

Abstract
The diversity of small non-coding RNAs (sRNA) is rapidly expanding and their roles in biological processes, including gene regulation, are emerging. Most interestingly, sRNAs are also found outside of cells and are stably present in all biological fluids. As such, extracellular sRNAs represent a novel class of disease biomarkers and are likely involved in cell signaling and intercellular communication networks. To assess their potential as biomarkers, sRNAs can be quantified in plasma, urine, and other fluids. Nevertheless, to fully understand the impact of extracellular sRNAs as endocrine signals, it is important to determine which carriers are transporting and protecting them in biological fluids (e.g., plasma), which cells and tissues contribute to extracellular sRNA pools, and cells and tissues capable of accepting and utilizing extracellular sRNA. To accomplish these goals, it is critical to isolate highly pure populations of extracellular carriers for sRNA profiling and quantification. We have previously demonstrated that lipoproteins, particularly high-density lipoproteins (HDL), transport functional microRNAs (miRNA) between cells and HDL-miRNAs are significantly altered in disease. Here, we detail a new protocol that utilizes tandem HDL isolation with density-gradient ultracentrifugation (DGUC) and fast-protein-liquid chromatography (FPLC) to obtain highly pure HDL for downstream profiling and quantification of all sRNAs, including miRNAs, using both high-throughput sequencing and real-time PCR approaches. This protocol will be a valuable resource for the investigation of sRNAs on HDL.
Keywords: Biochemistry, Issue 117, HDL, small non-coding RNAs, microRNAs, high-throughput sequencing, density-gradient ultracentrifugation, fast-protein liquid chromatography.
Introduction
Extracellular non-coding small RNAs (sRNAs) represent a new class of disease biomarkers and potential therapeutic targets and likely facilitate cell-to-cell communication1. The most widely studied type of sRNA are microRNAs (miRNA) which are approximately 22 nts in length and are processed from longer precursor forms and primary transcripts2. miRNAs have been demonstrated to post-transcriptionally regulate gene expression through suppression of protein translation and induction of mRNA degradation2. Nevertheless, miRNAs are just one of many types of sRNAs; as sRNAs can be cleaved from parent tRNAs (tRNA-derived sRNAs, tDR), small nuclear RNAs (sRNA-derived sRNAs, sndRNA), small nucleolar RNAs (snoRNA-derived sRNAs, snRNA), ribosomal RNAs (rRNA-derived sRNAs, rDR), Y RNAs (yDR), and other miscellaneous RNAs1. A few examples of these novel sRNAs have been reported to function similar to miRNAs; however, the biological functions of many of these sRNAs remains to be determined, although roles in gene regulation are likely3-6. Most interestingly, miRNAs and other sRNAs are stably present in extracellular fluids, including saliva, plasma, urine, and bile. Extracellular sRNAs are likely protected from RNases through their association with extracellular vesicles (EV), lipoproteins, and/or extracellular ribonucleoprotein complexes.
Previously, we reported that lipoproteins, namely high-density lipoproteins (HDL), transport miRNAs in plasma7. In this study, HDL were isolated using a sequential method of density-gradient ultracentrifugation (DGUC), fast-protein liquid chromatography (size-exclusion chromatography gel filtration, FPLC), and affinity chromatography (anti-apolipoprotein A-I (apoA-I) immunoprecipitation)7. Using both real-time PCR-based low-density arrays and individual miRNA assays, miRNA levels were quantified on HDL isolated from healthy and hypercholesterolemic subjects7. Using this approach, we were able to profile miRNAs and quantify specific miRNAs in highly pure HDL preparations. Since 2011, we have determined that although affinity chromatography enhances HDL purity, antibody saturation greatly limits yield, and can be cost-prohibitive. Currently, our protocol recommends a two-step sequential tandem method of DGUC followed by FPLC, which also produces high quality HDL samples for down-stream RNA isolation and sRNA quantification. Due to recent advances in high-throughput sequencing of sRNAs (sRNAseq), e.g., miRNAs, and the increased awareness of other non-miRNA sRNA classes, sRNAseq is the current state-of-the-art in miRNA and sRNA profiling. As such, our protocol recommends quantifying miRNAs and other sRNAs on HDL samples using sRNAseq. Nonetheless, total RNA isolated from HDL can also be used to quantify individual miRNAs and other sRNAs or validate sRNAseq results using real-time PCR approaches. Here we describe in detail a protocol for the collection, purification, quantification, data analysis, and validation of highly pure HDL-sRNAs.
The overall goal of this paper is to demonstrate the feasibility and process of sRNA quantification in highly pure HDL isolated from human plasma.
Protocol
1. HDL Purification (~ 5.5 days)
- Density-Gradient Ultracentrifugation (DGUC, ~ 5 days)
- Add 90 μL of 100x anti-oxidants to 9 mL of plasma isolated from fresh venous blood.
- Adjust plasma density with KBr from 1.006 g/mL to 1.025 g/mL by adding 0.251 g KBr to 9 mL of plasma from Step 1.1.1 (0.0278 g/mL KBr plasma). Rock the plasma until all the salt is dissolved at room temperature and transfer to ultracentrifuge tubes and ensure all bubbles rise to the top.
- Bend the tip of an 18-G needle to 90° 1 cm from the tip, bevel side facing up. Using a syringe and the 18-gauge needle, carefully place 3 mL of overlay solution #1 over the plasma (Table 1). NOTE: The bent needle ensures all the VLDL/IDL, LDL, and HDL are removed without mixing with the overlay.
- Remove the majority of the bubbles at the top, leaving 2 - 3 mm space from meniscus to the top of the tube and carefully place the tubes in SW-40Ti buckets.
- Weigh each bucket (with the caps) on the balance, and exactly balance with the opposite bucket (bucket + cap): #1 to #4, #2 to #5, and #3 to #6. NOTE: These buckets must be balanced and matched at all times.
- Place rotor in ultracentrifuge (List of Materials). Centrifuge at 274,400 x g for 24 h at 4 °C with a #4 intermediate brake.
- Carefully remove buckets from rotor and the tubes from buckets.
- Carefully remove 2 mL from the top layer of the overlay using a syringe and bent needle. This will be the VLDL/IDL fraction which can be stored at 4°C or -80°C.
- After removing the VLDL/IDL fraction, collect the remaining 7 mL of sample (plasma) and place it into a 15 mL conical tube. Bring the volume of the sample up to 9 mL with overlay solution #2 (Table 1).
- Adjust the sample density from 1.025 g/mL to 1.080 g/mL with KBr using approximately 0.746 g KBr in the 9 mL sample or 0.0828 g/mL KBr of sample. Gently rock the sample until all the KBr is dissolved (approximately 20 min) and transfer to ultracentrifuge tube while ensuring bubbles rise to the top.
- With syringe and needle, carefully place 3 mL of overlay solution #3 over the sample (Table 1). Leave 2 - 3 mm space from meniscus to the top of the tube.
- Remove the majority of any remaining bubbles at the top of the tube and carefully place the filled ultracentrifuge tubes in SW-40Ti buckets.
- Weigh each bucket (with the caps) on the balance, and exactly balance with the opposite bucket + cap, as in 1.1.5.
- Place rotor in ultracentrifuge (see List of Materials). Centrifuge at 274,400 x g for 24 h at 4 °C with a #4 intermediate brake.
- Carefully remove buckets from rotor and the tubes from buckets.
- Carefully remove 2 mL from the top layer of the overlay using a syringe and bent needle. This will be the LDL fraction which can be stored at 4°C or -80°C.
- After removing the LDL fraction, collect the remaining approximate 7 mL of sample (plasma) and place it into a new 15 mL conical tube. Bring the volume of the sample (plasma) up to 9 mL with 1.080 g/mL solution #4 (Table 1).
- Adjust the sample density from 1.080 g/mL to 1.30 g/mL with KBr using 3.33 g KBr in the 9 mL sample (plasma) or 0.37 g KBr for each mL of sample. Rock or slightly agitate the sample until all the KBr (salt) is dissolved and transfer to ultracentrifuge tube.
- With syringe and needle, carefully place 3 mL of overlay solution #5 over the sample (Table 1).
- Remove the majority of any remaining bubbles at the top of the tube, leaving 2- 3 mm space from meniscus to the top of the tube and carefully place the filled ultracentrifuge tubes in SW-40Ti buckets.
- Weigh each bucket (with the caps) on the balance, and exactly balance with the opposite bucket + cap, as in 1.1.5.
- Place rotor in ultracentrifuge (see List of Materials). Centrifuge at 274,400 x g for 48 h at 4 °C with a #4 brake.
- Carefully remove buckets from rotor and the tubes from buckets.
- Carefully remove approximately 2 mL from the top layer of the overlay using a syringe and bent needle. This is the HDL fraction, which can be stored at 4 °C or -80 °C. NOTE: Following DGUC HDL may be dialyzed to remove the high-salt solutions.
- To dialyze, place the collected HDL fraction in a sealed dialysis sleeve (10,000 m.w. cut-off) and dialyze overnight in 1 L 1x PBS. Change dialysis buffer (1x PBS) 3 times over 24 h. Gentle perturbation of the PBS with a magnetic stir-bar will improve dialysis.
- Determine the protein concentration of the DGUC-HDL using a BCA method8.
- Fast-Protein Liquid Chromatography (FPLC) or Size-Exclusion Chromatography (~ 6 h)
- Filter 1.2 mg of DGUC-HDL (total protein) in 500 µL through a 0.22 µm micro-centrifugal filter (see List of Materials), immediately prior to injection. NOTE: An additional filtering of the DGUC-HDL sample through a 0.45 µm micro-centrifugal filter prior to the 0.22 µm filter may be required for some samples.
- Collect the filtered DGUC-HDL sample, load the FPLC (fill) injection syringe, and ensure there are no bubbles in the syringe before injecting into the FPLC.
- Set the FPLC flow rate to 0.3 mL/min with a pressure limit at 2.6 MPa. Equilibrate columns with 0.2 column volumes (approximately 15 mL) of buffer, prior to sample injection. Inject the sample with 3 mL of buffer into the FPLC (injection loop) instrument and start the run. Collect 1.5 mL fractions for a total of 72 fractions.
2. High-throughput Small RNA Sequencing (sRNAseq, ~ 9 days)
- RNA Isolation (~ 1 day)
- Identify the FPLC fractions corresponding to DGUC-HDL by determining total cholesterol levels using a colorimetric kit according to the manufacturer's instructions. Using this FPLC set-up, expect to find 6 - 7 FPLC fractions containing DGUC-HDL.
- Collect all the volume (approximately 8 -1 0 mL pool of 1.45 mL fraction volumes) from DGUC-HDL FPLC fractions and concentrate using 10 kDa m.w. cut-off centrifugal filter units at 4,000 x g for 1 h at 4 °C or until HDL concentrate is approximately 100 µL.
- Collect the HDL concentrate and quantify HDL total protein concentration using the BCA method8.
- Determine the highest concentration of HDL total protein, up to 1 mg, that can be aliquoted from each sample in the set. For example, isolate RNA from the same amount of HDL total protein for each sample.
- Perform RNA Isolation using a modified protocol(see List of Materials).
- Add 10x volume of phenol lysis reagent (see List of Materials) sample and vortex for 1 min.
- Incubate at room temperature for 5 min.
- Add chloroform (20% of phenol lysis reagent volume used in step 2.1.5.1) and shake vigorously for 15 s.
- Incubate at room temperature for 2 - 3 min.
- Centrifuge for 15 min at 12,000 x g at 4 °C in a refrigerated centrifuge.
- Transfer the upper aqueous phase to a new tube, avoiding the interphase.
- Add 1.5x volume of 100% ethanol to the aqueous phase and vortex for 1 min.
- Store samples for at least 1 h or overnight at -80 °C.
- Continue with the RNA isolation protocol using the provided mini columns.
- Elute with 30 µL of RNase-free H2O. First add 15 µL of H2O directly to the frit, spin at low speed (2,000 x g) for 2 min, and then spin at high speed (max) for 1 min. Repeat this step with an additional 15 µL of H2O.
- Immediately proceed to sRNA library generation or store at -80 °C.
- Library Generation (~ 6 h)
- Prepare sRNA cDNA libraries using the sRNA library generation kit protocol, as per manufacturer's instructions with one modification to the PCR amplification as detailed below.
- Prepare the PCR mastermix on ice containing 0.5 µL DNase/RNase-free H2O, 15 µL PCR Mix (PML) and 1 µL RNA PCR Primer (RP1) per sample (include an additional 10% for pipetting error).
- Add 16.5 µL of the PCR mix to each (cDNA) sample.
- Assign an index/barcode number to each sample for multiplexing and add 1 µL PCR Primer Index (to the corresponding number) to the sample.
- Perform a quick spin using the microfuge. NOTE: Total volume should now be 30 µL per sample.
- Perform the cycling conditions for PCR amplification as detailed in Tables 2 and 3.
- Size Select sRNA Library to Remove Adapter:Adapters (~ 1.5 h)
- Perform library size-selection using automatic DNA size selector as per manufacturer's instructions with the following size parameters: Target: 156 bp, Start: 135 bp, End: 177 bp, Range Flag: Broad.
- DNA Clean Up (~0.5 h)
- Clean up size-selected sRNA cDNA according to the manufacturer's instructions.
- Elute clean and concentrated sRNA cDNA with 2 x 7 µL DNase/RNase-free H2O.
- Quantify and Assess sRNA cDNA Library Yield and Quality (~ 1 h)
- Assess sRNA cDNA library quality and size using a high-sensitivity DNA chip on a bioanalyzer (List of Materials) as per manufacturer's instructions.
- Quantify the individual sRNA cDNA library concentrations using a high-sensitivity DNA assay (List of Materials), as per manufacturer's instructions. NOTE: It is recommended to have all samples with different indices be run on the same lane of the flow cell if possible. If more than one lane is required, mix case and control samples appropriately.
- High-throughput sRNA Sequencing (~ 7 days)
- Pool individual indexed sRNA libraries based on equimolar concentrations.
- Screen pooled sRNA libraries for quality using high-sensitivity DNA chip on the bioanalyzer and determine library DNA concentration as in 2.5.1 - 2.5.2.
- Perform quantitative PCR (qPCR) of adapter content to ensure appropriate sequencing depth using the sequencing library qPCR quantification kit as per manufacturer's instructions (List of Materials).
- Perform sequencing using a single read, 50 bp protocol to reach a depth of approximately 20 - 25 M reads/sample, as per manufacturer's instructions.
3. Data Analysis (~ 1 day)
Use bcl2fastq2, as per user guide instructions, to preprocess raw fastq files of demultiplexed samples (List of Materials).
Use Cutadapt to trim adapters9 and remove reads < 16 nucleotides (nts) in length, as per user guide instructions (List of Materials).
Remove contamination sequences present in the sRNA libraries, including stop solution sequence (STP: CCACGTTCCCGTGG) and adapter carry-over (CTACAGTCCGACGATC) using removeSequenceInFastq function in NGSPERL framework, as per user guide instructions (List of Materials).
Perform quality control metrics by FastQC using Center for Quantitative Sciences (CQS) tools, as per user guide instructions (List of Materials).
Generate non-redundant list of "identical" reads (collapse redundant reads) and record copy numbers using CQSTools, as per user guide instructions (List of Materials).
Align reads to human genome using Bowtie1.1.2 allowing 1 mismatch (options: -a -m 100 --best --strata -v 1), as per user guide instructions (List of Materials).
Perform read alignment, counting, and result summarization tasks using NGSPERL, as per user guide instructions.
Convert exported count table to a spreadsheet file.
Normalize reads of a specific class (e.g., miRNAs) to total number of reads for that class (e.g., total number of miRNAs reads) and report signals as Reads Per Mapped miRNA (RPMM). (e.g., RPMM =(miR-X/Total # of miRNA reads)*1,000,000).
Input normalized count table as generic single color technology into software for low-level and high-level differential analyses as per user guide instructions (List of Materials). NOTE: Currently, there are no well-characterized housekeeping genes/sRNAs for HDL-sRNA. A spike-in control may be used, but can be problematic. Normalizing to HDL total protein input or volume is advised. Most importantly, it is recommended to validate sequencing results by one of the various strategies to quantify miRNAs and sRNAs using real-time PCR or quantitative PCR.
Representative Results
This protocol is a series of established methods linked together to allow for the quantification of sRNAs on highly pure HDL by high-throughput sequencing or real-time PCR (Figure 1). To demonstrate the feasibility and impact of this protocol, HDL was purified from human plasma by the tandem DGUC and FPLC method. Collected FPLC fractions corresponding to HDL (by cholesterol distribution) were concentrated and total RNA was isolated from 1 mg of HDL (total protein). sRNA libraries were generated with HDL collected from n = 4 healthy human subjects. All human blood/plasma samples were collected under active Institutional Review Board protocol (# 070416), and human subjects were enrolled with informed consent as approved by Vanderbilt University School of Medicine. Prepared sRNA libraries were sequenced at Vanderbilt Technologies for Advanced Genomics (VANTAGE) DNA sequencing center and data analyses were performed using in-house pipelines. Data visualizations were created by graphing software.
The critical need to perform a second method (FPLC) for HDL isolation after DGUC is due to possible co-purification of EVs. EVs are a generalized class of membrane-derived vesicles that originate from multivesicular bodies (exosomes), plasma membrane budding (microvesicles), and other processes, including apoptotic bodies10. EVs, namely exosomes, have been reported to have a similar density as small HDL (Figure 2A), and thus, could be present in the HDL density fraction (1.063 - 1.21 g/mL) after DGUC11,12. To remove EVs and other lipoproteins (e.g., LDL), from DGUC-HDL, FPLC was used to separate HDL from other vesicles and particles by size; HDL (7 - 12 nm in diameter) and EVs (40 - 220 nm in diameter) are easily fractionated due to their size differences (Figure 2A,B). HDL fractions were readily apparent due to the distribution of cholesterol, and DGUC-HDL FPLC fractions were pooled and concentrated using 10 kDa cut-off centrifugation filters. Concentrated HDL were collected and used for downstream RNA analysis (Figure 2B). To illustrate this point, plasma pre-DGUC was fractionated using FPLC and the distribution of phospholipids (phosphatidylcholine, PC); total cholesterol (TC), and protein levels were quantified and plotted across fractions. PC, TC, and proteins levels were quantified using three colorimetric kits. All three parameters clearly defined the HDL fractions (19 - 24) and VLDL/LDL fractions (14 - 18). In FPLC-fractionated plasma, protein and lipid signals were minimally detected or not detected at all in fractions corresponding to vesicle sizes greater than VLDL (left of orange box) (Figure 3A). To demonstrate DGUC separation of HDL, DGUC-HDL was also fractionated by FPLC and PC, TC, and proteins levels were quantified (Figure 3B). Plotting these values illustrates the co-fractionation of a small amount of LDL using DGUC and reinforces the need to use the tandem DGUC-FPLC approach to isolate highly pure HDL. Although EVs are predicted to co-fractionate with HDL during DGUC, there was little to none found in the lipid and protein in fractions corresponding to vesicle sizes greater than LDL (Figure 3B). The EV lipid and protein signature in the predicted size range is also minimally detectable or absent from DGUC-VLDL and DGUC-LDL when fractionated by FPLC (data not shown). For RNA analysis, total RNA was isolated from 1 mg of tandem purified HDL for sRNA library generation. Briefly, adapters were ligated to the 3' and then 5' terminal ends of sRNAs, including miRNAs and tDRs (Figure 4A). Ligated products were reverse transcribed (RT) and PCR amplified using PCR primers harboring specific indexes designed for multiplexing samples during downstream sequencing reactions (Figure 4A). Each adapter is 61 nts in length; therefore, a ligated miRNA (approximately 22 nts in length) would produce a product of 144 nts in length. Many non-miRNA sRNAs are slightly longer than miRNAs (e.g., 30 - 35 nts in length). As such, our targeted length of product was 156 bps in length. Amplified products were size-selected using an automatic DNA size selector and all cDNA products 135 - 177 bps in length were collected (Figure 4A). The qualities of size-selected libraries were assessed by high-sensitivity DNA chips using the bioanalyzer (Figure 4B). sRNA cDNA libraries of this size appeared slightly larger in size due to possible "bubbling" effects during the analysis. For multiplexed sequencing, equimolar concentrations of cDNA libraries were pooled, cleaned, concentrated, and reanalyzed using the bioanalyzer (Figure 4B). The multiplexed sample was then sequenced using a single read 50 bp protocol. In-house data analysis pipelines were used to demultiplex samples based on indexes, trim adapters, run quality control metrics, align to the human genome and count annotated sRNAs. The distribution of normalized reads (Reads Per Million, RPM), > 16 nts in length, illustrate the enrichment of sRNAs 20 - 22 nts in length and 34 - 35 nts in length on HDL (Figure 4C).
Alignment and counting analysis of the four HDL-sRNA libraries found that 38% of reads aligning to annotated regions of the human genome were miRNAs (Figure 5A). Equally abundant (37%) were sRNAs-derived from tRNAs (tDRs). sRNAs-derived from miscellaneous RNAs (e.g., Y RNAs), rRNAs, smRNAs, snoRNAs, and long non-coding RNAs (lincRNAs) were also present on HDL (Figure 5A). After normalization to the total number of reads for each class, the top 50 most abundant miRNAs (Reads Per Mapped miRNA, RPMM, Figure 5B), tDRs (Reads Per Mapped tDRs, RPMT, Figure 5C), and other non-miRNA non-tDR sRNAs (Reads Per Mapped sRNA, RPMS, Figure 5D) were organized by heatmaps. These data illustrate the similarities (e.g., HDL-miRNAs) and discrepancies (e.g., tDRs) of the most abundant HDL-sRNAs across healthy subjects (Figure 5B-D). These results demonstrate the power of this strategy and protocol to provide remarkable insights into the transport of sRNAs by HDL. Nevertheless, sRNA sequencing should not be relied on for absolute quantitation. sRNAs of interest should be validated by real-time PCR. Likewise, many investigators may only require the quantification of individual miRNAs on HDL. As such, total HDL RNA was isolated from each subject for relative quantification of HDL-miRNAs using real-time PCR. Five of the more abundant miRNAs detected on HDL by sRNAseq were used for real-time PCR assays to determine their levels on each HDL sample (Figure 5E). The relative quantitative values were calculated using an arbitrary housekeeping Ct of 32 (RQV= 2-ΔCtArb) and normalized to 1,000 µg of HDL protein input into the RNA isolation13. Results from the real-time PCR study demonstrate that this protocol is also valuable in detecting HDL-miRNAs and quantifying differences across individuals. Collectively, the presented schematics define the critical details of the methods, and the results from both high-throughput sequencing and real-time PCR approaches represent the deliverables of this protocol.
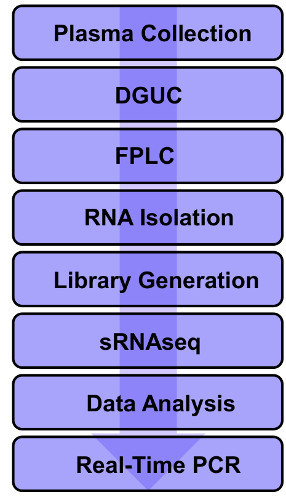 Figure 1. Representative Flow-chart of the Protocol. Pure HDL is isolated from density gradient ultracentrifugation and fast protein liquid chromatography. Total HDL RNA can then be utilized for small RNA (sRNA) sequencing. Please click here to view a larger version of this figure.
Figure 1. Representative Flow-chart of the Protocol. Pure HDL is isolated from density gradient ultracentrifugation and fast protein liquid chromatography. Total HDL RNA can then be utilized for small RNA (sRNA) sequencing. Please click here to view a larger version of this figure.
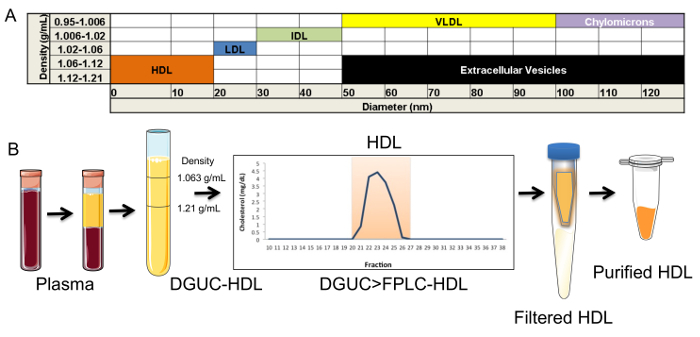 Figure 2. Isolation of HDL. (A) Chart of the density and diameter of lipoproteins and extracellular vesicles (EV). (B) Schematic of sequential tandem HDL isolation, including plasma separation, density-gradient ultracentrifugation (DGUC), fast-protein liquid chromatography (FPLC), filtration, and concentration. Please click here to view a larger version of this figure.
Figure 2. Isolation of HDL. (A) Chart of the density and diameter of lipoproteins and extracellular vesicles (EV). (B) Schematic of sequential tandem HDL isolation, including plasma separation, density-gradient ultracentrifugation (DGUC), fast-protein liquid chromatography (FPLC), filtration, and concentration. Please click here to view a larger version of this figure.
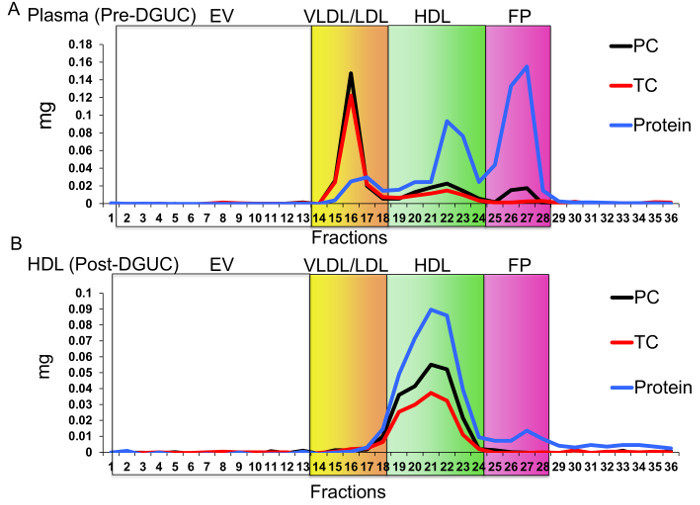 Figure 3. Fast-protein Liquid Chromatography (FPLC) Separation of Lipoproteins. EV, extracellular vesicles; VLDL, very low-density lipoproteins; LDL, low-density lipoproteins; HDL, high-density lipoproteins; FP, free protein; PC, phosphatidylcholine; and TC, total cholesterol. Red line, TC; Blue line, protein, and Black line, PC. Separation of lipoproteins from (A) human plasma prior to density-gradient ultracentrifugation (DGUC) and (B) HDL fraction after DGUC. Please click here to view a larger version of this figure.
Figure 3. Fast-protein Liquid Chromatography (FPLC) Separation of Lipoproteins. EV, extracellular vesicles; VLDL, very low-density lipoproteins; LDL, low-density lipoproteins; HDL, high-density lipoproteins; FP, free protein; PC, phosphatidylcholine; and TC, total cholesterol. Red line, TC; Blue line, protein, and Black line, PC. Separation of lipoproteins from (A) human plasma prior to density-gradient ultracentrifugation (DGUC) and (B) HDL fraction after DGUC. Please click here to view a larger version of this figure.
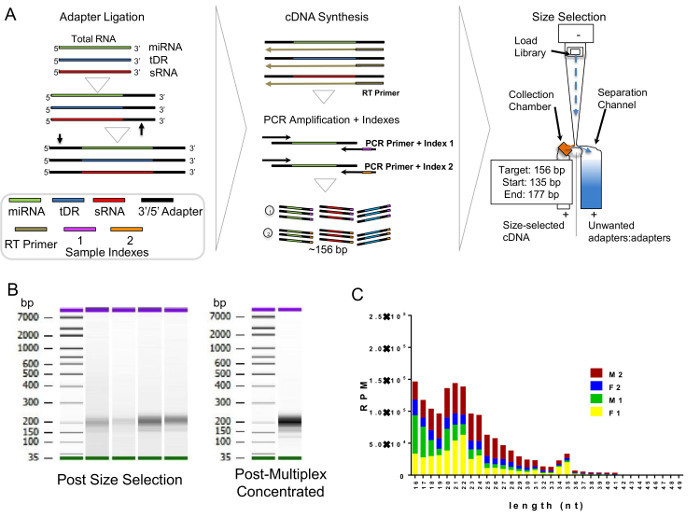 Figure 4. Generation of Small RNA Libraries. (A) Schematic of small RNA (sRNA) library construction and size-selection. miRNA, microRNA; tDR, tRNA-derived sRNAs; sRNA, non-miRNA non-tDR sRNAs; RT, reverse transcription; bp, base pair; and cDNA, cloned DNA. (B) Analysis of HDL-sRNA libraries before and after multiplexing using high-sensitivity DNA chips and bioanalyzer. (C) Length distribution of HDL-sRNA reads after sequencing. RPM, Reads Per Million; M, male subjects; F, female subjects; and nt, nucleotide. N=4. Please click here to view a larger version of this figure.
Figure 4. Generation of Small RNA Libraries. (A) Schematic of small RNA (sRNA) library construction and size-selection. miRNA, microRNA; tDR, tRNA-derived sRNAs; sRNA, non-miRNA non-tDR sRNAs; RT, reverse transcription; bp, base pair; and cDNA, cloned DNA. (B) Analysis of HDL-sRNA libraries before and after multiplexing using high-sensitivity DNA chips and bioanalyzer. (C) Length distribution of HDL-sRNA reads after sequencing. RPM, Reads Per Million; M, male subjects; F, female subjects; and nt, nucleotide. N=4. Please click here to view a larger version of this figure.
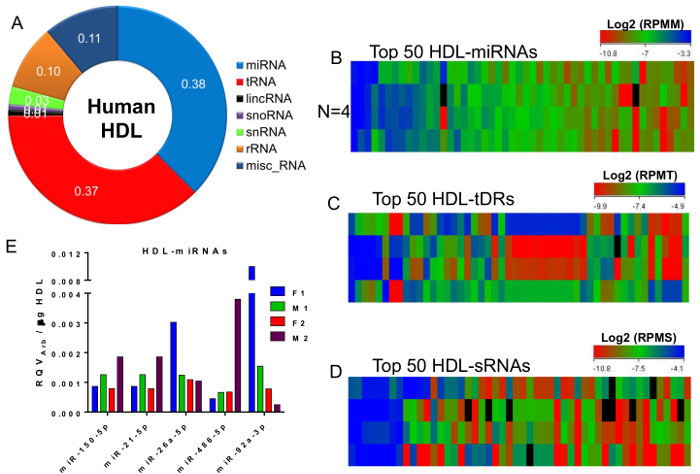 Figure 5. Diversity of HDL-sRNAs. (A) Distribution of HDL-sRNAs across sRNA classes. Fraction of reads aligned to annotated sRNAs in the human genome. (B-D) Heatmaps N = 4 HDL-sRNA samples. (B) Top 50 most abundant HDL-microRNAs (miRNA). Log2 Reads Per Mapped miRNA, RPMM. (C) Top 50 most abundant HDL-tRNA-derived sRNAs (tDRs). Log2 Reads Per Mapped tDR, RPMT. (D) Top 50 most abundant HDL-other non-miRNA and non-tDR sRNAs (sRNA). Log2 Reads Per Mapped sRNA, RPMS. (E) Real-time PCR quantification of HDL-miRNAs. M, male subjects; F, female subjects. Relative Quantitative Value determined by arbitrary housekeeping Ct = 32, normalized to 1,000 µg of HDL protein input13. N = 4. Please click here to view a larger version of this figure.
Figure 5. Diversity of HDL-sRNAs. (A) Distribution of HDL-sRNAs across sRNA classes. Fraction of reads aligned to annotated sRNAs in the human genome. (B-D) Heatmaps N = 4 HDL-sRNA samples. (B) Top 50 most abundant HDL-microRNAs (miRNA). Log2 Reads Per Mapped miRNA, RPMM. (C) Top 50 most abundant HDL-tRNA-derived sRNAs (tDRs). Log2 Reads Per Mapped tDR, RPMT. (D) Top 50 most abundant HDL-other non-miRNA and non-tDR sRNAs (sRNA). Log2 Reads Per Mapped sRNA, RPMS. (E) Real-time PCR quantification of HDL-miRNAs. M, male subjects; F, female subjects. Relative Quantitative Value determined by arbitrary housekeeping Ct = 32, normalized to 1,000 µg of HDL protein input13. N = 4. Please click here to view a larger version of this figure.
| 1. Anti-oxidants for Lipoprotein separation | Make stock solution at 100x |
| EDTA | Ethylenediaminetetraacetic acid (0.5 M stock) |
| AZT | 3-Amino-1,2,4-triazole: 42 mg/mL H2O |
| BHT | Butylated Hydroxytoluene: 25 mg/100 µL EtOH |
| TROLOX | (+)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid: 41.7 mg/500 µL EtOH |
| 2. Overlay Solutions | |
| Solution #1 | 1.019 g/mL in H2O |
| Solution #2 | 1.025 g/mL KBr/H2O |
| Solution #3 | 1.063 g/mL KBr/H2O |
| Solution #4 | 1.080 g/mL KBr/H2O |
| Solution #5 | 1.255 g/mL KBr/H2O |
| 3. FPLC Buffer | |
| 1 L Recipe | 50 mL 3M NaCl; 20 mL 0.5 Tris-HCl, pH 7.6; 2 mL of 10% Sodium Azide; 930 mL H2O |
Table 1. Special Reagents.
| Stage | Temp (°C) | Time | Cycles |
| A | 98 | 30 s | 1 |
| B | 98 | 10 s | 25 |
| 60 | 30 s | ||
| 72 | 15 s | ||
| C | 72 | 10 min | 1 |
Table 2. Library Generation Amplification Steps .
| Primer | Sequence |
| Primer 1.1 | AATGATACGGCGACCACCGAGAT |
| Primer 2.1 | CAAGCAGAAGACGGCATACGA |
Table 3. PCR Primers.
| Stage | Temp (°C) | Time | Cycles |
| A | 95 | 10 min | 1 |
| B | 95 | 10 s | 40 |
| 60 | 30 s |
Table 4. Library Quantification Amplification Steps.
Discussion
This protocol is designed to quantify miRNAs and other sRNAs by high-throughput sequencing or real-time PCR on highly pure HDL. As with any approach, special considerations should be given to each step in the process of purifying HDL and RNA and then quantifying sRNAs. This protocol is designed for projects starting with ≥ 2 mL of plasma. Nevertheless, high quality RNA analyses can successfully be completed with HDL purified from as little as 80 µL of human or mouse plasma using affinity chromatography; however, larger starting plasma volumes produce better data using the methods presented here. Sample processing and extraction methods can drastically alter miRNA/sRNA quality and yield. The use of anticoagulants, while essential for plasma collection, likely affects downstream extraction. For example, heparin is not easily removed during RNA extraction14,15, and can inhibit downstream enzymes utilized in PCR16-18. In addition, there are reports that sodium citrate may also interfere with qPCR measurements where miRNAs from EDTA samples are shown to have improved expression profile compared to sodium citrate samples16,19,20. These studies demonstrate the need for careful consideration when choosing an anticoagulant. Furthermore, due to increased potential for cell lysis during coagulation, serum samples are not recommended as lysed cellular miRNAs may artificially associate with lipoproteins. Likewise, plasma is ideally isolated immediately from whole blood to avoid contamination from other peripheral blood cells, including lysed leukocytes. As cold temperature is widely known to affect platelet activation, whole blood samples are spun away from erythrocytes, leukocytes and platelets at 3,000 x g for 15 min at room temperature. Hemolysis is also ill advised as ruptured erythrocytes have been reported to also influence miRNA outcome21,22, as they contain miRNAs that may be shed during hemolysis23,24. Plasma can then be stored at 4 °C for 2 - 4 weeks or alternatively plasma may be frozen at -80 °C where miRNA content is stable and viable in both short term and long term storage (max 4 years)25. To our current knowledge, freezing plasma samples is not predicted to harm HDL-miRNAs and frozen plasma samples are suitable to analyze. As lipoproteins and other whole blood components can be affected by food intake, fasting blood collection is preferred.
As mentioned above, plasma HDL can be isolated through a number of standard methods, including DGUC, FPLC, and affinity chromatography against apoA-I. Isolation of plasma lipoproteins by DGUC using KBr, as described by Redgraves et al.26, separates VLDL (d = 0.94 - 1.006 g/mL), LDL (d = 1.006 - 1.063 g/mL) and HDL (d = 1.063-1.21 g/mL) and is an ideal method for large volumes of plasma (2 - 60 mL per sample). The limitation to using this approach alone is that exosomes and other miRNA containing EVs are reported to have a similar density to HDL and may be present in DGUC HDL (Figure 2A). Other disadvantages include possible structural damage to proteins from high salt exposure in buffers and strong ultracentrifugation forces as well as differential stabilities of HDL sub-classes27,28. Despite this, DGUC is commonly used as it results in a high yield of HDL protein, producing close to 1 mg of HDL protein per 1 mL of plasma29. The FPLC approach, which involves size-exclusion gel filtration, requires less time, depending on the flow rate (approximately 6 h), and has greater precision separating HDL from apoB-containing lipoproteins (by size) and their sub-classes, including EVs that are much larger than HDL but have similar densities. It should be noted that using the methods detailed in this protocol, plasma vesicles between approximately 220 - 75 nm in diameter do not elicit a detectable lipid or protein signature when separated by FPLC. Depending on the set-up, FPLC requires relatively small input, 0.3 - 1 mL, which is useful for rodent plasma and small frozen aliquots. Quick colorimetric cholesterol assays are also important following FPLC to confirm the distribution of HDL or other lipoprotein fractions. Fractionation dilutes samples and lipoproteins; however, samples can then be concentrated using standard centrifugal size-filters (e.g., 10 kDa m.w. cut-off filter). HDL isolation by apoA-I IP using micro-spin columns are the quickest of the three approaches, but are limited by low input (0.08 - 1 µL) volume and low yield (approximately 0.1 mg). While apoA-I IP can be laborious, this method is ideal for low volume cell culture supernatants. While each of these methods has strengths and weaknesses, these can be reduced if used in tandem (e.g., DGUC followed FPLC). Using two approaches in tandem, results in greater purity of HDL for miRNA and sRNA analysis.
Here, we detailed the steps required to isolate highly pure HDL using the tandem method of DGUC followed by FPLC and outlined the steps used to quantify HDL-miRNAs and sRNAs using either high-throughput sequencing or real-time PCR. Although this protocol was designed for HDL, it has great applicability in quantifying sRNAs on other lipoproteins, including LDL and VLDL, based on their densities and sizes.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by awards from the National Institutes of Health, National Heart, Lung and Blood Institute to K.C.V. HL128996, HL113039, and HL116263. This work was also supported by awards from the American Heart Association to K.C.V. CSA2066001, D.L.M POST26630003, and R.M.A. POST25710170.
References
- Vickers KC, Roteta LA, Hucheson-Dilks H, Han L, Guo Y. Mining diverse small RNA species in the deep transcriptome. Trends Biochem Sci. 2015;40:4–7. doi: 10.1016/j.tibs.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Haussecker D, et al. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–695. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, et al. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012;40:6787–6799. doi: 10.1093/nar/gks307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens-Uzunova ES, Olvedy M, Jenster G. Beyond microRNA--novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013;340:201–211. doi: 10.1016/j.canlet.2012.11.058. [DOI] [PubMed] [Google Scholar]
- Maute RL, et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Chen C, Khaleel SS, Huang H, Wu CH. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med. 2014;9:8. doi: 10.1186/1751-0473-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers KC, Remaley AT. Lipid-based carriers of microRNAs and intercellular communication. Curr Opin Lipidol. 2012;23:91–97. doi: 10.1097/MOL.0b013e328350a425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening DW, Xu R, Ji H, Tauro BJ, Simpson RJ. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol. 2015;1295:179–209. doi: 10.1007/978-1-4939-2550-6_15. [DOI] [PubMed] [Google Scholar]
- Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015;6:7164. doi: 10.1038/ncomms8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Jung R, Lubcke C, Wagener C, Neumaier M. Reversal of RT-PCR inhibition observed in heparinized clinical specimens. Biotechniques. 1997;23 doi: 10.2144/97231bm03. [DOI] [PubMed] [Google Scholar]
- Johnson ML, Navanukraw C, Grazul-Bilska AT, Reynolds LP, Redmer DA. Heparinase treatment of RNA before quantitative real-time RT-PCR. Biotechniques. 2003;35:1140–1142. doi: 10.2144/03356bm03. [DOI] [PubMed] [Google Scholar]
- Garcia ME, Blanco JL, Caballero J, Gargallo-Viola D. Anticoagulants interfere with PCR used to diagnose invasive aspergillosis. J Clin Microbiol. 2002;40:1567–1568. doi: 10.1128/JCM.40.4.1567-1568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota M, Tatsumi N, Nathalang O, Yamada T, Tsuda I. Effects of heparin on polymerase chain reaction for blood white cells. J Clin Lab Anal. 1999;13:133–140. doi: 10.1002/(SICI)1098-2825(1999)13:3<133::AID-JCLA8>3.0.CO;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, et al. Predictive value of quantitative PCR-based viral burden analysis for eight human herpesviruses in pediatric solid organ transplant patients. J Mol Diagn. 2000;2:191–201. doi: 10.1016/S1525-1578(10)60637-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShine RL, Sibinga S, Brozovic B. Differences between the effects of EDTA and citrate anticoagulants on platelet count and mean platelet volume. Clin Lab Haematol. 1990;12:277–285. doi: 10.1111/j.1365-2257.1990.tb00038.x. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer S, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner MB, et al. Haemolysis during sample preparation alters microRNA content of plasma. PLoS One. 2011;6:e24145. doi: 10.1371/journal.pone.0024145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan M, Atreya C. Differential profiling of human red blood cells during storage for 52 selected microRNAs. Transfusion. 2010;50:1581–1588. doi: 10.1111/j.1537-2995.2010.02585.x. [DOI] [PubMed] [Google Scholar]
- Chen SY, Wang Y, Telen MJ, Chi JT. The genomic analysis of erythrocyte microRNA expression in sickle cell diseases. PLoS One. 2008;3:e2360. doi: 10.1371/journal.pone.0002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzano F, et al. miRNA Stability in Frozen Plasma Samples. Molecules. 2015;20:19030–19040. doi: 10.3390/molecules201019030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redgrave TG, Roberts DC, West CE. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal Biochem. 1975;65:42–49. doi: 10.1016/0003-2697(75)90488-1. [DOI] [PubMed] [Google Scholar]
- Cheung MC, Wolf AC. Differential effect of ultracentrifugation on apolipoprotein A-I-containing lipoprotein subpopulations. J Lipid Res. 1988;29:15–25. [PubMed] [Google Scholar]
- Kunitake ST, Kane JP. Factors affecting the integrity of high density lipoproteins in the ultracentrifuge. J Lipid Res. 1982;23:936–940. [PubMed] [Google Scholar]
- Laurent LC, et al. Meeting report: discussions and preliminary findings on extracellular RNA measurement methods from laboratories in the NIH Extracellular RNA Communication Consortium. J Extracell Vesicles. 2015;4:26533. doi: 10.3402/jev.v4.26533. [DOI] [PMC free article] [PubMed] [Google Scholar]