---|
| Total Genome Assembly Size (N’s removed) | 50,866,947 | 63,193,777 | 49,075,584 |
| Single Copy Portion | 42,075,489 | 46,160,404 | 42,288,585 |
| Total Alignable Single-Copy DNA: C. graminicola | NA | 23,584,235 | 9,885,330 |
| Total Alignable Single-Copy DNA: C. sublineola | 23,584,235 | NA | 9,919,326 |
| SNPS per 1 Mb alignable, single-copy DNA: C. graminicola | NA | 100,146 | 127,026 |
| SNPS per 1 Mb alignable, single-copy DNA: C. sublineola | 100,146 | NA | 129,126 |
Partial sequences of four genes have been used previously for multigene phylogenetic analysis of Colletotrichum [69]. These included portions of the ACT gene; the CHS gene; the HIS3 gene; and the TUB2 gene. These sequences from CgSl1 shared 100% identity with those of strain S.3001, the designated epitype specimen for C. sublineola [10, 69] (Additional file 4: Figure S3). The internal transcribed spacer (ITS) sequence from CgSl1 also shared 99.6% identity with the ITS sequence of S3.001 [10]. This confirms that CgSl1 belongs to the C. sublineola species as it is presently defined (Additional file 4: Figure S3).
Approximately 50% of the single-copy DNA sequence in the CgSl1 and M1.001 assemblies could be directly aligned by blastn (Table 2). In comparison, only about 23% of the assembly of C. higginsianum, a more distantly related species pathogenic on Brassicaceae, and belonging to a sister clade [69, 71], could be aligned with either of these two genomes (Table 2). As expected, there were also fewer single nucleotide polymorphisms (SNPs) per Mb of alignable single-copy DNA between C. graminicola and C. sublineola than between C. higginsianum and the other two genomes (Table 2).
Eighty-three percent of the C. graminicola genome assembly could be aligned with C. sublineola scaffolds based on the relative arrangement of conserved genes (Fig. 5a, Table 3). More than 80% of the C. graminicola and C. sublineola genes were syntenous (Table 3). Regions that appear to be translocated and/or inverted, and small “islands” that appeared to lack synteny, could be discerned embedded within the largely co-linear assemblies (Fig. 5b). No part of the C. sublineola assembly could be aligned with the three C. graminicola minichromosomes (Fig. 5a), which seem to be unique to this strain of C. graminicola [72].
Fig. 5.
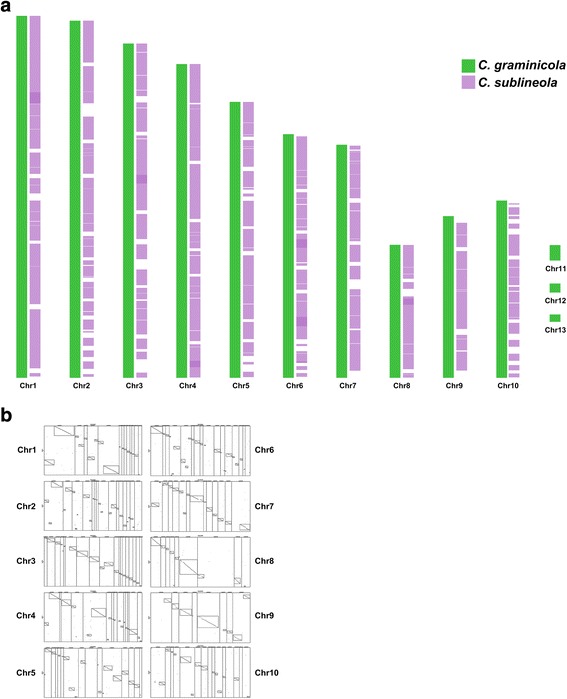
a C. sublineola scaffolds anchored to C. graminicola chromosomes (chromosome optical map of C. graminicola published in [69]. b Microsynteny between C. sublineola contigs and C. graminicola chromosomes. Each panel illustrates a different chromosome. The three C. graminicola minichromosomes are not included in the figure
Table 3.
Genome synteny between C. graminicola strain M1.001 and C. sublineola strain CgSl1
| Synteny blocks | % Coverage | Mean block length (Kb) a | Number of genes included in synteny blocks | % Genes included in synteny blocksb | Mean number of genes per synteny blockc | ||
|---|---|---|---|---|---|---|---|
| C. graminicola x C. sublineola | 182 | C. graminicola | 83 | 223 | 9874 | 83 | 54.25 |
| C. sublineola | 77 | 234 | 9884 | 86 | 54.31 |
a Calculated as SyMap Total Kb * % coverage/ # blocks
b Number of distinct genes that overlap a synteny anchor assigned to a synteny block
c Calculated as # genes hit/ # blocks
Colletotrichum graminicola and C. sublineola encode similar proteins and protein families
The Protein Family Database (Pfam) [73] was used to characterize and compare predicted proteins from C. graminicola and C. sublineola (Additional file 5: Table S1). Only 67% of C. graminicola proteins, and 62% of C. sublineola proteins, could be categorized into Pfam families. Most of these families were shared by both isolates, with relatively few differences in the number of family members across the strains. There were 13 families in which there was at least a three-fold expansion in one species versus the other (Additional file 5: Table S1). For example, C. sublineola appeared to be enriched in some SSM domains, and in one family of phosphotransferase enzymes, in comparison with C. graminicola. There were 82 Pfam families that were found only in C. graminicola, while 73 were found only in C. sublineola (Additional file 5: Table S1). Nearly all of these non-conserved families contained only a single protein, and relatively few (26% for C. sublineola and 13% for C. graminicola) included members that have been previously implicated in pathogenicity, based on comparisons to the Pathogen-Host Interactions database (PHI-base), which catalogs pathogenicity-associated genes that have been identified in a variety of pathogenic microbes [74, 75] (Additional file 5: Table S1).
The C. graminicola and C. sublineola annotations each include more than 1000 predicted proteins that are not shared between the two species
Ortho-MCL [76] was used initially to identify putative orthologous (aka. shared) proteins from C. graminicola and C. sublineola. Results indicated that C. graminicola and C. sublineola shared more than 90% of their proteins (Table 4, Additional file 5: Tables S2, S3). They shared fewer proteins with their more distant relative C. higginsianum, but all three species still had more than 85% of their proteins in common (Table 4, Additional file 5: Tables S2, S3).
Table 4.
Summarized data of Ortho-MCL and RBH analysis of predicted proteins from C. graminicola and C. sublineola
| Shared by: | C. graminicola | Shared by: | C. sublineola | ||
|---|---|---|---|---|---|
| OrthoMCL | RBH | OrthoMCL | RBH | ||
| CgramCsubChigg | 9281 | 9264 | Cgram:Csub:Chigg | 9323 | 9252 |
| Cgram:Csub | 1015 | 1018 | Csub:Cgram | 1036 | 1057 |
| Cgram:Chiggs | 473 | 560 | Csub:Chiggs | 333 | 500 |
| Cgram only | 134 | 1164 | Csub only | 456 | 2502 |
| Not Characterized | 1103 | 0 | Not Characterized | 2163 | 0 |
| Total | 12006 | 12006 | Total | 13311 | 13311 |
Approximately 9% of C. graminicola predicted proteins, and 16% of C. sublineola predicted proteins, were not assigned to ortholog groups by Ortho-MCL (Table 4, Additional file 5: Tables S2, S3). Thus, the Reciprocal BLAST Hits (RBH) approach [77] was also used to identify putative orthologous proteins. With this approach, all proteins could be accounted for. For more than 90% of the proteins, RBH gave the same result as Ortho-MCL (Additional file 5: Tables S2, S3). Because the RBH included all of the predicted proteins, these results were used for subsequent analyses. The results indicated that the C. graminicola annotation included 1724 proteins that were not found in C. sublineola (Table 4; Additional file 5: Table S2), while the CgSl1 annotation included 3002 proteins that were not shared with M1.001 (Table 4; Additional file 5: Table S3). These proteins will hereafter be referred to as non-conserved proteins (NCPs). Almost one third of the M1.001 NCPs, and 17% of the CgSl1 NCPs, were shared with the more distantly-related C. higginsianum, suggesting a role for loss as well as gain of genes in the evolutionary history of these species (Additional file 5: Tables S2, S3).
Mapping of the genes encoding NCPs of C. graminicola to the C. sublineola genome assembly, and vice versa, revealed that between one third and one half of them (48% in C. graminicola, and 30% in C. sublineola) matched sequences in the other genome assembly (Additional file 5: Tables S4, S5). These sequences might represent homologs that were not annotated due to assembly fragmentation or to differences in the gene-calling parameters of the two annotation programs. They could also represent mutant alleles (e.g. nonsense mutations) that were not recognized as ORFs. More detailed studies will be necessary to determine which of these possibilities applies to each sequence.
Characteristics of the C. graminicola and C. sublineola NCPs
The predicted proteins that were not shared between the two Colletotrichum species were relatively small, with an average size of less than 300 aa, compared with an average of more than 460 aa for all proteins (Additional file 5: Tables S4, S5). A majority in each case (60% of C. graminicola NCPs, and 70% of C. sublineola NCPs) were not classified by Ortho-MCL (Additional file 5: Tables S4, S5). Transcript data for C. sublineola are not available, but 50% of the NCPs of C. graminicola were supported by transcript evidence in planta (Additional file 5: Table S4) [78]. This could indicate that the rest of the predicted C. graminicola NCP genes are not really genes. It could also mean that NCP genes tend to be expressed at especially low levels, or under very specific circumstances that were not achieved in our in planta transcriptome analysis. Further studies will be necessary to address these different possibilities.
About half of the NCPs in both C. graminicola and C. sublineola were predicted to localize to either mitochondria or nuclei (Table 5; Additional file 5: Tables S4, S5). Only about 15% in each species were predicted to be secreted, and another 10% were predicted to localize to the plasma membrane.
Table 5.
Numbers of non-conserved proteins of C. graminicola and C. sublineola that are predicted to localize to various locations
| Predicted Location | C. graminicola | C. sublineola |
|---|---|---|
| Cyto-mito | 11 | 17 |
| Cyto-nucl | 62 | 116 |
| Cyto-pero | 2 | 3 |
| Cytoskeleton | 66 | 94 |
| Cytosol | 288 | 531 |
| Endoplasmic reticulum | 3 | 2 |
| Extracellular | 232 | 447 |
| Mito-nucl | 4 | 4 |
| Mitochondria | 449 | 715 |
| Nuclear | 420 | 827 |
| Peroxisome | 4 | 3 |
| Plasma membrane | 183 | 239 |
| No Prediction | 0 | 4 |
| Total | 1724 | 3002 |
The high number of predicted nuclear proteins among the NCPs may suggest that there have been shifts in the regulation of gene expression in these two species that have had important impacts on host specificity. Some of these NCPs may also specifically target the host nucleus: for example, one of the predicted nuclear proteins in C. graminicola was GLRG_04079, aka. CgEP1, recently characterized as an essential C. graminicola effector that is targeted to the plant nucleus, with both a secretion signal and a nuclear localization signal (NLS) [79] (Additional file 5: Table S4). In our study, neither SignalP nor WoLF PSORT indicated the presence of a signal peptide in this protein. A second candidate nuclear effector identified in [79], GLRG_03517, was similarly not predicted to have a signal peptide in our study. A third putative NLS effector from that study (GLRG_08510) was on our list of NCPs as a predicted SSP, but not as a nuclear protein. These differences in predicted locations probably relate to differences in the localization prediction protocols that we used. This illustrates why localization predictions should be experimentally confirmed. The rest of the NLS effectors identified in [79] are conserved in CgSl1, and thus they were not among the NCPs.
Approximately a quarter of the NCPs in each species were predicted to be localized in the mitochondria (Table 5). Mitochondrial proteins have been implicated in several important animal disease mechanisms [80–82]. In animal cells, some transcription factors and receptors are known to translocate to the mitochondria in response to extracellular signals, where they promote cell death or cell survival [83]. The high number of predicted mitochondrial proteins among the Colletotrichum NCP may point to an important role for mitochondrial functions in host adaptation and specificity in these two species. However, the locations of these proteins in the mitochondria should be confirmed by more direct methods before drawing any definitive conclusions.
The NCPs were further evaluated by blastx against the NCBI nr database, and also against the predicted proteomes of the C. sublineola epitype strain, and of five other closely related species of Colletotrichum isolated from gramineaceous hosts [10]. The latter can be accessed from the Joint Genome Institute (JGI) genome portal (http://genome.jgi.doe.gov/). Based on this analysis, about 20% (361/1724) of the NCPs in C. graminicola, and about 25% (736/3002) of the C. sublineola NCPs, appeared to be lineage-specific (LS). Although the number of LS genes may decrease as new fungal genomes are added to the databases, the lack of homologs in the five closely related species should make this less likely.
A majority (>65%) of the NCPs in both strains did not match any Pfam categories (Table 6). About 10% of these non-classified NCPs in each case were putative SSPs. Among the minority of NCPs with Pfam classifications, the largest groups consisted of transporters; cytochrome P450s; SSM-associated proteins; carbohydrate-active enzymes (CAZymes); and transcription factors (Table 6). There was also a large group of proteins in each case categorized as heterokaryon incompatibility factors, and a number of other proteins that could potentially be involved in signaling (e.g. protein kinases and protein phosphatases), and pathogenicity, e.g. proteins with LysM chitin-binding domains [84]; necrosis-inducing NPP domains [85]; NUDIX domains [86, 87]; and Common in Fungal Extracellular Membrane (CFEM) domains [88]. Seventeen percent of the C. sublineola NCPs, and 20% of the C. graminicola NCPs, matched entries in the PHI database. The NCPs for each species were comprised of similar classes, but the CgSl1 annotation generally included more members of each class than the M1.001 annotation, accounting for the larger number of NCPs predicted overall in the C. sublineola strain (Table 6).
Table 6.
Numbers of non-conserved proteins in C. graminicola and C. sublineola in various categories
| Protein Category | C. graminicola | C. sublineola | |
|---|---|---|---|
| No Pfam | 1123 | 2180 | |
| Pfam category | Transcription Factors | 22 | 25 |
| Pfam category | Glycosyl Hydrolases | 19 | 22 |
| Pfam category | Heterokaryon Incompatibility Proteins | 13 | 24 |
| Pfam category | Transporters (MFS, ABC, and other) | 54 | 55 |
| Pfam category | Cytochrome P450s | 23 | 56 |
| Pfam category | NUDIX domain | 3 | 1 |
| Pfam category | CFEM domain | 2 | 0 |
| Pfam category | Necrosis inducing NPP domain | 2 | 5 |
| Pfam category | Protein kinase domain | 3 | 15 |
| Pfam category | Secondary Metabolism | 13 | 33 |
| SSP Putative Effectors | 143 | 301 | |
| Cysteine-rich SSP | 64 | 111 | |
| Cazymes Database | 62 | 73 | |
| Members of Secondary Metabolism Clusters | 78 | 13 | |
| Hits to PHI-base | 343 | 503 | |
| Hits to MEROPS Secreted Peptidase Database | 14 | 13 | |
| Hits to Transporters Database | 127 | 161 | |
Transporters represented a major category of the NCPs with Pfam designations, and included members of several different superfamilies (Additional file 5: Tables S4, S5). The largest group belonged to the Major Facilitator Superfamily (MFS). MFS transporters are the most common category of secondary carrier proteins. Members of this group are involved in the uptake of essential minerals and nutrients, also serving in many cases as nutrient sensors [89]. Many of the other overrepresented categories of MFS transporters function in the transport of various drugs and toxins [90], and include members that are homologs of known toxin-associated genes in other fungi (Additional file 5: Tables S4, S5). Another well-represented group of NCP transporters, the ATP-Binding Cassette (ABC) Superfamily, are also known to have important functions in the transport of toxic substances [91]. The relative abundance of these two categories among the NCPs suggests an important role for detoxification and/or production of toxic SSMs in host-species adaptation. The additional presence of SSM-associated proteins and cytochrome P450s as highly represented NCPs reinforces this conclusion. In addition to MFS, several other categories of NCP transporters are known to be involved in sensing of nutritional and other environmental factors. For example, the largest single category of NCP transporters was the Ankyrin-B class, which functions to link the cytoskeleton to a variety of membrane proteins, some of which may act as receptors for plant signals [92]. The prominence of these classes among the NCP receptors suggests a necessity for adaptive changes in the sensory receptors of the pathogens to variations in the signals provided by each host plant.
Transcription factors (TFs) were another conspicuous category among the NCPs. Both species encoded non-conserved (NC) TFs belonging to two Pfam categories: PF00172 (fungal Zn(2)-Cys(6) binuclear cluster domain); and PF04082 (fungal specific transcription factor domain). A little over one third of the NC TFs were predicted to localize to mitochondria, and most of the rest to the nuclei. In C. graminicola, one of the predicted nuclear NC TFs was related to DEP6, which is part of the depudecin PKS gene cluster in Alternaria brassicicola. When DEP6 was knocked out it resulted in a small reduction in virulence on cabbage [93]. This TF gene in C. graminicola is part of a PKS SSM gene cluster (Cluster 28) that produces an unknown product. NC TFs in C. sublineola included two additional types, a bZIP transcription factor (PF00170), and two nuclear PF11951 proteins. Nearly all of these also had hits in the PHI database. One of the PF00172 proteins in C. sublineola was related to the CTB8 regulator of cercosporin biosynthesis in Cercospora nicotianae, which is part of the cercosporin gene cluster. A knock out of that gene resulted in an inability to produce cercosporin and a reduction in virulence [94]. There is a second ortholog of CTB8 in C. sublineola that is shared with C. graminicola. In C. graminicola, that gene is part of a PKS cluster (cluster 18) [69, 78]. However, C. sublineola doesn’t appear to share cluster 18, and the C. sublineola-specific ortholog of CTB8 was a part of a PKS cluster (cluster 11), which is not conserved in C. graminicola (Additional file 5: Table S6).
A third prominent category of NCPs were CAZYmes (Additional file 5: Tables S4, S5). Specific enzyme categories that were over-represented included pectinases, ligninases, and lignocellulases. Wall structures of maize and sorghum do not appear to differ very much [95, 96], so it is possible that some of these enzymes are targeted by plant defense mechanisms, which has driven their diversification [97]. Similar categories of CAZYmes were also evolving rapidly among a larger group of more distantly related genera of Colletotrichum fungi [25, 64].
Colletotrichum graminicola and C. sublineola each encode non-conserved SSM-associated genes and gene clusters that may produce novel metabolites
Identification of SSM-associated genes in C. sublineola strain CgSl1
The program Ortho-MCL and the refiner COCO-CL were used to identify genes in C. sublineola that were orthologous to the previously identified SSM-associated genes of C. graminicola and C. higginsianum [69]. Using this approach, combined with manual annotation, 31 PKS genes, eight NRPS genes, six PKS-NRPS hybrid genes, 14 TS genes, and eight DMAT genes, were identified in C. sublineola (Table 7). Pfam analysis of the C. sublineola protein predictions identified 172 putative SSM domains. All of the SSM-associated genes that were identified by Ortho-MCL and COCO-CL (above) were included among the SSM genes identified after manual annotation of the Pfam domains. However, the Pfam analysis identified additional genes in some classes (three TSs, and one DMAT) encoded by C. sublineola that were not found in either C. graminicola or C. higginsianum (Table 7).
Table 7.
Ortho-MCL prediction of shared secondary metabolism-associated genes for the three species of Colletotrichum
| Type | C. sublineola | C. graminicola a | C. higginsianum a |
|---|---|---|---|
| PKS | 31 (31) | 36 | 36 |
| NRPS | 8 (8) | 7 | 12 |
| PKS-NRPS | 6 (6) | 4 | 3 |
| DMAT | 8 (9) | 7 | 10 |
| TS | 14 (17) | 14 | 17 |
PKS polyketide synthases, NRPSs non-ribosomal peptide synthetases, PKS-NRPS hybrids contain at least one PKS and one NRPS domain, DMAT dimethylallyl transferases, and TS terpene synthases. The numbers in parentheses represent the total number of genes in each category based on Pfam predictions for C. sublineola strain CgSl1. a Manual annotation was performed on data retrieved from [69]
Phylogenetic analysis of the SSM-associated proteins
A phylogenetic analysis was performed to address the relationships among the putative SSM-associated proteins in C. graminicola and C. sublineola. The more distantly-related species C. higginsianum was also included for comparison. SSM-associated genes in C. graminicola and C. higginsianum were previously published [69]. After manual annotation and identification of overlapping gene models, the 58 PKS genes that were previously identified in C. higginsianum [69] were reduced to 36 complete genes for analysis (Table 7). The adenylation domain (A domain) of NRPS proteins and PKS-NRPS hybrids [98, 99], the keto-synthase (KS) N-terminal and C-terminal domains of PKS proteins and PKS-NRPS hybrids [100], and the entire DMAT and TS protein sequences, were used for the phylogenetic analyses.
Results of the analysis revealed a high degree of diversity, with relatively few SSM-associated protein ortholog families that were conserved across all three Colletotrichum species (Figs. 6, 7, 8 and 9). As expected, C. graminicola and C. sublineola shared more ortholog families than either shared with C. higginsianum, consistent with a more recent common ancestor. The presence of some ortholog families only in C. higginsianum and C. graminicola, or only in C. higginsianum and C. sublineola, suggested that some members of these families may have been lost since the divergence of C. higginsianum from the other two species. The PKS proteins were the largest and most diverse group of SSM-associated proteins, with 79 proteins or protein ortholog families across the three species. The NRPS proteins comprised the smallest group, with only 15 different proteins or ortholog families. Colletotrichum graminicola and C. sublineola shared about half of their PKS proteins, and also about half of their PKS-NRPS hybrid and TS proteins. The DMAT and NRPS proteins were more highly conserved, with about two thirds represented in both species. Searches of the NCBI nr database, and of the predicted proteomes of five close relatives in the JGI database, revealed that there were no SSM-associated protein genes in either C. sublineola or in C. graminicola that were unique to either species (Additional file 5: Tables S4, S5).
Fig. 6.
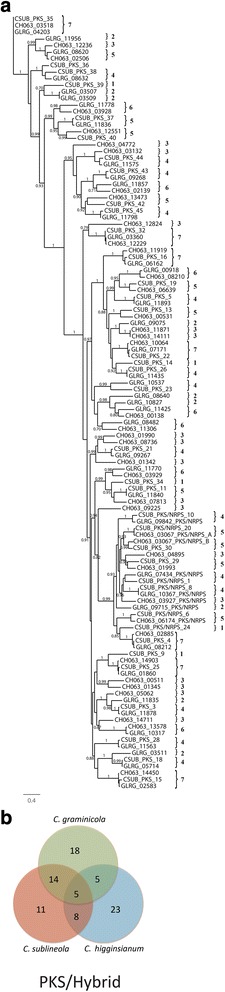
a Phylogenetic tree of the ketoacyl CoA synthetase domain amino acid sequences of putative PKSs and PKS-NRPS hybrids. Sequences were aligned by using MUSCLE version 3.7, and phylogenies were inferred by maximum-likelihood using PhyML version 3-0 Statistical. The numbers on the branch nodes indicate support values above 50%, calculated by aLRT. Sequences present in (1) C. sublineola only; (2) C. graminicola only; (3) C. higginsianum only; (4) C. sublineola and C. graminicola; (5) C. sublineola and C. higginsianum; (6) C. graminicola and C. higginsianum; and (7) C. sublineola, C. graminicola and C. higginsianum are indicated by the numbered brackets on the figure. b Venn diagram summarizing the numbers of conserved and non-conserved sequences among the three species
Fig. 7.
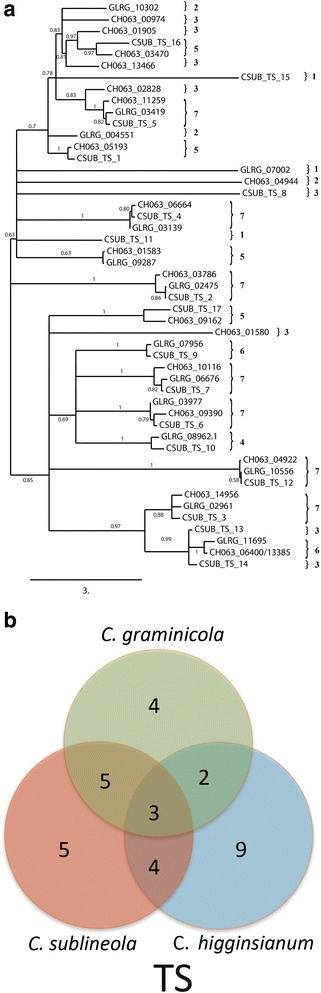
a Phylogenetic tree of the terpene synthase amino acid sequences. Sequences were aligned by using MUSCLE version 3.7, and phylogenies were inferred by maximum-likelihood using PhyML version 3-0 Statistical. The numbers on the branch nodes indicate support values above 50%, calculated by aLRT. Sequences present in (1) C. sublineola only; (2) C. graminicola only; (3) C. higginsianum only; (4) C. sublineola and C. graminicola; (5) C. sublineola and C. higginsianum; (6) C. graminicola and C. higginsianum; and (7) C. sublineola, C. graminicola and C. higginsianum are indicated by the numbered brackets on the figure. b Venn diagram summarizing the numbers of conserved and non-conserved sequences among the three species
Fig. 8.
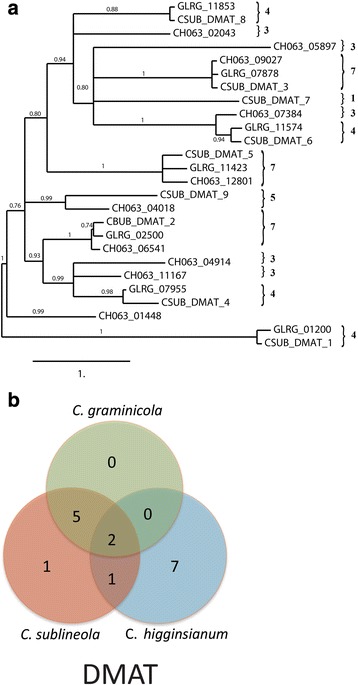
a Phylogenetic tree of the dimethylallyl transferase amino acid sequences. Sequences were aligned by using MUSCLE version 3.7, and phylogenies were inferred by maximum-likelihood using PhyML version 3-0 Statistical. The numbers on the branch nodes indicate support values above 50%, calculated by aLRT. Sequences present in (1) C. sublineola only; (2) C. graminicola only; (3) C. higginsianum only; (4) C. sublineola and C. graminicola; (5) C. sublineola and C. higginsianum; (6) C. graminicola and C. higginsianum; and (7) C. sublineola, C. graminicola and C. higginsianum are indicated by the numbered brackets on the figure. b Venn diagram summarizing the numbers of conserved and non-conserved sequences among the three species
Fig. 9.
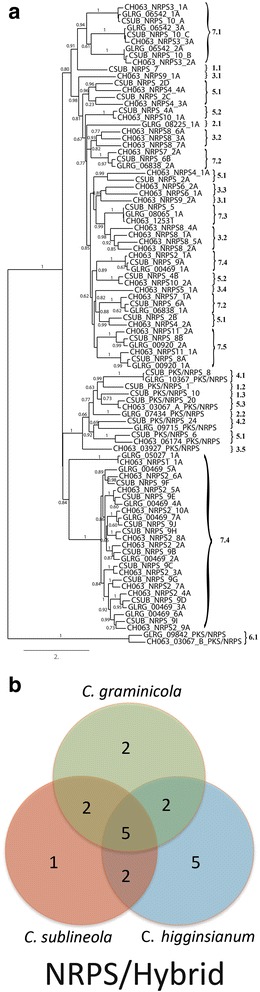
a Phylogenetic tree of the AMP binding domain amino acid sequences of putative NRPS and PKS-NRPS hybrids. Sequences were aligned by using MUSCLE version 3.7, and phylogenies were inferred by maximum-likelihood using PhyML version 3-0 Statistical. The numbers on the branch nodes indicate support values above 50%, calculated by aLRT. Sequences present in (1) C. sublineola only; (2) C. graminicola only; (3) C. higginsianum only; (4) C. sublineola and C. graminicola; (5) C. sublineola and C. higginsianum; (6) C. graminicola and C. higginsianum; and (7) C. sublineola, C. graminicola and C. higginsianum are indicated by the numbered brackets on the figure. The number after the period indicates modules of the same gene. b Venn diagram summarizing the numbers of conserved and non-conserved sequences among the three species
Conservation of gene clusters
Gene clusters in C. sublineola were identified by manual analysis of the genes located on either side of the “backbone” SSM-associated genes (ie. the genes encoding PKS, NRPS, TS, DMAT, and PKS-NRPS hybrids) that had been identified by using Ortho-MCL/COCO-CL and Pfam. A total of 67 putative SSM-associated gene clusters in the C. sublineola genome (Additional file 5: Table S6), were compared with the 42 clusters that were previously identified from C. graminicola [69]. There were 25 PKS gene clusters that appeared to be shared (with more than 50% of the genes in common) between C. sublineola and C. graminicola. One of these is the melanin cluster (Fig. 10) [69], and another is likely to be responsible for the production of monorden because it is identical in gene structure and content with the RADS cluster of Pochonia chlamydospora (Fig. 11) [78]. Colletotrichum sublineola and C. graminicola also shared five DMAT clusters, five NRPS gene clusters, and thirteen TS gene clusters (Additional file 5: Table S6). One of these conserved TS clusters is probably involved in the production of carotenoids [69].
Fig. 10.
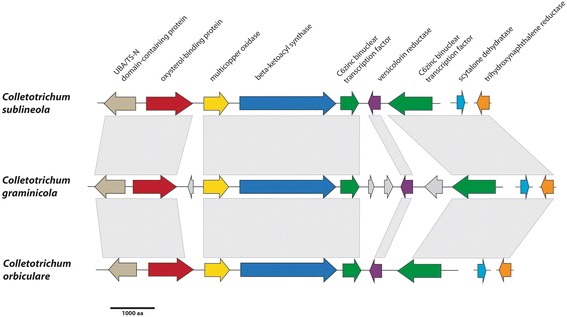
The organization of the conserved melanin gene clusters from C. sublineola, C. graminicola and C. orbiculare are shown, with orthologous genes depicted in the same color. The predicted genes shown in gray encode hypothetical proteins. Microsynteny among the clusters is indicated by the gray bars
Fig. 11.
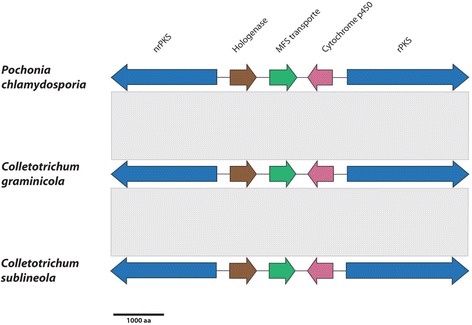
The organization of radicicol (RADS) gene clusters from Pochonia chlamydospora, C. graminicola and C. sublineola are shown, with orthologous genes depicted in the same color. Microsynteny among the clusters is indicated by the gray bars
Colletotrichum graminicola and C. sublineola each encode unique putative secreted proteins and SSPs
Identification of SSP genes in C. sublineola and C. graminicola
The primary characteristic for bioinformatic identification of an effector protein is that it includes an N-terminal sequence that targets it for processing and secretion. About 14% of the predicted proteins in C. graminicola and in C. sublineola had canonical signal peptides. Secreted effector proteins are usually described as small, but various sources have defined “small” differently, ranging from < 400 amino acids [101] to < 100 amino acids [102]. We chose a cutoff of 300 amino acids for our definition of SSPs. Colletotrichum graminicola is predicted to encode 687 small secreted proteins (SSPs) of 40 to 300 amino acids in size, with or without predicted functional domains. The number for C. sublineola is 824. The level of amino acid similarity of homologous secreted proteins is less than that of non-secreted proteins (Fig. 12). If only SSPs are considered, versus all secreted proteins, the level of similarity is even lower (Fig. 12).
Fig. 12.
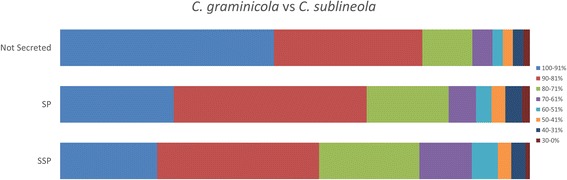
Percent similiarity among proteins that are predicted to be secreted, versus among all predicted proteins, in C. graminicola and C. sublineola
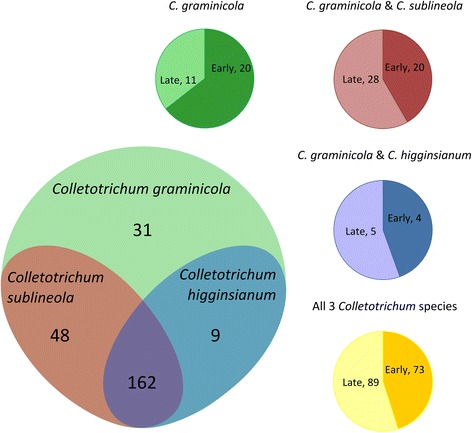
Colletotrichum graminicola and C. sublineola have more SSPs in common than either share with their more distant relative C. higginsianum (Fig. 13). Colletotrichum graminicola M1.001 encodes 143 predicted SSPs that are not found in C. sublineola strain CgSl1, while C. sublineola has 301 that are not shared with C. graminicola (Additional file 5: Tables S4, S5). The majority of these NC SSPs from both species (67% in C. graminicola, and 66% in C. sublineola) were similar to predicted proteins in other fungi in the NCBI database, although in most cases these were classified as hypothetical proteins (Additional file 5: Tables S4, S5). The remainder in each case did not match predicted protein sequences from any other species in the NCBI nr database. Analysis with the EffectorP prediction tool [103] revealed that about 60% of the NC SSPs in each species had a probability of at least 50% of being fungal effectors (Additional file 5: Tables S4 and S5). After additional comparisons with the available genome data from a group of five close relatives of C. graminicola and C. sublineola (http://genome.jgi.doe.gov/), there appeared to be only 32 C. graminicola LS-SSPs, and 21 C. sublineola LS-SSPs (Fig. 14). Interestingly, C. sublineola shares more SSPs with C. eremochloae than it does with any of the other close relatives included in the JGI database. Colletotrichum eremochloae is a pathogen of centipedegrass, and it was previously shown to be very closely related to C. sublineola [104].
Fig. 13.
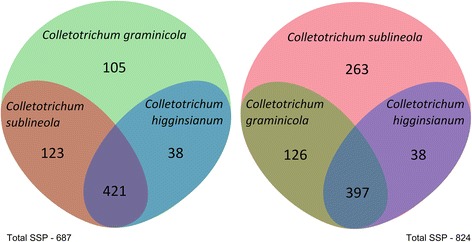
Venn diagrams summarizing conservation of predicted small-secreted proteins among C. graminicola, C. sublineola, and C. higginsianum. Shared proteins were identified by the Reciprocal Best Hit (RBH) method
Fig. 14.
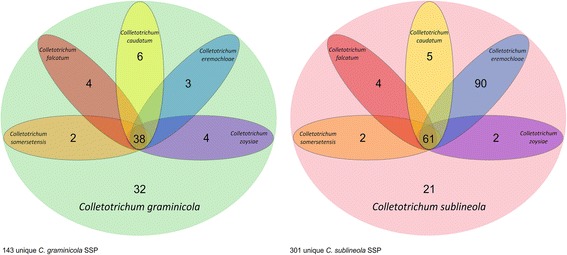
Venn diagrams summarizing conservation of predicted small-secreted proteins of C. graminicola strain M1.001 and C. sublineola strain CgSl1 with five close relatives. Shared proteins were identified through a combination of blastp and tblastn searches of the predicted proteomes and translated assemblies (respectively) of these five relatives with the protein sequences from C. graminicola and C. sublineola
Analysis of C. graminicola in planta transcriptome data [78] revealed that a majority of the transcribed C. graminicola NC SSP genes were more highly expressed in the early stages of infection (appressoria and/or biotrophy), whereas less than half of the genes shared with C. sublineola and/or with C. higginsianum were expressed during these early stages (Additional file 5: Table S4, Fig. 15).
Fig. 15.
Expression patterns of SSP-encoding genes that matched transcripts from the in planta C. graminicola transcriptome [78]
Characterized effector classes among NC SSPs
Several classes of fungal effectors described in the literature from other organisms are included among the NC SSPs of C. graminicola and C. sublineola.
The CFEM proteins have an eight cysteine-containing domain of around 66 amino acids [88]. Some CFEM proteins have important roles in pathogenesis [105, 106]. There are 11 CFEM SSPs in C. graminicola M1.001, and C. sublineola CgSl1 has homologs for 10 of these (Additional file 5: Tables S1 and S2). The C. sublineola epitype strain S3.001 has a homolog for the eleventh (http://genome.jgi.doe.gov/).
Effectors with chitin-binding domains [107] are thought to bind to chitin present in fungal cell walls, thus protecting the pathogen from plant chitinases [108]. Colletotrichum graminicola and C. sublineola share two SSP genes that encode chitin binding domains (Additional file 5: Table S1). Colletotrichum graminicola encodes one additional NC chitin-binding SSP (Additional file 5: Table S4).
Genes containing lysin motifs (LysM) are conserved in pathogenic and nonpathogenic fungi [109]. They appear to be highly divergent among species, and thus to be evolving rapidly [102]. LysM effectors, eg. Ecp6 from Cladosporium fulvum, are believed to sequester fungal chitin fragments, thus preventing host detection [110–112]. In C. lindemuthianum, a LysM protein called ClH1 was localized specifically to the surface of biotrophic hyphae by using a monoclonal antibody [113, 114]. There are two predicted LysM-domain SSP genes in C. graminicola, one of which is a homolog of ClH1. Both of these are expressed during the early stages of fungal colonization in the WT strain (Additional file 5: Table S4). Colletotrichum sublineola has four predicted LysM-domain SSP genes. Two of these are shared with C. graminicola, including a homolog of C1H1.
There are five predicted C. graminicola proteins, and nine in C. sublineola, that belong to the conserved NEP1-like protein (NLP) family [85], which also includes the NPP1 family of Phytophthora effectors [115]. This family induces apoptosis in host plant tissues, and members are believed to play roles in the induction of necrotrophy [116–118]. Four NLPs are conserved in the two Colletotrichum species, and also have homologs in C. higginsianum [102]. In C. higginsianum, two of five NLPs (ChNLP3 and ChNLP5) lacked crucial amino acids and were not able to induce necrosis in N. benthamiana [102]. There are two putative C. sublineola homologs of ChNLP3 and three of ChNLP5, but C. graminicola has only a single homolog for each of these proteins. Two additional SSPs containing NPP1 domains in C. sublineola are not conserved in C. graminicola (Additional file 5: Table S5).
Only 21 C. graminicola NC SSPs, and 46 C. sublineola NC SSPs, matched Pfam categories. The vast majority (117 in C. graminicola and 225 in C. sublineola) did not have Pfam classifications, and this group included all of the LS-SSP proteins.
SSP families
The existence of gene families was explored by using blastp to identify potential orthologs and paralogs among the SSPs from C. graminicola and C. sublineola. The 1511 SSPs from the two species could be grouped into 789 families of related sequences (Additional file 5: Table S7). Most of the 325 conserved families that included members from both species were comprised of only one member in C. graminicola and one in C. sublineola. About 1/3 of the conserved families consisted of more than one putative paralog in one or both species. The largest conserved family included 29 predicted glycosyl hydrolase genes; 14 paralogs in C. graminicola, and 15 in C. sublineola.
C. graminicola had 189 NC SSP gene families that were not found in C. sublineola, and C. sublineola had 275 that were not found in C. graminicola (Additional file 5: Table S7). Among these NC families, nine included two paralogs, while the rest were each represented by only a single member. None of the NC families included more than two members. These results suggest that there has been relatively little duplication of SSP proteins within these two species.
SSP and SSM diversity among isolates
We sequenced the genome of a second strain of C. graminicola, M5.001, which was isolated from maize with anthracnose symptoms in the late 1980s in Brazil. This strain is sexually compatible with M1.001 [119]. Assembly and annotation statistics are included in Table 1, and predicted protein sequences are provided in Additional file 6. Only 73 out of the 12006 M1.001 predicted gene sequences (~1%) had no match in the M5.001 assembly (Additional file 5: Table S4). Only five of those genes were predicted to encode SSPs, while one was a putative SSM-associated gene. Of the 73 predicted M1.001 strain-specific genes, only seven had no matches to any other sequences in the NCBI nr database or the JGI databases (Additional file 5: Table S4). None of these seven had Pfam descriptions, and none were predicted to encode SSPs or SSM-associated proteins. There was transcript evidence for only one of them (Additional file 5: Table S4). The apparent low number of strain-specific SSPs in C. graminicola is consistent with an earlier report [120] that suggested that differences in expression may be more important than presence-absence polymorphisms for pathotype identity.
Two other genome assemblies are available for C. sublineola. The TX430BB strain was isolated in Texas in the late 1980s, and was sequenced by Baroncelli et al. [25]. The S3.001 strain is the epitype for the species [10, 104], and its genome assembly can be accessed from JGI (http://genome.jgi.doe.gov/). This strain was isolated in the late 1980s in Burkina Faso [5].
C. sublineola isolate CgSl1 has 117 predicted gene sequences (<1%), including 23 SSP genes, that are not found in the TX430BB assembly (Additional file 5: Table S5). It has 147 gene sequences (~1%) that are not found in S3.001, only 7 of which encode SSPs. Only 39 gene sequences are not found in either of the other two other strains, including 2 SSPs. All of the SSM-associated genes in CgSl1 appear to have matches in both other strains of C. sublineola. Of the 39 CgSl1 strain-specific genes, only four had no matches to any other sequences in the NCBI nr database or the JGI databases (Additional file 5: Table S5). None of these genes encodes an SSP, and only one has a Pfam domain (PF12511, a protein of unknown function).
The apparent rarity of strain-specific SSP gene sequences differs from some other fungal species, eg. Magnaporthe oryzae, where the deletion of secreted effector genes seems to be common, and to play an important role in the evolution of new races [121, 122]. However, comparisons with genome assemblies of the five closely related species within the graminicolous clade, accessed from JGI (http://genome.jgi.doe.gov/), suggests a more important role for deletion of effector genes, as well as other classes of genes, in speciation and host species adaptation, a finding that has also been reported by others based on comparative analyses of a wider range of Colletotrichum genera [25, 64].
Conclusions
In this work we have compared gene models from two contemporaneous, co-occurring strains of the sibling species C. graminicola and C. sublineola, and identified those that do not appear to be conserved as potential candidates for involvement in host specificity. Our approach was based on previous studies that have shown that gene gain and loss is associated with host range in many plant pathogens, including Colletotrichum [25, 64]. However, we do not mean to suggest that products of conserved genes don’t also play important roles, either alone or in combination with non-conserved gene products, in host specialization. The list of non-conserved genes identified in this work is a function of how we defined them, including the level of similarity that we considered significant, and the ability to accurately assign orthologs.
Our analysis confirmed that the genomes of the C. graminicola and C. sublineola strains were very similar to one another in both gene content and gene order, consistent with a relatively recent common ancestor. We also confirmed that each strain was able to successfully colonize its own living host (maize and sorghum, respectively), while the closely related non-host underwent an apparent hypersensitive response upon challenge. After applying our chosen parameters, we found that 14% of the C. graminicola gene models, and 22% of the C. sublineola gene models, were not conserved in the other species. Certain categories of genes were especially likely to be non-conserved including, as expected, genes that were predicted to encode SSPs and SSM-associated proteins that may play important roles in early events related to host recognition and the induction of compatibility. A relatively small number of the NC SSP gene sequences were also not conserved among different strains within each species, especially C. sublineola, which suggested the possibility of selection within the population and a potential Avr function. Races of both C. sublineola and C. graminicola have been reported to occur [123–130].
The majority of NCPs were not SSPs or SSM-associated proteins. Transporters, cytochrome P450s, and signaling proteins were well-represented, suggesting an important role for these functions in adaptation to varying aspects of each host environment, and in the secretion or evasion of toxic secondary metabolites. Transcription factors were also particularly abundant, suggesting that changes in gene expression patterns may be more important than the presence/absence of individual genes. Transcriptome and proteome comparisons would help us to address this hypothesis. CAZYmes were another common category, in spite of similarity of cell wall structure in maize and sorghum. It is known that some plant defenses target some CAZYmes in the apoplast [97] so it may be that these CAZYmes have diversified as a result of selection against host specific defenses. A relatively large number of the NCPs in both species were not categorized by either Ortho-MCL or Pfam. Many of these genes appeared to be conserved in other fungi, where they are predicted to encode hypothetical proteins of unknown function. Many are predicted to be secreted, or targeted to the nucleus or the mitochondria, and may interact with specific host factors to suppress or avoid host defenses, or to establish biotrophic hyphae or nutritional access. Similar categories of proteins were found to be rapidly evolving among several more distantly related Colletotrichum genera, suggesting that these categories play important roles in niche adaptation across the entire genus [64].
Our findings indicate that host specificity in these closely related pathosystems is not only a matter of recognition of, and response to, particular pathogenicity factors at the point of attempted penetration. Differences in fungal gene content reflect a much broader adaptation to the living host environment across the entire course of pathogen development, which has presumably developed during co-evolution of the host and its pathogen.
We found that the quality of the available assemblies and annotations had an important impact on our findings. We compared the published Broad annotation of C. graminicola with our MAKER annotation of C. sublineola. According to these data, C. sublineola had more genes than C. graminicola. As an exercise, we re-annotated C. graminicola with MAKER, and 14,419 genes were predicted, 1,108 more than MAKER predicted for C. sublineola. Comparison of the two annotations of C. graminicola (MAKER and Broad) using blastp revealed that they had about 10,000 genes in common, while the rest of the gene models were specific to each annotation (Additional file 5: Table S8). Some of the genes that were found in only one annotation were predicted to encode SSPs or SSM-associated proteins (Additional file 5: Table S8). We conclude from this exercise that the total number of potential SSP and SSM-associated genes we have reported here for C. graminicola and C. sublineola might be under-estimated, while the numbers of unique SSPs and SSM-associated proteins could be somewhat inflated. When we mapped the potential unique genes from each species against the genome assemblies of the other, between 50 and 70% of these genes did not hit the assembly of the other strain at all, and thus do appear to be truly non-conserved sequences. Among the apparently NC genes that did have hits to the assembly, our preliminary investigations suggest that many were not annotated due to fragmentation, which may be related to the different assembly qualities. The C. graminicola assembly, which was produced by using a combination of Sanger and 454 sequencing, includes fewer contigs and scaffolds than the C. sublineola assembly, which was done by using 454 alone. This fragmentation effect is expected to become progressively more significant as methods providing shorter reads (eg Illumina) are increasingly used for genome sequencing in fungi. Although it has not been widely acknowledged in previous comparative studies, it is clear that the use of datasets from diverse sources that have been developed by using different assembly and annotation programs and program parameters will have an impact on the results. Because of this, we emphasize the importance of confirming these data with other methods (e.g. amplification and cloning of entire genes, and confirmation of absence by hybridization or sequencing analysis), before proceeding with any additional studies focused on individual genes.
This work has provided important clues to functions (i.e. detoxification and transport, regulation of host and pathogen gene expression, and signaling and recognition) that are important in the determination of host preference among these two closely related and economically important pathogens. The data included here will provide a useful foundation for further studies to explore the basis for non-host recognition, with the goal of using this information to develop improved varieties of maize and sorghum for management of anthracnose diseases.
Methods
Plant and fungal growth and inoculation
Strains M1.001 and CgSl1 were originally obtained from Drs. Ralph Nicholson and Bob Hanau (Purdue University) and preserved on silica gel at −80 °C [131]. They are available from the corresponding author by request. Strains were cultured on potato dextrose agar (PDA, BD Difco, Franklin Lakes, NJ) under continuous fluorescent light at 23 °C. Spores were harvested from 2-week-old culture plates by gently scraping them from the surface, and washed three times before use.
Sweet sorghum variety Sugar Drip was obtained from Dr. Todd Pfeiffer (University of Kentucky). Maize inbred Mo17 was obtained from the North Central Regional Plant Introduction Station. Seeds were sown in a mixture of two parts sterile topsoil and three parts of Pro-Mix BX (Premiere Horticulture, Ltd, Riviere du Loup, PQ, Canada). Seedlings were maintained in the greenhouse with 14 h of light, watered every other day to saturation using an automated overhead irrigation system, and fertilized beginning 1 week after emergence two or three times per month as needed with a solution of 150 ppm of Peters 20-10-20 (Scotts-Sierra Horticultural Product Co., Marysville, OH).
Maize leaf sheaths were inoculated with a suspension of 5 × 105 spores per ml as described in [16]. Sorghum leaf sheaths were inoculated with a similar protocol, but instead of applying a single drop of inoculum, the leaf sheaths were entirely filled with the spore suspensions. Maize and sorghum seedlings at the V6 stage were inoculated with a suspension of 5 × 106 spores per ml by using a compressed-air spray applicator (Preval Model 267 Paint Spray Gun). After inoculation, the plants were incubated for 18 h in the dark at 25 °C in a dew chamber at 100% relative humidity before being returned to the greenhouse bench.
Sequencing and assembly of fungal genomes
Genomic DNA was extracted from fungal cultures by using the method described in [69] Shotgun Libraries were prepared according to the “Rapid Library Preparation Method Manual” (2010) for the GS FLX Titanium Series, using the Library Prep Kit with Rapid Library Rgt/Adaptors (Roche, Pleasanton CA). Paired-End 3000 Libraries were prepared according to the “GS FLX Titanium 3 kb Span Paired End Library Preparation Method Manual” using a Library Prep Kit with General Library Reagents and the GS FLX Titanium Paired End Adaptor Set (Roche). Emulsion PCR and enrichment was performed according to the “GS FLX emPCR Method Manual“ using the emPCR Kit Reagents (Lib-L) (Roche). Beads were loaded onto a PicoTiterPlate (70 × 75) for sequencing with the Sequencing Kit Reagents XLR70 (Roche). The genomes of C. graminicola strain M5.001 and C. sublineola strain CgSl1 were sequenced to 29X, and 43X coverage, respectively. Genome assembly was done by using Newbler version 2.9. The M5.001 Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank as BioProject SAMN06043298, under the accession number MRBI00000000. The version described in this paper is MRBI01000001. The CgSl1 Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank as BioProject PRJNA356071, under the accession number MQVQ00000000. The version described in this paper is MQVQ01000001.
The genome assemblies for C. graminicola strain M1.001 and for C. sublineola strain TX430BB were downloaded from the NCBI BioProjects database (BioProjects PRJNA37879 and PRJNA246670, respectively). Genome assemblies for C. sublineola strain S3.001 and for C. falcatum, C. somersetensis, C. caudatum, C. eremochloae, and C. zoysiae were downloaded from the Joint Genomes Institute Genome Portal (http://genome.jgi.doe.gov/).
Comparative analysis of genome assemblies
The genome assemblies were repeat-masked using a filtering algorithm previously implemented in TruMatch [132] The masked genomes were then aligned with one another in reciprocal pairwise using blastn with an e-value cutoff of 1e-200. The resulting blast reports were pre-screened to filter out aligned regions that contain hidden paralogs and single nucleotide polymorphisms were then identified. Finally, the SNP totals were divided by the total length of uniquely aligned sequence and multiplied by one million to provide a standard measure of genetic distance (SNPs/Mb). All steps in the analysis are implemented in a package of perl scripts known as SNPcounts.pl (available on request).
Genome annotation
The C. sublineola CgSl1 genome was annotated by using MAKER version 2.03 (http://www.yandell-lab.org/software/maker.html). Assembled contigs were filtered against RepBase model organism “fungi” with RepeatMasker version open-3.2.8. The MAKER analysis used the ab initio gene predictors AUGUSTUS version 2.3.1 (Fusarium model), GeneMark-ES version bp 2.3a (self-trained, see below), and SNAP version 2006-07-28 (self-trained, see below). Supporting evidence provided to MAKER consisted of protein sequences from Colletotrichum graminicola M1.001, as previously published [69]; and normalized unigenes from C. graminicola M1.001 as alternate organism EST evidence. To allow identification of previously-unannotated genes, MAKER was instructed to retain ab initio predictions that were not concordant with this evidence. MAKER was also instructed to extend coding sequences to include start and stop codons.
The C. graminicola M5.001 and M1.001 genomes were annotated by using MAKER version 2.28 (http://www.yandell-lab.org/software/maker.html). Assembled contigs were filtered against RepBase model organism “fungi” with RepeatMasker version open-3.2.8. The MAKER analysis used the ab initio gene predictors AUGUSTUS version 2.3.1 (Fusarium model), FGENESH version 3.1.1 (Fusarium model), GeneMark-ES version bp 3.9e (self-trained, see below), and SNAP version 2006-07-28 (self-trained, see below). Supporting evidence provided to MAKER included all complete protein sequences from Colletotrichum in the NCBI non-redundant protein database. As with C. sublineola annotations, MAKER was instructed to retain ab initio predictions. MAKER was also instructed to take additional steps to find alternatively spliced transcripts, and to extend coding sequences to include start and stop codons.
The two self-trained ab initio predictors were trained on the gene annotations produced by a preliminary MAKER run which did not include these two predictors (that is, using only AUGUSTUS, protein evidence, and alternate organism EST evidence for C. sublineola; and AUGUSTUS, FGENESH, and protein evidence for C. graminicola). To produce annotations more suitable for training SNAP and GeneMark-ES, this preliminary MAKER run was instructed to disregard ab initio predictions not concordant with protein evidence, to disregard single-exon evidence, and not to take additional steps to find alternatively-spliced transcripts. Other than these exceptions, the preliminary training run used the same inputs and parameters as the final MAKER run.
The predicted protein sequences for C. sublineola strain CgSl1 that were used for this work are included as supplementary data (Additional file 3). The predicted protein sequences for C. graminicola strain M5.001 are included in (Additional file 6).
Comparative analyses of genome annotations
To identify M1.001 gene sequences that were not present in the C. sublineolum assembly, (Additional file 5: Table S4), nucleotide sequences from Broad gene annotations of M1.001 published previously [69] were aligned against the C. sublineolum genome using exonerate version 2.2.0 (model est2genome) [133] (Additional file 5: Table S4). A gene sequence was considered non-unique if there was an alignment with at least 40% of the possible score for a sequence of that length. The same procedure was used to compare C. sublineolum MAKER annotations to the C. graminicola genome assembly (Additional file 5: Table S5).
As an exercise, the MAKER annotation for M1.001 (see above) was compared with the Broad annotation published previously [69]. The set of inferred protein sequences of the MAKER annotations were aligned against the set of inferred protein sequences of the Broad annotations using NCBI BLAST version 2.2.18 in protein-to-protein (blastp) mode.
For each protein sequence P, the best alignment against the set of sequences annotated by the other procedure (MAKER or Broad), as determined by blastall -b 1, was selected. High-scoring pairs (HSPs) with an e-value of 1e-10 or higher were discarded, and a percent identity ID A for the alignment was obtained by weighted average of the percent identities of the remaining HSPs, with the alignment length of the HSP as the weight. The total alignment length L A was taken to be the sum of the alignment lengths of the (non-discarded) HSPs.
A gene was considered to be a unique annotation if the percent identity, weighted by the ratio of total alignment length to query or to target length, was less than 70%. That is, an annotation was considered unique if either ID A × L A /L P < 70%, or ID A × L A /L H < 70%, where L P denotes the length of the query sequence P and L H denotes the length of the sequence that was selected as the best hit among annotations produced by the other method.
Genome synteny
Genome synteny was analyzed by using the Synteny Mapping and Analysis Program (Symap) v4.2 [134] and default parameters. Colletotrichum sublineola scaffolds were aligned to the 13 previously published chromosomes of C. graminicola strain M1.001 [69].
Identification of orthologous and unique genes
Fungal protein sequences used in this study were downloaded from the Broad Institute (C. graminicola, C. higginsianum, Fusarium graminearum, F. oxysporum, Verticillium dahliae, Aspergillus flavus) and the Joint Genome Institute (Trichoderma reesei, C. falcatum, C. somersetensis, C. caudatum, C. eremochloae, C. zoysiae, C. sublineola strain S3.001). Protein sequences from Epichloë festucae were the FGENESH gene predictions previously used in the Clavicipitaceae analysis [135]. Putative orthologs were identified by using two methods. The first method was application of Ortho-MCL and COCO-CL (COrrelation COefficient-based CLustering) to the annotations [76, 136], following a procedure previously used for ortholog identification within the Clavicipitaceae [135]. The species included for comparison in the Ortho-MCL/COCO-CL analysis were: C. graminicola; C. higginsianum; C. sublineola CgSl1; M. oryzae; E. festucae; F. graminearum; F. oxysporum; T. reesei; V. dahlia; and A. flavus. The second method used for ortholog identification was Reciprocal Best Hit (RBH) with an expect-value cutoff of 1e-5 [77, 137]. This method was used to compare proteins from C. graminicola, C. sublineola, and C. higginsianum.
Protein characterization
Predicted proteins were compared by blastp with the non-redundant protein sequence database from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with an expect-value cutoff of 1e-5 [138]. Predicted proteins were assigned to functional families by comparing to the Protein Family (Pfam) database (http://pfam.sanger.ac.uk/) version 29.0 (December 2015) by using pfamScan software version 1.5 (October 2013), with an e-value cutoff of 1e-5 [139]. Transporters were predicted by using the Transporters Classification Database (http://www.tcdb.org) (2016) with an e-value cutoff of 1e-5 [140]. CAZymes were characterized by using dbCAN HMMs version 5.0 (http://csbl.bmb.uga.edu/dbCAN/annotate.php), which is based on the classification scheme of CAZyDB [141, 142]. Predicted proteins were compared with the Pathogen-Host Interaction (PHI) database (www.phi-base.org) Version 4.1 (May 2016) [74, 75] using blastp and an e-value cutoff of 1e-5. To predict protein localizations, WoLF-PSORT for fungi [143] version 0.2 (August 2006) was used, as described in [69]. For the classification of putative secreted proteases, the sequences of predicted secreted proteins were submitted to MEROPS release 10.0 batch blast analysis (http://merops.sanger.ac.uk) [144] also as described [69]. For prediction of fungal effectors, predicted secreted proteins were analyzed by using the EffectorP prediction tool (http://effectorp.csiro.au) (December 2015) [103].
SSM-associated proteins
The five classes of candidate SSM-associated genes (PKS, NRPS, PKS-NRPS hybrid, DMAT, and TS) were identified from C. sublineola by applying a process that included Pfam and Ortho-MCL/COCO-CL analysis; followed by manual annotation and validation of domains using the Conserved Domain Database (CDD) (http://www.ncbi.nlm.nih.gov/cdd/); blastp comparisons with the NCBI nr database; and InterproScan analysis. This protocol has been described in more detail previously [69].
Colletotrichum sublineola SSM gene clusters were manually annotated by evaluating Ortho-MCL/COCO-CL results for the genes that were located upstream and downstream of the SSM-associated backbone genes. Genes that had no or few orthologs were considered to belong to the clusters, while genes that were conserved in most or all of the ten species included in the analysis defined the outside boundaries of the clusters.
Phylogenetic analysis of SSM-associated proteins
Phylogenetic analysis of SM genes was performed by using phylogeny.fr (http://www.phylogeny.fr/index.cgi) (2003). The A and KS N-terminal and C-terminal domains of the NRPS, PKS, and NRPS-PKS hybrids were identified by using the NCBI CCD. Amino acid sequences were aligned by using MUSCLE version 3.8.31 (May 2010) [145] and default parameters, and phylogenies were inferred by maximum-likelihood using PhyML version 3.0. Statistical branch support was provided by an approximation to the standard likelihood ratio test, aLRT [146].
Acknowledgements
The authors are very grateful to Etta Nuckles, Doug Brown, Jola Jaromczyk, Harrison Inocencio, and Sarah Holton for excellent technical support. The information reported in this paper (No. 16-12-109) is part of a project of the Kentucky Agricultural Experiment Station and is published with the approval of the Director.
Funding
This work was partially supported by the U.S. Department of Agriculture Cooperative State Research, Education, and Extension Service (USDA-CSREES) grant #20093445720125 (LJV); by the National Institute of Food and Agriculture, U.S. Department of Agriculture Hatch project 0231781 (LJV); and by a University of Kentucky College of Agriculture, Food, and Environment Research Activity Award (LJV).
Availability of data and materials
The assemblies and annotations generated for the current study are available in the Genbank repository, as BioProjects SAMN06043298 and PRJNA356071. Biological materials from the current study are available from the corresponding author on reasonable request.
Authors’ contributions
Co-first authors EASB and KVX performed all the laboratory experiments. KVX did manual analysis and phylogenetics of specialized secondary metabolite genes and gene clusters, with the assistance of MFT and CLS. KVX did the cytological analyses of non-host reactions of maize and sorghum to Colletotrichum fungi. EASB did manual and computational analyses of the putative SSP genes and characterization of the fungal protein families. NM and EASB did the bioinformatic analysis of the C. graminicola and C. sublineola genomes and predicted proteomes. MLF conducted the blastn and SNP comparisons of the genome assemblies of Colletotrichum graminicola, C. higginsianum, and C. sublineola. LJV conceived and managed the project and conducted manual confirmations of the data. EASB, KVX, NM, MLF, and LJV wrote and revised the manuscript, and CLS and MFT helped with revisions. All authors have read and approve of the final version of the manuscript.
Authors information
The first two authors (EASB and KVX) contributed equally to this work and are listed in alphabetical order.
Competing interests
The authors declare that they have no competing interests. EASB is currently an employee of Monsanto Company, Brazil, but Monsanto was not involved in any of the work described in this manuscript, which was done prior to her employment there, and the current position of EASB does not affect the authors’ adherence to BMC Genomics policies on sharing data and materials.
Consent for publication
NA.
Ethics approval and consent to participate
NA.
Additional files
{kind=link}
M1.001 on Sugar Drip sorghum, 48 hpi, cells beneath appressoria (white arrows) plasmolyzed (result not typical). Scale bars equal to 50 μm. (JPG 302 kb)
{kind=link}
Plasmolysis controls. A: Maize leaf sheath, 72 h after mock inoculation, most cells still plasmolyze. B: Sugar Drip leaf sheath, 72 h after mock inoculation, most cells still plasmolyze. Scale bars equal to 50 μm. (JPG 645 kb)
FASTA predicted proteins of C. sublineola strain CgSl1. (TXT 6816 kb)
{kind=link}
Alignments of sequences of CgSl1 with species type S3.001. A: actin, B: chitin synthase, C: histone H3, D: beta-tubulin, E: ITS. Alignments done with MUSCLE version 3.7 and default parameters. (JPG 300 kb)
EXCEL file including detailed analysis of the genes of C. graminicola M1.001 and C. sublineola CgSl1. (XLSX 1703 kb)
FASTA predicted proteins of C. graminicola strain M5.001. (TXT 7838 kb)
Contributor Information
E. A. S. Buiate, Email: esterbuiate@uky.edu
K. V. Xavier, Email: katiaxavier@uky.edu
N. Moore, Email: neil@cs.uky.edu
M. F. Torres, Email: mariaftorres@gmail.com
M. L. Farman, Email: mark.farman@uky.edu
C. L. Schardl, Email: chris.schardl@uky.edu
L. J. Vaillancourt, Email: vaillan@uky.edu
References
- 1.Crouch J, O’Connell R, Gan P, Buiate E, Torres MF, Beirn L, Shirasu K, Vaillancourt L. The genomics of Colletotrichum. In: Genomics of Plant-Associated Fungi: Monocot Pathogens. Berlin-Heidelberg: Springer; 2014. 69-102.
- 2.Dean R, Van Kan JA, Pretorius ZA, Hammond‐Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J. The Top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 2012;13(4):414–30. doi: 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyde K, Cai L, Cannon P, Crouch J, Crous P, Damm U, Goodwin P, Chen H, Johnston P, Jones E. Colletotrichum—names in current use. Fungal Divers. 2009;39(1):147–82. [Google Scholar]
- 4.Sutton B. The appressoria of Colletotrichum graminicola and C. falcatum. Can J Bot. 1968;46(7):873–6. doi: 10.1139/b68-115. [DOI] [Google Scholar]
- 5.Vaillancourt LJ, Hanau RM. Genetic and morphological comparisons of Glomerella (Colletotrichum) isolates from maize and from sorghum. Exp Mycol. 1992;16(3):219–29. doi: 10.1016/0147-5975(92)90030-U. [DOI] [Google Scholar]
- 6.Jamil F, Nicholson R. Susceptibility of corn to isolates of Colletotrichum graminicola pathogenic to other grasses. Plant Dis. 1987;71(9):809–10. doi: 10.1094/PD-71-0809. [DOI] [Google Scholar]
- 7.Sherriff C, Whelan M, Arnold G, Bailey J. rDNA sequence analysis confirms the distinction between Colletotrichum graminicola and C. sublineolum. Mycol Res. 1995;99(4):475–8. doi: 10.1016/S0953-7562(09)80649-7. [DOI] [Google Scholar]
- 8.Du M, Schardl CL, Nuckles EM, Vaillancourt LJ. Using mating-type gene sequences for improved phylogenetic resolution of Colletotrichum species complexes. Mycologia. 2005;97(3):641–58. doi: 10.3852/mycologia.97.3.641. [DOI] [PubMed] [Google Scholar]
- 9.Crouch JA, Clarke BB, Hillman BI. Unraveling evolutionary relationships among the divergent lineages of Colletotrichum causing anthracnose disease in turfgrass and corn. Phytopathology. 2006;96(1):46–60. doi: 10.1094/PHYTO-96-0046. [DOI] [PubMed] [Google Scholar]
- 10.Crouch JA, Clarke BB, White JF, Hillman BI. Systematic analysis of the falcate-spored graminicolous Colletotrichum and a description of six new species from warm-season grasses. Mycologia. 2009;101(5):717–32. doi: 10.3852/08-230. [DOI] [PubMed] [Google Scholar]
- 11.Swigoňová Z, Lai J, Ma J, Ramakrishna W, Llaca V, Bennetzen JL, Messing J. Close split of sorghum and maize genome progenitors. Genome Res. 2004;14(10a):1916–23. doi: 10.1101/gr.2332504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dale J. Corn anthracnose. Plant Dis Rep. 1963;47:245–9. [Google Scholar]
- 13.LeBeau F. The eradicant action of a fungicide on the Colletotrichum-Lilii in lily bulbs. St. Paul: American Phytopathological Society 3340 pilot knob road; 1946. pp. 391–3. [Google Scholar]
- 14.Williams L, Willis G. Disease of corn caused by Colletotrichum graminicolum. Phytopathology. 1963;53(3):364–5. [Google Scholar]
- 15.Venard C, Vaillancourt L. Penetration and colonization of unwounded maize tissues by the maize anthracnose pathogen Colletotrichum graminicola and the related nonpathogen C. sublineolum. Mycologia. 2007;99(3):368–77. doi: 10.3852/mycologia.99.3.368. [DOI] [PubMed] [Google Scholar]
- 16.Torres MF, Cuadros DF, Vaillancourt LJ. Evidence for a diffusible factor that induces susceptibility in the Colletotrichum–maize disease interaction. Mol Plant Pathol. 2014;15(1):80–93. doi: 10.1111/mpp.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury SC. A disease of Zea mays caused by Colletotrichum graminicola [Ces.] Wils. Indian J Agric Sci. 1936;6:833–43. [Google Scholar]
- 18.Wheeler H, Politis D, Poneleit C. Pathogenicity, host range, and distribution of Colletotrichum graminicola on corn. Phytopathology. 1974;64(3):293–6. [Google Scholar]
- 19.Kroken S, Glass NL, Taylor JW, Yoder O, Turgeon BG. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci. 2003;100(26):15670–5. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Condon BJ, Leng Y, Wu D, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, MohdZainudin N. Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 2013;9(1):e1003233. doi: 10.1371/journal.pgen.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito K, Tanaka T, Hatta R, Yamamoto M, Akimitsu K, Tsuge T. Dissection of the host range of the fungal plant pathogen Alternaria alternata by modification of secondary metabolism. Mol Microbiol. 2004;52(2):399–411. doi: 10.1111/j.1365-2958.2004.04004.x. [DOI] [PubMed] [Google Scholar]
- 22.Tyler BM. Entering and breaking: virulence effector proteins of oomycete plant pathogens. Cell Microbiol. 2009;11(1):13–20. doi: 10.1111/j.1462-5822.2008.01240.x. [DOI] [PubMed] [Google Scholar]
- 23.de Jonge R, Bolton MD, Thomma BP. How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr Opin Plant Biol. 2011;14(4):400–6. doi: 10.1016/j.pbi.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Donofrio NM, Raman V. Roles and delivery mechanisms of fungal effectors during infection development: common threads and new directions. Curr Opin Microbiol. 2012;15(6):692–8. doi: 10.1016/j.mib.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Baroncelli R, Amby DB, Zapparata A, Sarrocco S, Vannacci G, Le Floch G, Harrison RJ, Holub E, Sukno SA, Sreenivasaprasad S. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genomics. 2016;17(1):1. doi: 10.1186/s12864-016-2917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bentley S, Chater K, Cerdeno-Tarraga A-M, Challis G, Thomson N, James K, Harris D, Quail M, Kieser H, Harper D. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2) Nature. 2002;417(6885):141–7. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 27.Birch A. Biosynthesis of polyketides and related compounds. Science. 1967;156(3772):202–6. doi: 10.1126/science.156.3772.202. [DOI] [PubMed] [Google Scholar]
- 28.Lee S-L, Floss HG, Heinstein P. Purification and properties of dimethylallylpyrophosphate: Tryptophan dimethylallyl transferase, the first enzyme of ergot alkaloid biosynthesis in Claviceps. sp. SD 58. Arch Biochem Biophys. 1976;177(1):84–94. doi: 10.1016/0003-9861(76)90418-5. [DOI] [PubMed] [Google Scholar]
- 29.McAlpine JB, Bachmann BO, Piraee M, Tremblay S, Alarco A-M, Zazopoulos E, Farnet CM. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J Nat Prod. 2005;68(4):493–6. doi: 10.1021/np0401664. [DOI] [PubMed] [Google Scholar]
- 30.Martin JF, Liras P. Organization and expression of genes involved in the biosynthesis of antibiotics and other secondary metabolites. Annu Rev Microbiol. 1989;43(1):173–206. doi: 10.1146/annurev.mi.43.100189.001133. [DOI] [PubMed] [Google Scholar]
- 31.Ellis JG, Rafiqi M, Gan P, Chakrabarti A, Dodds PN. Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens. Curr Opin Plant Biol. 2009;12(4):399–405. doi: 10.1016/j.pbi.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 32.van der Hoorn RA, Kamoun S. From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell. 2008;20(8):2009–17. doi: 10.1105/tpc.108.060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Djamei A, Schipper K, Rabe F, Ghosh A, Vincon V, Kahnt J, Osorio S, Tohge T, Fernie AR, Feussner I. Metabolic priming by a secreted fungal effector. Nature. 2011;478(7369):395–8. doi: 10.1038/nature10454. [DOI] [PubMed] [Google Scholar]
- 34.Dou D, Kale SD, Wang X, Jiang RH, Bruce NA, Arredondo FD, Zhang X, Tyler BM. RXLR-mediated entry of Phytophthora sojae effector Avr1b into soybean cells does not require pathogen-encoded machinery. Plant Cell. 2008;20(7):1930–47. doi: 10.1105/tpc.107.056093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kemen E, Kemen AC, Rafiqi M, Hempel U, Mendgen K, Hahn M, Voegele RT. Identification of a protein from rust fungi transferred from haustoria into infected plant cells. Mol Plant-Microbe Interact. 2005;18(11):1130–9. doi: 10.1094/MPMI-18-1130. [DOI] [PubMed] [Google Scholar]
- 36.Khang CH, Berruyer R, Giraldo MC, Kankanala P, Park S-Y, Czymmek K, Kang S, Valent B. Translocation of Magnaporthe oryzae effectors into rice cells and their subsequent cell-to-cell movement. Plant Cell. 2010;22(4):1388–403. doi: 10.1105/tpc.109.069666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Phytopathology. 2006;44(1):41. doi: 10.1146/annurev.phyto.44.070505.143436. [DOI] [PubMed] [Google Scholar]
- 38.van der Does HC, Rep M. Virulence genes and the evolution of host specificity in plant-pathogenic fungi. Mol Plant-Microbe Interact. 2007;20(10):1175–82. doi: 10.1094/MPMI-20-10-1175. [DOI] [PubMed] [Google Scholar]
- 39.Vleeshouwers VG, Oliver RP. Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol Plant-Microbe Interact. 2014;27(3):196–206. doi: 10.1094/MPMI-10-13-0313-IA. [DOI] [PubMed] [Google Scholar]
- 40.Rep M, Van Der Does HC, Meijer M, Van Wijk R, Houterman PM, Dekker HL, De Koster CG, Cornelissen BJ. A small, cysteine‐rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I‐3‐mediated resistance in tomato. Mol Microbiol. 2004;53(5):1373–83. doi: 10.1111/j.1365-2958.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 41.Mosquera G, Giraldo MC, Khang CH, Coughlan S, Valent B. Interaction transcriptome analysis identifies Magnaporthe oryzae BAS1-4 as biotrophy-associated secreted proteins in rice blast disease. Plant Cell. 2009;21(4):1273–90. doi: 10.1105/tpc.107.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444(7117):323–9. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 43.Schulze-Lefert P, Panstruga R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 2011;16(3):117–25. doi: 10.1016/j.tplants.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Tosa Y. A model for the evolution of formae speciales and races. Phytopathology. 1992;82(7):728–30. [Google Scholar]
- 45.Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, Kusaba M, Yoshida K, Terauchi R, Fujita Y, Nakayashiki H. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog. 2011;7(7):e1002147. doi: 10.1371/journal.ppat.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami J, Tosa Y, Kataoka T, Tomita R, Kawasaki J, Chuma I, Sesumi Y, Kusaba M, Nakayashiki H, Mayama S. Analysis of host species specificity of Magnaporthe grisea toward wheat using a genetic cross between isolates from wheat and foxtail millet. Phytopathology. 2000;90(10):1060–7. doi: 10.1094/PHYTO.2000.90.10.1060. [DOI] [PubMed] [Google Scholar]
- 47.Scoles G, Nga N, Hau V, Tosa Y. Identification of genes for resistance to a Digitaria isolate of Magnaporthe grisea in common wheat cultivars. Genome. 2009;52(9):801–9. doi: 10.1139/G09-054. [DOI] [PubMed] [Google Scholar]
- 48.Takabayashi N, Tosa Y, Oh H, Mayama S. A gene-for-gene relationship underlying the species-specific parasitism of Avena/Triticum isolates of Magnaporthe grisea on wheat cultivars. Phytopathology. 2002;92(11):1182–8. doi: 10.1094/PHYTO.2002.92.11.1182. [DOI] [PubMed] [Google Scholar]
- 49.Tosa Y, Tamba H, Tanaka K, Mayama S. Genetic analysis of host species specificity of Magnaporthe oryzae isolates from rice and wheat. Phytopathology. 2006;96(5):480–4. doi: 10.1094/PHYTO-96-0480. [DOI] [PubMed] [Google Scholar]
- 50.Valent A, Bénard J, Clausse B, Barrois M, Valteau-Couanet D, Terrier-Lacombe M-J, Spengler B, Bernheim A. In vivo elimination of acentric double minutes containing amplified MYCN from neuroblastoma tumor cells through the formation of micronuclei. Am J Pathol. 2001;158(5):1579–84. doi: 10.1016/S0002-9440(10)64112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang S, Sweigard JA, Valent B. The PWL host specificity gene family in the blast fungus Magnaporthe grisea. Mol Plant Microbe Interact. 1995;8(6):939–48. doi: 10.1094/MPMI-8-0939. [DOI] [PubMed] [Google Scholar]
- 52.Matsumura K, Tosa Y. The rye mildew fungus carries avirulence genes corresponding to wheat genes for resistance to races of the wheat mildew fungus. Phytopathology. 1995;85(7):753–6. doi: 10.1094/Phyto-85-753. [DOI] [Google Scholar]
- 53.de Wit PJ, Van Der Burgt A, Ökmen B, Stergiopoulos I, Abd-Elsalam KA, Aerts AL, Bahkali AH, Beenen HG, Chettri P, Cox MP. The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012;8(11):e1003088. doi: 10.1371/journal.pgen.1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nemri A, Saunders DG, Anderson C, Upadhyaya NM, Win J, Lawrence GJ, Jones DA, Kamoun S, Ellis JG, Dodds PN. The genome sequence and effector complement of the flax rust pathogen Melampsora lini. Front Plant Sci. 2014;5:98. doi: 10.3389/fpls.2014.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantu D, Segovia V, MacLean D, Bayles R, Chen X, Kamoun S, Dubcovsky J, Saunders DG, Uauy C. Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics. 2013;14(1):1. doi: 10.1186/1471-2164-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brefort T, Tanaka S, Neidig N, Doehlemann G, Vincon V, Kahmann R. Characterization of the largest effector gene cluster of Ustilago maydis. PLoS Pathog. 2014;10(7):e1003866. doi: 10.1371/journal.ppat.1003866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schirawski J, Mannhaupt G, Münch K, Brefort T, Schipper K, Doehlemann G, Di Stasio M, Rössel N, Mendoza-Mendoza A, Pester D. Pathogenicity determinants in smut fungi revealed by genome comparison. Science. 2010;330(6010):1546–8. doi: 10.1126/science.1195330. [DOI] [PubMed] [Google Scholar]
- 58.Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, Jiang RH, Zody MC, Kunjeti SG, Donofrio NM. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science. 2010;330(6010):1540–3. doi: 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 59.Rafiqi M, Ellis JG, Ludowici VA, Hardham AR, Dodds PN. Challenges and progress towards understanding the role of effectors in plant–fungal interactions. Curr Opin Plant Biol. 2012;15(4):477–82. doi: 10.1016/j.pbi.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 60.Lee HA, Kim SY, Oh SK, Yeom SI, Kim SB, Kim MS, Kamoun S, Choi D. Multiple recognition of RXLR effectors is associated with nonhost resistance of pepper against Phytophthora infestans. New Phytol. 2014;203(3):926–38. doi: 10.1111/nph.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Win J, Morgan W, Bos J, Krasileva KV, Cano LM, Chaparro-Garcia A, Ammar R, Staskawicz BJ, Kamoun S. Adaptive evolution has targeted the C-terminal domain of the RXLR effectors of plant pathogenic oomycetes. Plant Cell. 2007;19(8):2349–69. doi: 10.1105/tpc.107.051037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stüber K, van Themaat EVL, Brown JK, Butcher SA, Gurr SJ. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science. 2010;330(6010):1543–6. doi: 10.1126/science.1194573. [DOI] [PubMed] [Google Scholar]
- 63.Dong S, Stam R, Cano LM, Song J, Sklenar J, Yoshida K, Bozkurt TO, Oliva R, Liu Z, Tian M. Effector specialization in a lineage of the Irish potato famine pathogen. Science. 2014;343(6170):552–5. doi: 10.1126/science.1246300. [DOI] [PubMed] [Google Scholar]
- 64.Gan P, Narusaka M, Kumakura N, Tsushima A, Takano Y, Narusaka Y, Shirasu K. Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome Biol Evol. 2016;8(5):1467–81. doi: 10.1093/gbe/evw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saunders DG, Win J, Cano LM, Szabo LJ, Kamoun S, Raffaele S. Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS One. 2012;7(1):e29847. doi: 10.1371/journal.pone.0029847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forgey W, Blanco M, Loegering W. Differences in pathological capabilities and host specificity of Colletotrichum graminicola on Zea mays [maize]. Plant Dis Rep. 1978;62(7-12):573.
- 67.Snyder BA, Nicholson RL. Synthesis of phytoalexins in sorghum as a site-specific response to fungal ingress. Science. 1990;248(4963):1637–9. doi: 10.1126/science.248.4963.1637. [DOI] [PubMed] [Google Scholar]
- 68.Mims C, Vaillancourt L. Ultrastructural characterization of infection and colonization of maize leaves by Colletotrichum graminicola, and by a C. graminicola pathogenicity mutant. Phytopathology. 2002;92(7):803–12. doi: 10.1094/PHYTO.2002.92.7.803. [DOI] [PubMed] [Google Scholar]
- 69.O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Damm U, Buiate EA, Epstein L, Alkan N. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet. 2012;44(9)1060–65. [DOI] [PMC free article] [PubMed]
- 70.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 71.Damm U, O’Connell R, Groenewald J, Crous P. The Colletotrichum destructivum species complex–hemibiotrophic pathogens of forage and field crops. Stud Mycol. 2014;79:49–84. doi: 10.1016/j.simyco.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rollins JA. The characterization and inheritance of chromosomal variation in Glomerella graminicola. West Lafayette: Purdue University; 1996. [Google Scholar]
- 73.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J. Pfam: the protein families database. Nucleic Acids Res. 2013;42:gkt1223. [DOI] [PMC free article] [PubMed]
- 74.Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(suppl 1):D459–64. doi: 10.1093/nar/gkj047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack KE. The Pathogen-Host Interactions database (PHI-base): additions and future developments. Nucleic Acids Res. 2014;43:gku1165. [DOI] [PMC free article] [PubMed]
- 76.Li LSCJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wall D, Fraser H, Hirsh A. Detecting putative orthologs. Bioinformatics. 2003;19(13):1710–1. doi: 10.1093/bioinformatics/btg213. [DOI] [PubMed] [Google Scholar]
- 78.Torres MF, Ghaffari N, Buiate EA, Moore N, Schwartz S, Johnson CD, Vaillancourt LJ. A Colletotrichum graminicola mutant deficient in the establishment of biotrophy reveals early transcriptional events in the maize anthracnose disease interaction. BMC Genomics. 2016;17(1):1. doi: 10.1186/s12864-016-2546-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vargas WA, Sanz-Martín JM, Rech GE, Armijos-Jaramillo VD, Rivera LP, Echeverria MM, Díaz-Mínguez JM, Thon MR, Sukno SA. A fungal effector with host nuclear localization and DNA-binding properties is required for maize anthracnose development. Mol Plant Microbe Interact. 2016;29:83–95. doi: 10.1094/MPMI-09-15-0209-R. [DOI] [PubMed] [Google Scholar]
- 80.Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–59. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin K, Musso G, Vlasblom J, Jessulat M, Deineko V, Negroni J, Mosca R, Malty R, Nguyen-Tran D-H, Aoki H. Yeast mitochondrial protein–protein interactions reveal diverse complexes and disease-relevant functional relationships. J Proteome Res. 2015;14(2):1220–37. doi: 10.1021/pr501148q. [DOI] [PubMed] [Google Scholar]
- 83.Lee J, Sharma S, Kim J, Ferrante RJ, Ryu H. Mitochondrial nuclear receptors and transcription factors: who’s minding the cell? J Neurosci Res. 2008;86(5):961–71. doi: 10.1002/jnr.21564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Jonge R, Thomma BP. Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol. 2009;17(4):151–7. doi: 10.1016/j.tim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 85.Gijzen M, Nürnberger T. Nep1-like proteins from plant pathogens: recruitment and diversification of the NPP1 domain across taxa. Phytochemistry. 2006;67(16):1800–7. doi: 10.1016/j.phytochem.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 86.Bhadauria V, Banniza S, Vandenberg A, Selvaraj G, Wei Y. Overexpression of a novel biotrophy-specific Colletotrichum truncatum effector, CtNUDIX, in hemibiotrophic fungal phytopathogens causes incompatibility with their host plants. Eukaryot Cell. 2013;12(1):2–11. doi: 10.1128/EC.00192-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dong S, Wang Y. Nudix effectors: a common weapon in the arsenal of plant pathogens. PLoS Pathog. 2016;12(8):e1005704. doi: 10.1371/journal.ppat.1005704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kulkarni RD, Kelkar HS, Dean RA. An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem Sci. 2003;28(3):118–21. doi: 10.1016/S0968-0004(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 89.Pao SS, Paulsen IT, Saier MH. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saier Jr MH, Paulsen IT. Phylogeny of multidrug transporters. In: Seminars in cell & developmental biology. Academic Press. 2001;12(3):205–13. [DOI] [PubMed]
- 91.Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009;14(1):3–9. doi: 10.1007/s10911-009-9109-9. [DOI] [PubMed] [Google Scholar]
- 92.Rao PV, Maddala R. Ankyrin-B in lens architecture and biomechanics: Just not tethering but more. BioArchitecture. 2016;6(2):39–45. doi: 10.1080/19490992.2016.1156284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wight WD, Kim K-H, Lawrence CB, Walton JD. Biosynthesis and role in virulence of the histone deacetylase inhibitor depudecin from Alternaria brassicicola. Mol Plant-Microbe Interact. 2009;22(10):1258–67. doi: 10.1094/MPMI-22-10-1258. [DOI] [PubMed] [Google Scholar]
- 94.Chen H, Lee MH, Daub ME, Chung KR. Molecular analysis of the cercosporin biosynthetic gene cluster in Cercospora nicotianae. Mol Microbiol. 2007;64(3):755–70. doi: 10.1111/j.1365-2958.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 95.Carpita NC, McCann MC. Maize and sorghum: genetic resources for bioenergy grasses. Trends Plant Sci. 2008;13(8):415–20. doi: 10.1016/j.tplants.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 96.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–6. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 97.Misas-Villamil JC, Van der Hoorn RA. Enzyme–inhibitor interactions at the plant–pathogen interface. Curr Opin Plant Biol. 2008;11(4):380–8. doi: 10.1016/j.pbi.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 98.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6(8):493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 99.Brakhage AA, Schroeckh V. Fungal secondary metabolites–strategies to activate silent gene clusters. Fungal Genet Biol. 2011;48(1):15–22. doi: 10.1016/j.fgb.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 100.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Tolerance and specificity of polyketide synthases. Annu Rev Biochem. 1999;68(1):219–53. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 101.Bowen JK, Mesarich CH, Rees-George J, Cui W, Fitzgerald A, Win J, Plummer KM, Templeton MD. Candidate effector gene identification in the ascomycete fungal phytopathogen Venturia inaequalis by expressed sequence tag analysis. Mol Plant Pathol. 2009;10(3):431–48. doi: 10.1111/j.1364-3703.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kleemann J, Rincon-Rivera LJ, Takahara H, Neumann U, van Themaat EVL, van der Does HC, Hacquard S, Stüber K, Will I, Schmalenbach W. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathog. 2012;8(4):e1002643. doi: 10.1371/journal.ppat.1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sperschneider J, Gardiner DM, Dodds PN, Tini F, Covarelli L, Singh KB, Manners JM, Taylor JM. EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2015;210:743–61. [DOI] [PubMed]
- 104.Crouch JA, Tomaso-Peterson M. Anthracnose disease of centipedegrass turf caused by Colletotrichum eremochloae, a new fungal species closely related to Colletotrichum sublineola. Mycologia. 2012;104(5):1085–96. doi: 10.3852/11-317. [DOI] [PubMed] [Google Scholar]
- 105.DeZwaan TM, Carroll AM, Valent B, Sweigard JA. Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell. 1999;11(10):2013–30. doi: 10.1105/tpc.11.10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Choi W, Dean RA. The adenylate cyclase gene MAC1 of Magnaporthe grisea controls appressorium formation and other aspects of growth and development. Plant Cell. 1997;9(11):1973–83. doi: 10.1105/tpc.9.11.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Raikhel N, Lee H, Broekaert W. Structure and function of chitin-binding proteins. Annu Rev Plant Biol. 1993;44(1):591–615. doi: 10.1146/annurev.pp.44.060193.003111. [DOI] [Google Scholar]
- 108.van Esse HP, Bolton MD, Stergiopoulos I, de Wit PJ, Thomma BP. The chitin-binding Cladosporium fulvum effector protein Avr4 is a virulence factor. Mol Plant-Microbe Interact. 2007;20(9):1092–101. doi: 10.1094/MPMI-20-9-1092. [DOI] [PubMed] [Google Scholar]
- 109.Kombrink A, Thomma BP. LysM effectors: secreted proteins supporting fungal life. PLoS Pathog. 2013;9(12):e1003769. doi: 10.1371/journal.ppat.1003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MH, Thomma BP. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science. 2010;329(5994):953–5. doi: 10.1126/science.1190859. [DOI] [PubMed] [Google Scholar]
- 111.Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, Hammond-Kosack KE, Thomma BP, Rudd JJ. Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 2011;156(2):756–69. doi: 10.1104/pp.111.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, Saitoh H, Terauchi R, Nishizawa Y, Shibuya N, Thomma BP. Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell. 2012;24(1):322–35. doi: 10.1105/tpc.111.092957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pain RH. In: Mechanisms of protein folding. PAIN RH, editor. 1994. [Google Scholar]
- 114.Perfect SE, O’Connell RJ, Green EF, Doering‐Saad C, Green JR. Expression cloning of a fungal proline‐rich glycoprotein specific to the biotrophic interface formed in the Colletotrichum–bean interaction. Plant J. 1998;15(2):273–9. doi: 10.1046/j.1365-313X.1998.00196.x. [DOI] [PubMed] [Google Scholar]
- 115.Fellbrich G, Romanski A, Varet A, Blume B, Brunner F, Engelhardt S, Felix G, Kemmerling B, Krzymowska M, Nürnberger T. NPP1, a Phytophthora‐associated trigger of plant defense in parsley and Arabidopsis. Plant J. 2002;32(3):375–90. doi: 10.1046/j.1365-313X.2002.01454.x. [DOI] [PubMed] [Google Scholar]
- 116.Qutob D, Kamoun S, Gijzen M. Expression of a Phytophthora sojae necrosis‐inducing protein occurs during transition from biotrophy to necrotrophy. Plant J. 2002;32(3):361–73. doi: 10.1046/j.1365-313X.2002.01439.x. [DOI] [PubMed] [Google Scholar]
- 117.Qutob D, Kemmerling B, Brunner F, Küfner I, Engelhardt S, Gust AA, Luberacki B, Seitz HU, Stahl D, Rauhut T. Phytotoxicity and innate immune responses induced by Nep1-like proteins. Plant Cell. 2006;18(12):3721–44. doi: 10.1105/tpc.106.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bae H, Kim MS, Sicher RC, Bae H-J, Bailey BA. Necrosis-and ethylene-inducing peptide from Fusarium oxysporum induces a complex cascade of transcripts associated with signal transduction and cell death in Arabidopsis. Plant Physiol. 2006;141(3):1056–67. doi: 10.1104/pp.106.076869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vaillancourt LJ, Hanau RM. A method for genetic analysis of Glomerella graminicola (Colletotrichum graminicola) from maize. Phytopathology. 1991;81(5):530–4. doi: 10.1094/Phyto-81-530. [DOI] [Google Scholar]
- 120.Rech GE, Sanz-Martín JM, Anisimova M, Sukno SA, Thon MR. Natural selection on coding and noncoding DNA sequences is associated with virulence genes in a plant pathogenic fungus. Genome Biol Evol. 2014;6(9):2368–79. doi: 10.1093/gbe/evu192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xue M, Yang J, Li Z, Hu S, Yao N, Dean RA, Zhao W, Shen M, Zhang H, Li C. Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet. 2012;8(8):e1002869. doi: 10.1371/journal.pgen.1002869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yoshida K, Saunders DG, Mitsuoka C, Natsume S, Kosugi S, Saitoh H, Inoue Y, Chuma I, Tosa Y, Cano LM. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genomics. 2016;17(1):1. doi: 10.1186/s12864-016-2690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Boora KS, Frederiksen R, Magill C. DNA-based markers for a recessive gene conferring anthracnose resistance in sorghum. Crop Sci. 1998;38(6):1708–9. doi: 10.2135/cropsci1998.0011183X003800060048x. [DOI] [Google Scholar]
- 124.Rosewich U, Pettway R, McDonald B, Duncan R, Frederiksen R. Genetic structure and temporal dynamics of a Colletotrichum graminicola population in a sorghum disease nursery. Phytopathology. 1998;88(10):1087–93. doi: 10.1094/PHYTO.1998.88.10.1087. [DOI] [PubMed] [Google Scholar]
- 125.Valerio H, Resende M, Weikert-Oliveira R, Casela C. Virulence and molecular diversity in Colletotrichum graminicola from Brazil. Mycopathologia. 2005;159(3):449–59. doi: 10.1007/s11046-005-0373-y. [DOI] [PubMed] [Google Scholar]
- 126.Chala A, Tronsmo A, Brurberg M. Genetic differentiation and gene flow in Colletotrichum sublineolum in Ethiopia, the centre of origin and diversity of sorghum, as revealed by AFLP analysis. Plant Pathol. 2011;60(3):474–82. doi: 10.1111/j.1365-3059.2010.02389.x. [DOI] [Google Scholar]
- 127.Ali MEK, Warren HL. Physiological races of Colletotrichum graminicola on Sorghum. Plant Dis. 1987;71(5):402–4. doi: 10.1094/PD-71-0402. [DOI] [Google Scholar]
- 128.da Costa R, Cota L, da Silva D, Parreira D, Casela C, Landau E, Figueiredo J. Races of Colletotrichum graminicola pathogenic to maize in Brazil. Crop Prot. 2014;56:44–9. doi: 10.1016/j.cropro.2013.10.005. [DOI] [Google Scholar]
- 129.Nicholson R, Warren H. The issue of races of Colletotrichum graminicola pathogenic to corn. Plant Dis. 1981;65:143–45.
- 130.White D, Yanney J, Anderson B. Variation in pathogenicity, virulence, and aggressiveness of Colletotrichum graminicola on corn. Phytopathology. 1987;77(7):999–1001. doi: 10.1094/Phyto-77-999. [DOI] [Google Scholar]
- 131.Tuite J. Plant pathological methods. Fungi and bacteria. Minneapolis: Burgess Publishing Co.; 1969. [Google Scholar]
- 132.Li W, Rehmeyer CJ, Staben C, Farman ML. TruMatch—a BLAST post-processor that identifies bona fide sequence matches to genome assemblies. Bioinformatics. 2005;21(9):2097–8. doi: 10.1093/bioinformatics/bti257. [DOI] [PubMed] [Google Scholar]
- 133.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6(1):31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Soderlund C, Nelson W, Shoemaker A, Paterson A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006;16(9):1159–68. doi: 10.1101/gr.5396706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schardl CL, Young CA, Hesse U, Amyotte SG, Andreeva K, Calie PJ, Fleetwood DJ, Haws DC, Moore N, Oeser B. Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013;9(2):e1003323. doi: 10.1371/journal.pgen.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jothi R, Zotenko E, Tasneem A, Przytycka TM. COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics. 2006;22(7):779–88. doi: 10.1093/bioinformatics/btl009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24(3):319–24. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- 138.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Punta M, Coggill P, Eberhardt R, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saier MH, Tran CV, Barabote RD. TCDB: the transporter classification database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34(suppl 1):D181–6. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(suppl 1):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(W1):W445–51. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier C, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(suppl 2):W585–7. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38(suppl 1):D227–33. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36(suppl 2):W465–9. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The assemblies and annotations generated for the current study are available in the Genbank repository, as BioProjects SAMN06043298 and PRJNA356071. Biological materials from the current study are available from the corresponding author on reasonable request.