

This Update is concerned with the mechanism of synthesis of heavy metal-binding thiol peptides, phytochelatins (PCs), by the enzyme PC synthase (EC 2.3.2.15). The bulk of the considerations in this review centers on what has been learned recently of the fundamental mechanics of PC synthesis, the domain organization and phylogenetic distribution of PC synthases, and PC synthase-like enzymes, and what this tells us about the chemistry underlying and the enzyme residues necessary for PC synthesis. It was decided to prepare a review of this type rather than aim at a more comprehensive treatment of heavy metal homeostasis and detoxification in plants for two reasons. The first is that there are already several contemporary reviews dealing with the more global aspects of plant heavy metal physiology. Excellent examples are Cobbett (2000), Clemens (2001), and Cobbett and Goldsbrough (2002). Readers who have not already read these are encouraged to do so. The second reason is that some of the most fascinating and unexpected developments for our understanding in this area of late derive from investigations of the catalytic mechanism and distribution of PC synthases, facets of this field of research that have yet to be reviewed in detail.
HEAVY METAL TOXICITY
Heavy metals, metals whose densities exceed 5 g/cm3 (Elmsley, 2001), come in two varieties—those that are and those that are not essential to organisms. Essential heavy metals, such as copper and zinc, are required as cofactors in redox reactions and ligand interactions, as well as for charge stabilization, charge shielding, and water ionization during biocatalysis (Elmsley, 2001; Voet and Voet, 2004). Nonessential heavy metals, such as arsenic, cadmium, lead, and mercury, are not required as cofactors, but instead interfere with those that are and/or simulate the effects of supraoptimal levels of essential heavy metals.
Despite a huge literature on the environmental, veterinary, and clinical phenomenology of heavy metal toxicity, its mechanistic basis is not well understood and has seldom been addressed directly. Notwithstanding this lack of precise details, in most cases, the consensus is that supraoptimal levels of essential heavy metals, and trace or higher levels of nonessential heavy metals, undergo aberrant capping reactions with the thiol groups of proteins and some thiol-containing coenzymes, displace endogenous metal cofactors (heavy or otherwise) from their cellular binding sites, and promote the formation of active oxygen species (Stadtmann, 1993). Cadmium ions, for example, are considered to displace Zn2+, Fe2+, and/or Cu2+ and/or other (nonheavy) metals, such as Ca2+ from proteins that require these metals as cofactors, thus abolishing their activity (Stohs and Bagchi, 1995). In the case of the release of free iron and/or copper, a transition metal and redox-active heavy metal, respectively, this is thought to elicit the generation of highly reactive hydroxyl (OH.) radicals (Halliwell and Gutteridge, 1984) by Fenton's reaction (Fe3+ + O2˙− → Fe2+ + O2; Fe2+ + H2O2 → Fe3+ + ·OH + OH·− and Cu2+ + O2·− → Cu+ + ·OH + OH−; Cu+ + H2O2 → Cu2+ + ·OH + OH−). What is not so clear, however, is whether the strict association between metal toxicity and the imposition of oxidative stress (Brennan and Schiestl, 1996), as exemplified by lipid peroxidation (Stohs and Bagchi, 1995), is an immediate or secondary consequence of metal action and necessarily capable of explaining the extreme toxicity of heavy metals in all, or even in most, cases.
LONG-CHAIN GLUTATHIONE DERIVATIVES
The inherent reactivity of heavy metals toward thiol groups is not only a major factor in their toxicity, but also a common thread in their homeostasis and detoxification. Indeed, this is the crux of the action of PCs and their immediate precursors, glutathione (GSH) and its derivatives. The tripeptide GSH (γ-Glu-Cys-Gly) and in some species, such as legumes, its variant homoglutathione, h-GSH (γ-Glu-Cys-β-Ala), are considered to influence the form and toxicity of heavy metals in several ways. These include direct metal binding, promotion of the transfer of heavy metals to other ligands, such as metallothioneins and/or PCs (Freedman et al., 1989), removal of active oxygen species formed as a result of the exposure of cells to heavy metals (Inzé and Van Montagu, 1995), and/or the formation of transport-active metal complexes (Li et al., 1997). First discovered in the fission yeast Schizosaccharomyces pombe and originally termed cadystins (Kondo et al., 1984), PCs, long-chain GSH derivatives (poly-[γ-Glu-Cys]n-Xaa polymers), are thought to serve a similar function by mediating the high-affinity binding and promoting the vacuolar sequestration of heavy metals. PC-dependent vacuolar Cd2+ sequestration is perhaps best understood in S. pombe, in which the hmt1+ gene product, a half-molecule ATP-binding cassette transporter, contributes to the transport of cadmium-PC complexes (Cd.PCs) and apo-PCs from the cytosol into the vacuole at the expense of ATP (Ortiz et al., 1992, 1995).
All known PCs fall into five main classes. These are canonical PCs (Fig. 1), homo-PCs [iso(PC)(β-Ala)], hydroxymethyl-PCs [iso(PC)(Ser)], iso-PCs [iso-PC(Glu)], and des-Gly-PCs [des(Gly)PC], containing n γ-Glu-Cys repeats capped C-terminally by a Gly, β-Ala, Ser, Glu, or no residue, respectively (Zenk, 1996). With the exception of canonical PCs and possibly des(Gly)PCs, which are probably ubiquitous in PC-containing organisms, the distribution and abundance of these classes differ between species (Rauser, 1999).
Figure 1.

PC synthase-catalyzed synthesis of PC3 from GSH and PC2 by dipeptidyl transfer or tripeptidyl transfer. In dipeptidyl transfer, PC chain extension proceeds in the C to N direction and is not associated with the production of des(Gly)PCs, according to the general equation PCn + PCm → PCn+1 + PCm−1, where PC1 = GSH. In tripeptidyl transfer, PC chain extension proceeds in the N to C direction and is associated with the production of des(Gly)PCs according to the general equation PCn + PCm → PCn+1 + des(Gly)PCm−1 + G, where des(Gly)PC1 = γ-Glu-Cys. Also shown is a space-filling model of PC3—gray, white, red, blue, and yellow spheres denote C, H, O, N, and S atoms, respectively.
The strongly nucleophilic sulfhydryl groups of the Cys substituents of PCs and their immediate precursors confer on these compounds the capacity to react with a broad spectrum of agents, ranging from free radicals, active oxygen species, and cytotoxic electrophilic organic xenobiotics to heavy metals (Rabenstein, 1989). Their N-terminal and downstream γ-peptidyl bonds, on the other hand, probably serve to protect these thiol peptides from general protease action, except when salvage is to be initiated through the specific action of γ-glutamyltranspeptidases. Contrary to what might be anticipated from the context in which they are found, γ-peptidyl bonds do not appear to be an essential prerequisite for high-affinity metal binding in that (EC)nG and (γEC)nG peptides have indistinguishable Cd2+-binding properties (Satofuka et al., 2001).
MOLECULAR IDENTITY OF PC SYNTHASE
PC synthases catalyze the net synthesis of PCs from GSH, from GSH and previously synthesized PCs, or from previously synthesized PCs to generate polymers containing 2 to 11 γ-Glu-Cys repeats. Although it is now almost a decade and a half since pioneering investigations by Meinhardt Zenk, Erwin Grill, and colleagues yielded partially purified preparations of an enzyme capable of catalyzing these reactions (Grill et al., 1989), it is only in the last several years that its molecular identity has been determined by the independent cloning and characterization of genes encoding PC synthases (Clemens et al., 1999; Ha et al., 1999; Vatamaniuk et al., 1999). Originally isolated from Arabidopsis, S. pombe, and wheat (Triticum aestivum), these genes (designated AtPCS1, SpPCS, and TaPCS1, respectively) encode 50- to 55-kD polypeptides bearing 40% to 50% sequence similarity to each other (Clemens et al., 1999; Ha et al., 1999; Vatamaniuk et al., 1999). By all criteria—genetic, molecular, and biochemical—these genes encode PC synthases. Arabidopsis cad1 mutants, which are Cd-hypersensitive and PC-deficient (Howden et al., 1995), are mutated in AtPCS1 (Ha et al., 1999), SpPCS gene disruptants are hypersensitive to heavy metals and deficient in cellular PCs (Ha et al., 1999), and heterologous expression of AtPCS1 in Saccharomyces cerevisiae, an organism that lacks PCS homologs and does not otherwise synthesize PCs, confers increased heavy metal tolerance concomitant with Cd2+-dependent intracellular PC accumulation (Vatamaniuk et al., 1999). The capacity of cell-free extracts from AtPCS1- or SpPCS-transformed cells of Escherichia coli (Ha et al., 1999) and immunopurified epitope-tagged AtPCS1 for the heavy metal-activated synthesis of both short- and long-chain PCs from GSH in vitro (Vatamaniuk et al., 1999) establishes that AtPCS1 and SpPCS, and by implication TaPCS1, and their equivalents from other sources, are not only necessary but also sufficient for PC biosynthesis.
FROM WEEDS TO WORMS
A striking outcome of the cloning of PC synthase from plants and S. pombe was the detection of a similar gene in an animal. Database searches disclosed a homologous single-copy gene in the genome of the model nematode Caenorhabditis elegans. Designated ce-pcs-1, this gene encodes a hypothetical 40.8-kD polypeptide (CePCS1) bearing 30% identity (45% similarity) to AtPCS1 in an overlap of 367 amino acid residues (Clemens et al., 1999; Ha et al., 1999; Vatamaniuk et al., 1999).
Functional analyses of this homolog establish that it is a PC synthase. Heterologous expression of CePCS1 in S. cerevisiae confers increased Cd2+ tolerance and intracellular PC biosynthesis (Vatamaniuk et al., 2001), and expression of the same clone in S. pombe PC synthase-deficient mutants suppresses Cd2+ hypersensitivity and restores Cd2+-elicited PC accumulation (Clemens et al., 2001). As is the case for its plant and fungal equivalents, the tolerance conferred by CePCS1 is not limited to Cd2+ but extends to other soft metals and metalloids, including Hg and As. Critical for elucidation of the determinative role played by PC synthase in metal detoxification in the intact organism is the finding that targeted suppression of ce-pcs-1 by the double-stranded RNA interference technique confers a Cd2+-hypersensitive phenotype (Vatamaniuk et al., 2001). The progeny of worms injected with double-stranded ce-pcs-1 RNA show severe growth retardation, developmental arrest, necrosis, and sterility, and eventually die when exposed to Cd2+ concentrations tolerated by wild-type worms.
It is exhilarating to think that the involvement of PCs in the detoxification of heavy metals in animals had not been even cursorily mentioned or speculated on in the literature before the discovery, which owes its origins to fundamental research on plants, of CePCS1 (Vatamaniuk et al., 2002). By the same token, it is salutatory to learn that while other gene products have been inferred to contribute to Cd2+ tolerance in C. elegans, CePCS1 is actually the first for which there is a firm biochemical basis for the effects seen at the level of the whole organism.
GLUTATHIONE SYNTHETASE: A BIFUNCTIONAL ENZYME?
To unclutter the discussion that follows, it should be pointed out that, coincident with the original cloning of the PC synthase genes from plant and fungal sources, results were presented consistent with a direct role for the terminal enzyme of GSH biosynthesis, glutathione synthetase (GSH2), in the elaboration of PCs (Al-Lahman et al., 1999). In suppression screens of an S. pombe Cd2+-hypersensitive mutant (M379) that seemed able to sustain GSH at levels corresponding to 40% or more of wild type but was deficient in PC synthesis, it was found that the genes encoding GSH synthetase from S. pombe and Arabidopsis partially restore PC biosynthesis and Cd2+ tolerance. Since the mutant GSH2 allele in strain M379 contains a single base-pair substitution that appeared to impair PC synthesis more than GSH synthesis, it was proposed that the GSH2 genes of S. pombe (SpGSH2) and Arabidopsis (AtGSH2) encode bifunctional enzymes competent in the synthesis of both GSH and PCs (Al-Lahman et al., 1999). Tantalizing as they were at the time, especially from the conceptual standpoint of a bifunctional enzyme competent not only in the ATP-dependent glycylation of γ-Glu-Cys (γEC + G + ATP → γECG + ADP + Pi) but also in the γ-glutamylcysteinylation of GSH [γECG + γEC → (γEC)2G or γECG + γECG → (γEC)2G + G], all attempts to reproduce or extend these findings were unsuccessful. Neither SpGSH2 nor AtGSH2 confer Cd2+ tolerance when heterologously expressed in S. cerevisiae, and neither yield translation products able to catalyze the synthesis of PCs in vitro from either GSH or γ-Glu-Cys in the presence or absence of heavy metals, Gly, and/or ATP (R. Sánchez-Fernández and P.A. Rea, unpublished data). The reason for the discrepancy between these results and those of Al-Lahman et al. (1999) is not known, but it is suspected that the chromatographic peak that was attributed to GSH in strain M379 in the latter work was principally γ-Glu-Cys, thus giving the impression of selective abolition of the capacity for PC synthesis, despite the intracellular availability of GSH when, in fact, GSH was the missing factor for net PC synthesis for want of the activity responsible for the glycylation of γ-Glu-Cys.
A DIPEPTIDYL TRANSFERASE
In most studies of partially purified preparations of the PC synthase from plant sources, GSH-dependent PC synthesis has been assumed to proceed by the transpeptidation of a γ-Glu-Cys unit from one GSH molecule to another to form PC2, and after the accumulation of sufficient (substrate) levels of PCs, by the transpeptidation of a γ-Glu-Cys unit from GSH to a PC (PCn) molecule to generate PCn+1 (Grill et al., 1989). Appropriately, the enzyme has been defined as a γ-Glu-Cys dipeptidyl transpeptidase and presumed to catalyze a reaction of the type:
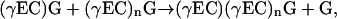 |
(1) |
in which chain extension proceeds C → N with cleavage of the Cys-Gly peptide bond of the donor, not the acceptor (Fig. 1). Surprising as it might seem, it is only very recently that this has been confirmed directly. Hitherto there were no published data to refute a scheme in which PC synthase catalyzes the transfer of γ-Glu-Cys-Gly, rather than γ-Glu-Cys, units in a tripeptidyl transpeptidase reaction of the type:
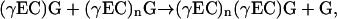 |
(2) |
in which chain extension proceeds N → C, not C → N, after cleavage of the Cys-Gly peptide bond of the acceptor, not the donor (Fig. 1).
This is not a trivial issue. It is critical in determining the classes of PCs that PC synthase is capable of manufacturing, as well as being vital to understanding the precise stoichiometry of the reaction catalyzed, an essential prerequisite for the dissection of the catalytic mechanism. Specifically, one implication of the possibility that PC synthase is a tripeptidyl transferase, in combination with its capacity for the synthesis of PCs from other PCs without the direct participation of GSH, is a simple mechanism for the synthesis of des(Gly)PCs. If γ-Glu-Cys is construed as the limiting case of a des(Gly)PC (= des(Gly)PC1), and GSH as the limiting case of a PC (= PC1), Equation 1 assumes the general form:
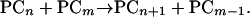 |
(3) |
And Equation 2 assumes the general form:
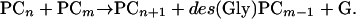 |
(4) |
The question of whether AtPCS1 is a dipeptidyl or tripeptidyl transferase has been addressed according to two criteria by determining whether: (1) During the synthesis of PC3 from PC2 and GSH, the Gly residue of GSH is or is not retained in PC3; and (2) des(Gly)PCs are or are not an immediate by-product of PC synthesis from PCs (Vatamaniuk et al., 2004). With regard to criterion 1, it has been demonstrated that, for short incubation times, one-third of the thiols in the PC3 product are 35S labeled when synthesis is from [35S-Cys]GSH and unlabeled PC2, and that when the precursors are [3H-Gly]GSH and PC2, the PC3 product remains unlabeled over the same time span. That is, the pattern of synthesis and radioincorporation is uniquely attributable to a dipeptidyl transpeptidation reaction.
The dipeptidyl model predicts that the net synthesis of PC3 from PC2 and [3H-Gly]GSH would result in the total elimination of radiolabel concomitant with the liberation of free [3H]Gly, while the initial synthesis of PC3 from PC2 and [35S-Cys]GSH would result in a 1:1 molar ratio of [35S]Cys:PC3, indicating that one in three of the thiols in the product are labeled (Fig. 1):
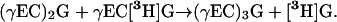 |
(5) |
The tripeptidyl transpeptidase model, by contrast, predicts the 1:1 stoichiometric incorporation of [3H]Gly from [3H-Gly]GSH and of [35S]Cys from [35S-Cys]GSH into PC3 concomitant with the liberation of unlabeled Gly from the outset of the reaction (Fig. 1):
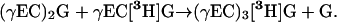 |
(6) |
With regard to criterion 2, attractive as it is as a simple and direct mechanism for the synthesis of des(Gly)PCs, in no case are these PC derivatives detectable when AtPCS1 catalyzes the synthesis of PCs from PCs in vitro, whether it be the synthesis of PC4 (and PC2) from PC3 or PC5 (and PC3) from PC4. This observation further substantiates the dipeptidyl transferase model and refutes the tripeptidyl transferase model.
ACTIVATION BY HEAVY METALS
A physiologically critical but biochemically confounding property of PC synthase is its susceptibility to activation by heavy metals. It is by virtue of the activation of PC synthase-catalyzed PC biosynthesis by agents, heavy metal ions that poison most enzymes, that plants, fungi, and some invertebrates are able to mount a PC-based response to heavy metals. A notable exception is the Cd-dependent carbonic anhydrase of diatoms (Lane and Morel, 2000). This being the case, few investigators have considered explicitly how heavy metals activate PC synthase, but those who have, have assumed that activation is a consequence of the direct binding of metal ions to the enzyme. One of the initially most-appealing models for PC synthase action invokes direct metal binding at several sites in the enzyme. In this model, it is proposed that the strongly conserved N-terminal half of the enzyme is responsible for core catalysis and that activation arises from the binding of metal ions to residues, possibly Cys residues, in this domain (Cobbett, 2000). The presence of five conserved Cys residues, two of which are vicinal, and consequently optimally disposed for the coordination of ions such as Cd2+, Cu2+, and/or Hg2+ in the N-terminal halves of eukaryotic PC synthases, is at least consistent with this notion (Fig. 2A), as is the observation that the three most extreme Arabidopsis cad1 alleles have amino acid substitutions in this region (Ha et al., 1999). An extension of this model, proposed to ascribe a role to the more sequence-divergent C-terminal half of the molecule and to account for the properties of the least extreme cad1 allele, cad1-5—a nonsense mutation causing premature termination and deletion of the C-terminal segment (Fig. 2A)—is the concept of a C-terminal metal-sensing domain whose multiple Cys residues bind heavy metals and bring them into contact with the putative activation site within the N-terminal, catalytic half of the molecule.
Figure 2.

A, Comparison of PC synthase polypeptides and their derivatives from different organisms. The examples shown are the full-length PC synthase polypeptides from Arabidopsis (AtPCS1), S. pombe (SpPCS), C. elegans (CePCS1), and Nostoc sp. PCC 7120 (Alr0975) and the two truncated derivatives of V8 protease-digested native AtPCS1 (PCS_Nt1 and PCS_Nt2). The approximate positions of all Cys residues are indicated by white vertical bars, and of the conserved His and Asp residues in the N-terminal domain by blue and red bars, respectively. AtPCS1 residues Cys-56, His-162, and Asp-180 are the three residues that are conserved in all known PC synthases and align with the catalytic triad residues of members of the papain family of Cys proteases. Also shown is the position of the Arabidopsis cad1-5 nonsense mutation. Numbers on the right denote total number of residues in each polypeptide. This figure is essentially an update of Figure 2 from Cobbett (2000). B, Alignment of papain superfamily polypeptides of known structure (papain, PDB code 1PE6; cruzain, 1AIM; staphopain, 1CV8) with AtPCS1, CePCS1, SpPCS, Alr0975, and the Microbulbifer degradans PC synthase homolog (GenBank ID 48861977) in the vicinity of the catalytic triad residues shown in A. Other residues that are also reasonably conserved among all of the sequences are shaded in gray. Shown above the alignment is the actual secondary structure of staphopain A. Shown below the alignment is the predicted secondary structure for AtPCS1. Cylinders, α-helix; arrows, β-sheet.
This model has been and will continue to be important in prompting structure-function investigations of PC synthases, but in its original form it probably does not account for the facility of heavy metals to activate core catalysis (Vatamaniuk et al., 2000). First, although AtPCS1 binds Cd2+ at moderate affinity (KL = 0.54 ± 0.20 μm) and high capacity (BL = 7.09 ± 0.94), its affinity for other metal ions such as Cu2+, which are equally effective activators in vitro, is much lower. Second, the metals that elicit net PC synthesis by AtPCS1 in vitro do so despite the presence of a 66- to 132-fold molar excess of GSH and/or PC-associated thiols in the reaction media. Given that the complexes formed between heavy metals and thiol compounds are among the most stable known (Rabenstein, 1989), this means that, under the conditions in which PC synthase catalyzes high rates of PC synthesis from GSH, the concentration of free Cd2+ is only of the order of 10−13 m (some 6 orders of magnitude lower than the KL for direct binding of this metal ion to the enzyme), and more than 98% of the total Cd2+ added to the reaction medium is associated with GSH as the bidentate thiolate, bis(glutathionato) cadmium (Cd.GS2). Third, analyses of the steady-state kinetics of AtPCS1-catalyzed PC synthesis demonstrate that activity is strictly dependent on metal thiolate and free GSH concentration, not free metal concentration, and approximates a bisubstrate-substituted enzyme reaction in which Cd.GS2 and GSH serve as high-affinity and low-affinity substrates, respectively. Fourth, when assayed in media devoid of metal salts, AtPCS1 catalyzes the net synthesis of S-alkyl-PCs from S-alkyl glutathione derivatives at rates similar to the synthesis of unsubstituted PCs from equivalent concentrations of GSH in media containing heavy metals.
The most straightforward explanation of these properties is that PC synthases catalyze a bisubstrate transpeptidation reaction in which both free GSH and its corresponding metal thiolate are cosubstrates. Moreover, although both free GSH and its metal thiolate are ordinarily required for maximal activity, other compounds, for instance S-substituted GSH derivates, can substitute for both in such a way as to overcome the enzyme's otherwise near-obligatory requirement for heavy metals for activity, implying that the decisive factor for core catalysis is the provision of glutathione-like substrate peptides containing blocked thiol groups.
It is important to note that this scheme does not necessarily preclude the augmentation of activity by direct metal ion binding to the enzyme. Indeed, when the reaction conditions are designed so as to be compatible with the availability of not only sufficient substrate but also adequate concentrations of free metal ions, by exploiting the capacity of S-alkylglutathiones to act as substrates, despite their inability to form metal thiolates, promotion of S-alkyl-PC synthesis up and above that conferred by the provision of substrate containing blocked thiol groups is readily detectable (Vatamaniuk et al., 2000). The meaning of this property remains to be resolved, but it does seem unlikely that direct modulation of catalytic turnover by heavy metal binding to the enzyme would be appreciable when the dominant thiol peptide is unsubstituted GSH. The free Cd2+ concentrations required for half-maximal stimulation of S-alkyl-PC2 synthesis, albeit equivalent to the KL for equilibrium binding, are more than 5 orders of magnitude greater than those that prevail when the rates of synthesis of PCs from GSH are maximal (Vatamaniuk et al., 2000).
On the basis of their investigations of AtPCS1 and the homo-PC synthase of soybean [Glycine max; GmhPCS1], Oven et al. (2002) challenge several of the conclusions drawn by Vatamaniuk et al. (2000). Principal among the concerns they cite are two, one of which appears to derive from a misunderstanding of the conclusions drawn by Vatamaniuk et al. (2000), and another which stems from studies of the enzyme under extreme conditions. Their first concern is that, when measured as a function of increasing Cd2+ concentration, in the range 0.01 to 0.5 mm at a fixed concentration of 1 mm GSH, the activity of AtPCS1 is minimal when Cd2+ is greatest. While this is indeed the case, it is not because, as Oven et al. (2002) contend, Cd.GS2 is not a substrate for the enzyme, but instead because, as Vatamaniuk et al. (2000) explain, the enzyme catalyzes a bisubstrate reaction in which not only Cd.GS2 but also free GSH participate directly. In other words, generation of the first substrate, Cd.GS2, at the expense of the second, free GSH, stalls the reaction. Oven et al.'s other concern is the finding that not only heavy metal-GSH complexes but also other heavy metal-thiol complexes, for instance, those of Cd2+ with 2-mercaptoethanol or Cys that are not substrates for the enzyme, contribute to the activation of GmhPCS1. While this finding is of potential interest and might be consistent with the participation of thiols, regardless of their precise chemical identity, in the shuttling of heavy metal ions to other, substrate-active thiols or to the auxiliary metal activation domain of the enzyme, it does not preclude the participation of Cd.GS2 as a cosubstrate. It is more likely that activation of GmhPCS1 exerted by thiols other than GSH under these conditions are attributable to the unusually high (millimolar) concentrations of heavy metal ions that were used, concentrations that would necessitate appreciable chelation by thiols, regardless of whether they could or could not act as substrates in order to alleviate the inhibitory thiol capping to which the enzyme would otherwise be subject.
SELF-TERMINATION OF PC BIOSYNTHESIS
Two notable physiological implications follow from investigations of the metal dependence of PC synthase-catalyzed PC synthesis in vitro. The first is that, contrary to earlier models, (Loeffler et al., 1989; Zenk, 1996), the reactions catalyzed by PC synthases probably do not self-terminate through the chelation of activating metal ions by the PC products per se because GSH and PC complexes containing heavy metal are active substrate species. Instead, termination of the reaction more likely results from substrate depletion. Diminution of the substrate-active thiolate pool, whether it be by the incorporation of heavy metals into higher order, substrate-inactive metal-PC complexes or by the removal of metal-PC complexes from the cytosolic pool into the vacuole, is probably the determining factor for ensuring that PC synthesis meets, but does not exceed, needs.
The second physiological implication is that the cytosolic concentration of free metal ions need not increase even transitorily for net PC synthesis. Given the high values of the stability constants of heavy metal-GSH complexes and the high prevailing concentration of GSH, any soft metal that gains access to the cytosol would be expected to be incorporated into the corresponding thiolate. The GSH thiolates so formed because of the moderately high and constitutive expression of PCS genes (Cobbett and Goldsbrough, 2002) would, in turn, be incorporated into derivatives, PCs, that also bind heavy metals but at higher affinity (Zenk, 1996).
The notion that the intracellular concentrations of free heavy metals need not increase except when the mechanisms for coping with them are exceeded, and necrotic or apoptotic cell death ensues, raises the question of whether other systems or components of the PC-dependent system for heavy metal detoxification, for instance, heavy metal transporters, interact directly with metal thiolates rather than with free metal ions. This clearly applies to the S. cerevisiae multidrug resistance-associated protein-type ATP-binding cassette transporter yeast cadmium factor 1 (YCF1), originally identified according to its ability to confer resistance to Cd2+ salts (Szczypka et al., 1994). Detailed investigations of the substrate requirements, kinetics, and Cd:GS stoichiometry of Cd2+ uptake and the mass of the transport-active substrate complex, demonstrate that YCF1 catalyzes the MgATP-energized transport of Cd.GS2 into the vacuole (Li et al., 1997). Analogous studies of As3+ transport by YCF1 indicate the same basic mechanism, transport of bis(glutathionato) arsenic (As.GS3; Ghosh et al., 1999). YCF1 and PC synthase therefore appear to recognize the same metal thiolates as substrates, which in turn introduces the possibility that a similar principle, or at least the shuttling of heavy metals to their active sites, might also apply to some of the other transporters implicated in heavy metal transport. Such a mechanism, if it were operative, would reconcile a fundamental problem, namely, that most considerations of the transporters concerned, members of the CAX, ZIP, Nramp, and CDF families in plants (Maser et al., 2001, and references therein), quote Km values for Cd2+ and other heavy metal ions in the micromolar range, yet their likely free concentrations are several orders of magnitude lower in vivo because of thiol coordination.
ENZYME ACYLATION
Implicit in the finding that the steady-state kinetics of PC synthase-catalyzed PC synthesis from GSH in media containing heavy metals approximate a scheme in which heavy metal thiolate and GSH interact via a substituted enzyme intermediate, not a ternary complex, is the formation of an enzyme covalent intermediate during catalysis. What is more, given that at least one peptide bond must be cleaved and at least one new peptide bond formed for each molecule of PC2 synthesized, and initial attack on the carbonyl carbon of the peptide bond to be cleaved must be by a nucleophile, it is almost inevitable that the substituted enzyme intermediate formed is a γ-Glu-Cys acyl intermediate. This is indeed the case. PC synthase undergoes acylation at two sites during catalysis, albeit with different ligand requirements, and acylation at at least one of these sites appears to be necessary for net PC synthesis (Vatamaniuk et al., 2004). The provision of free GSH in media devoid of metal ions is sufficient for acylation of the first site, a reaction that is not accompanied by appreciable net PC synthesis, whereas acylation at the second site necessitates the provision of heavy metal ions, as exemplified by Cd2+, and is accompanied by the net synthesis of PCs.
There are two corollaries that follow from these findings. The first is that initial nucleophilic attack on the scissile bond of the dipeptidyl donor is by an enzyme hydroxyl-derived oxyanion or thiol-derived thiolate anion to generate a γ-Glu-Cys-enzyme oxyester or thioester, respectively. The second is that at least some of the energy required for subsequent condensation of the two substrate molecules is derived from acylation of the enzyme coincident with cleavage of the first substrate. A mechanism for PC synthase analogous to those of Ser proteases (Kraut, 1977), Cys proteases (Kamphuis et al., 1985), and Cys hydrolases (Cheah et al., 1993; Humm et al., 1997a, 1997b) is therefore invoked except that, instead of mediating a dissipative hydrolysis reaction, at least some of the energy required for condensation of the γ-Glu-Cys unit from one substrate with the other substrate during the second, PC synthetic, phase of the catalytic cycle is derived from an enzyme oxyester of intermediate energy or an enzyme thioester of high energy formed during the first phase of the cycle.
The results of mutational analyses directed at determining which Cys or Ser residues, if any, in AtPCS1 might participate in initial nucleophilic attack on the γ-Glu-Cys donor are in agreement with this contention (Vatamaniuk et al., 2004). Of the five conserved Cys residues in the N-terminal catalytic sector of AtPCS1, only one, Cys-56, when substituted by a Ser or Ala residue, abolishes the capacity of the heterologously expressed enzyme to suppress Cd2+hypersensitivity, catalyze PC synthesis, and undergo direct acylation by free GSH. Cys-56 therefore satisfies the requirements of an active site residue responsible for initial nucleophilic attack on the scissile bond of the GSH (or PC) γ-Glu-Cys donor, in accord with the Cys protease model, such that AtPCS1-mediated catalysis is initiated by the formation of a high-energy thioester intermediate (Fig. 3). By contrast, substitution of any one of the conserved Ser residues in this portion of the enzyme has little or no effect on catalytic activity, thus excluding the Ser protease model from further consideration, at least in this restricted context.
Figure 3.

Tentative catalytic mechanism of PC synthase based on the results of kinetic analyses, measurements of acylation of the enzyme, site-directed mutagenesis, and the mechanism of distantly homologous Cys proteases of the papain superfamily. Refer to the main body of the text for detailed description. ∼∼, γ-peptidyl bond between Glu and Cys residues in GSH substrate.
A remarkable feature of PC synthase is that, while the C56S and C56A substitutions are strictly associated with the abolition of PC synthetic activity, they are not accompanied by abolition of acylation at the second site. The implication, but one that has yet to be deciphered mechanistically, is that the initial Cys-56-dependent, Cd2+-independent first-site acylation is not a prerequisite for the subsequent Cys-56-independent second-site acylation of AtPCS1, but it is essential for net PC synthesis which, in turn, might imply that the condensation of enzyme-bound γ-Glu-Cys with GSH for the synthesis of PC2 is contingent on acylation of the enzyme at the first site and possibly, but not necessarily, both sites (see below).
In considering these findings, account should also be taken of the alternate or auxiliary role proposed for Cys residues. This is the possibility that the activation of catalysis depends on the binding of heavy metal ions, such as Cd2+, to essential Cys-containing metal-binding sites in the N-terminal catalytic sector of the enzyme. In support of this, Maier et al. (2003) show that when synthetic libraries, consisting of overlapping 13-residue peptides whose sequences are based on those of SpPCS and TaPCS1, are screened for 109Cd2+ binding, several clusters of contiguous peptides with binding activity are discernible. Of particular relevance to the mutational studies of AtPCS1, most of the sequences that the contiguous overlapping peptides delimit, so-called core peptides, encompass the Cys residues that are subject to conservation between different PCS sequences. These are the SpPCS and TaPCS1 equivalents of AtPCS1 Cys-56, Cys-90, Cys-91, Cys-109, and Cys-113. And, in at least a subset of these core peptides, the Cys residues concerned are capable of contributing to binding insofar as Cys to Ala substitutions at these positions abolish or diminish 109Cd2+ binding in vitro.
Though elegant and in many ways informative, it cannot be decided whether the Cd2+ binding detected in these experiments would be operative under the conditions that prevail in vivo or even in vitro when PC synthetic activity is maximal because they were performed at free Cd2+ concentrations (10 μm) far in excess of those that are achieved cytosolically under physiological conditions or can be achieved when GSH is present at the millimolar concentrations necessary for net PC synthesis. Likewise, while intact AtPCS1 binds 109Cd2+ at high capacity—incidentally at a stoichiometry congruent with the total of seven putative binding sites identified by the peptide-mapping studies of TaPCS1 under identical buffer conditions (Maier et al., 2003)—it is suspected that the Cd2+ binding measured by Maier et al. (2003) is more a reflection of the capacity of heavy metals to augment PC synthase activity in the presence of substrate-active S-alkyl derivatives rather than a strict requirement for heavy metals for core catalysis.
PC SYNTHASE-LIKE HALF-MOLECULES IN PROKARYOTES
PC synthase-like polypeptides are much more widespread phylogenetically than was once thought. Systematic sequence database searches disclose 30 putative PC synthase homologs. Of these, 20 are canonical PC synthases from plants and three are their equivalents from other eukaryotes—one each from S. pombe, C. elegans, and C. briggsae. Nothing new here. What is new, however, is that the remaining sequences are from bacterial sources and all are approximately one-half the length of their cognates from eukaryotes (220–237 compared to 421–506 residues) because they lack the more variable C-terminal domain. Four of the bacterial sequences are from cyanobacteria (two Nostoc species, Prochlorococcus marinus and Trichodesmium erythraeum; Tsuji et al., 2004), and three are from proteobacteria (Burkholderia fungorum, Microbulbifer degradans, and Bradyrhizobium japonicum; D.J. Rigden, O.K. Vatamaniuk, A. Lang, and P.A. Rea, unpublished data). Despite their relatively low (22%–36%) sequence identity with the eukaryotic enzymes in the N-terminal catalytic domain, which in and of itself is insufficient to guarantee a strict catalytic equivalence, the one prokaryotic PC synthase homolog that has been assayed for activity, the alr0975 protein from Nostoc sp. PCC 7120, does indeed have PC synthase activity. This protein catalyzes the deglycylation of GSH to γ-Glu-Cys at a high rate and the synthesis of PC2 at a relatively low rate, and these activities are seen regardless of whether micromolar Cd2+ is or is not included in the reaction medium (Tsuji et al., 2004). Protein alr0975 and presumably other half-molecule PC synthase homologs from bacterial sources, therefore, have the characteristics of thiol peptide transpeptidases or minimally peptidases that antedate the canonical eukaryotic PC synthases.
Incisive investigations of AtPCS1 after its C-terminal truncation by limited proteolysis (Ruotolo et al., 2004) reinforce these deductions and verify the sufficiency of the N-terminal domain of eukaryotic PC synthases for core catalysis. When native AtPCS1 is subjected to limited proteolysis with V8 protease, two N-terminal fragments, ending at positions 372 (PCS_Nt1) and 283 (PCS_Nt2), are generated (Fig. 2A), both of which are competent in PC synthesis from GSH in the presence of Cd2+ at rates only about 5-fold lower than those of the full-length polypeptide (Ruotolo et al., 2004). Ablation of the C-terminal domain, however, substantially decreases the thermal stability of the enzyme and causes a greater than 100-fold impairment of PC synthesis when Hg2+ or Zn2+, instead of Cd2+ or Cu2+, are the sole metal ion cofactors. Whereas the N-terminal domain of AtPCS1, the domain shared with the prokaryotic half-molecule homologs, is responsible for core catalysis, the C-terminal domain appears to play a more subsidiary role by contributing to the stability of the full-length polypeptide and participating in defining the range of metals to which the enzyme is responsive (Ruotolo et al., 2004).
PC SYNTHASE IS A PAPAIN SUPERFAMILY MEMBER
Recognition of the fundamental equivalence between the N-terminal catalytic half of the eukaryotic PC synthases and their half-molecule cognates from prokaryotes has been instrumental in the assembly of a representative subset of sequences for deeper and wider scans of sequence databases. In particular, deployment of this expanded PC synthase sequence dataset for application of the FFAS03 method for sequence profile matching (Rychlewski et al., 2000) has yielded important fresh insights into the structure-function characteristics of PC synthases. FFASO3 is a high-sensitivity method for the detection of distant homologies wherein a PSI-BLAST profile (alignment) for the target protein is first generated by database searching and similar profiles are precalculated for all of the proteins of known structure identified in the searches. Using this information, FFASO3 then aligns the target profile with each of those derived from the proteins of known structure, while optimizing the scoring function for sensitivity and reliability (Rychlewski et al., 2000). Application of this algorithm to the extended PC synthase family reveals clear matches with Clan CA Cys proteases, as exemplified by the archetype, papain, from Carica papaya fruit and latex, staphopain A, an extracellular enzyme from Staphylococcus aureus, and several of the lysosomal cathepsins from animal sources (Fig. 2B).
The need for caution is inevitable when basing conclusions on sequence-matching algorithms of this type, but several other considerations support the notion of a distant homology between PC synthases and Clan CA Cys proteases. First, there is a fundamental catalytic equivalence between PC synthases, dipeptidyl transferases, and Cys proteases in terms of the partial reactions they catalyze (Vatamaniuk et al., 2000). Second, both classes of enzyme have a strict requirement for a nucleophilic Cys residue that undergoes peptidyl acylation during catalysis, residue Cys-56 in AtPCS1 and its equivalent in the Cys proteases, residue Cys-24 in staphopain A (Barrett and Rawlings, 2001; Vatamaniuk et al., 2004; Fig. 2B). Third, the nucleophilicity of the active-site Cys residues of Cys proteases is contingent on their immediate proximity to a His residue in the native enzyme, and it is the Cys and His residues of the papain-like peptidases, residues 24 and 120 in staphopain A, that precisely align with the conserved Cys and His residues of PC synthases, residues 56 and 162, respectively, in AtPCS1. Fourth, the third Cys protease catalytic triad residue, an Asn at position 141 in staphopain A, aligns with a conserved Asp residue in the PC synthase family, residue 180 in AtPCS1 (Fig. 2B). The relevance of this alignment is that the replacement of the triad Asn in proteases with an Asp in PC synthases is in principle acceptable functionally because the third catalytic residue can, as is the case for several members of Clan CA (Barrett and Rawlings, 2001), be either Asp or Asn.
Site-directed mutagenesis substantiates these inferences by confirming that not only Cys-56, but also His-162 and Asp-180, are essential for catalysis by AtPCS1 (D.J. Rigden, O.K.Vatamaniuk, A. Lang, and P.A. Rea, unpublished data). Of the 19 Asp, Cys, His, Ser, Thr, and Tyr residues conserved among the N-terminal domains of eukaryotic PC synthases, only three, Cys-56, His-162, and Asp-180, when substituted, abolish the capacity of AtPCS1 to confer Cd2+ tolerance and catalyze PC synthesis.
PC SYNTHASE IS A CYS TRANSPEPTIDASE
Although other transpeptidases acting on GSH and related thiols bear an evolutionary relationship with other hydrolases (Inoue et al., 2000), PC synthases are the first transpeptidases associated with the papain superfamily. Two broadly significant ramifications therefore follow. The first concerns the prokaryotic PC synthase-like half-molecules. The second concerns the mechanism of PC synthase-mediated catalysis.
With respect to the prokaryotic half-molecules, when account is taken of the overall degree of sequence similarity in the PC synthase family, in combination with conservation of the catalytic triad, it is likely even if the bacterial sequences are not those of a true PC synthase that their substrates resemble those of their eukaryotic homologs. One possibility, other than PC synthesis per se, is that the bacterial enzymes serve to cleave glutathione S-conjugates (GS-conjugates). Knowing that GS-conjugate cleavage is a major auxiliary capability of eukaryotic PC synthases (Beck et al., 2003; Vatamaniuk et al., 2004) and that glutathione S-transferases (GSTs) and/or their homologs are represented in the genomes of all of the bacteria possessing PC synthase homologs hints at the possibility that bacterial PC synthase homologs participate in GS-conjugate degradation. Pathways for the processing of bacterial GS-conjugates entailing the same deglycylation reaction as that catalyzed by eukaryotic PC synthases (Beck et al., 2003; Vatamaniuk et al., 2004) and proposed to function in the detoxification of GS-conjugable xenobiotics in plants (Beck et al., 2003) might be envisaged. The properties of the Nostoc alr0975 protein (Tsuji et al., 2004) are consistent with this and the subsidiary contention that the prokaryotic enzymes are more rudimentary, more peptidase- than transpeptidase-like, in their action. Conceivably, the deglycylation reaction of the eukaryotic PC synthases is an evolutionary relic of their prokaryotic origins.
TENTATIVE CATALYTIC MECHANISM
With respect to catalytic mechanism, the assignment of the PC synthase family to the papain superfamily, combined with conservation of the catalytic triad of the former in the latter, and the facility of both classes of enzyme to mediate reactions that approximate substituted enzyme kinetics necessitating the formation of a cysteinyl enzyme acyl-intermediate, is consistent with a Cys protease-type reaction scheme for PC synthesis (Fig. 3). Implicit in the finding that the time and concentration dependence of PC synthesis approximates substituted-enzyme kinetics (Vatamaniuk et al., 2000) and that AtPCS1 forms a γ-Glu-Cys acyl intermediate during catalysis (Vatamaniuk et al., 2004) is initiation of the overall biosynthetic reaction by cleavage of the Cys-Gly bond of the first substrate. By analogy with papain, therefore, nucleophilic attack on the scissile bond of the first substrate, the donor, by a Cys-derived thiolate anion, specifically that of Cys-56, and formation of a γ-Glu-Cys thioester, concomitant with the liberation of substrate Gly, is invoked for the first step (Step 1, Fig. 3). As in Cys proteases in general, the nucleophilicity of Cys-56 is thought to be enhanced by its proximity to His-162 and Asp-180. The sulfhydryl proton from Cys-56 is transferred to the imidazole ring of His-162, whose electrophilicity is enhanced by its immediate adjacency to the β-carboxylate on Asp-180. In the second step (Step 2, Fig. 3), the Gly released from the first substrate dissociates from the enzyme and is replaced by the second substrate, the acceptor. In the simplest case, the synthesis of PC2 from two molecules of GSH, the acceptor is another molecule of GSH. The N terminus of the acceptor GSH then nucleophilically attacks the enzyme thioester intermediate, resulting in the formation of PC2 and regeneration of the free thiol group of Cys-56 (Step 3, Fig. 3). The dissociation of PC2 from the enzyme and its replacement by a new molecule of donor GSH completes the catalytic cycle (Step 4, Fig. 3). The same mechanism can explain the formation of longer chain PCs if, instead of GSH, PCn is bound at Step 2 to yield PCn+1 as product.
The key difference between this mechanism and that of the papain family is that, during net PC synthesis, nucleophilic attack on the thioester intermediate is not by water but instead by a second substrate molecule, thus resulting in transpeptidation rather than net hydrolysis. The facility of not only prokaryotic PC synthases (Tsuji et al., 2004) but also eukaryotic PC synthases (Beck et al., 2003; Vatamaniuk et al., 2004) for the deglycylation of GS conjugates is especially pertinent in this context. The precise significance of this phenomenon is not known, but it does provide further support for the mechanism proposed here.
Pending data to the contrary, it is suspected that the second, non-Cys-56-coupled site of acylation of AtPCS1 (Vatamaniuk et al., 2004) is not located in the catalytic core of the enzyme but in the C-terminal extension. For this reason, and because systematic site-directed mutagenesis of the N-terminal domain discloses only one nucleophilic residue, Cys-56, whose substitution abolishes catalytic activity (Vatamaniuk et al., 2004; D.J. Rigden, O.K. Vatamaniuk, A. Lang, and P.A. Rea, unpublished data), the scheme depicted in Figure 3 is based on acylation at this one site.
CONCLUDING REMARKS
Considerable progress has been made in understanding the mechanism of heavy metal activation of PC synthesis and the catalytic mechanism of PC synthase. On the basis of the results of recent investigations, it can be inferred that the enzyme is a γ-glutamylcysteine dipeptidyl transpeptidase that mediates a bisubstrate reaction in which the thiol group(s) of at least one of the substrates is(are) blocked, usually, but not necessarily, through the formation of a heavy metal thiolate. The reaction approximates substituted enzyme kinetics and is strictly associated with γ-glutamylcysteinyl acylation of the enzyme at two sites, one of which corresponds to or is closely coupled to AtPCS1 Cys-56 or its equivalents in other PC synthases. In agreement with earlier speculations, the N-terminal domain of eukaryotic PC synthases, the portion that is represented in the half-molecule homologs from prokaryotes, is responsible for core catalysis. It is this domain that bears a distant homology to papain superfamily proteases and encompasses the putative Cys protease-type catalytic triad that is essential for catalysis, AtPCS1 residues Cys-56, His-162, and Asp-180. That is what is known, at least to a first approximation. What is not known is the identity of the second site of acylation and of the residues and structural determinants responsible for substrate selectivity, and the precise role played by the C-terminal domain in eukaryotic PC synthases other than in contributing to the range of metal ions capable of stimulating enzyme activity. These are eminently tractable short- to medium-term research objectives, ones that would be greatly expedited when the enzyme or one or more of its nearest homologs has been crystallized and its three-dimensional structure solved. Less tractable, but no less important, is the question of what precisely the enzyme does—why does the enzyme exist? Other review authors, Cobbett (2000), for instance, present cogent arguments to the effect that PC synthases may play a role in heavy metal detoxification even in unpolluted environments, but it is still a mystery as to why the enzyme is expressed constitutively and at such relatively high levels regardless of mineral status. From what is known of its domain organization, it is as if the enzyme is bifunctional and one of the functions, the non-PC synthetic one required for basic maintenance activity, has yet to be defined or at least defined knowingly. Perhaps this will be the topic of a future review.
This work was supported by the National Science Foundation (grant no. MCB–0077838 to P.A.R).
This review is dedicated to Professor Emmanuel Epstein, whose seminal work on ion transport by plants was a major factor in prompting one of us (P.A.R.) to venture into the world of mechanistic plant research. Professor Epstein's facility for incisive and inventive dissection of ostensibly daunting phenomena was a thrill to share through his many formative publications.
References
- Al-Lahman A, Rohde V, Heim P, Leughter R, Veeck J, Wunderlich C, Wolf K, Zimmermann M (1999) Biosynthesis of phytochelatin in the fission yeast. Phytochelatin synthesis: a second role for the glutathione synthetase gene of Schizosaccharomyces pombe. Yeast 15: 385–396 [DOI] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND (2001) Evolutionary lines of cysteine proteases. Biol Chem 382: 727–733 [DOI] [PubMed] [Google Scholar]
- Beck A, Lendzian K, Oven M, Christmann A, Grill E (2003) Phytochelatin synthase catalyzes key step in turnover of glutathione conjugates. Phytochemistry 62: 423–431 [DOI] [PubMed] [Google Scholar]
- Brennan RJ, Schiestl RH (1996) Cadmium is an inducer of oxidative stress in yeast. Mutat Res 356: 171–178 [DOI] [PubMed] [Google Scholar]
- Cheah E, Austin C, Ashley GW, Ollis D (1993) Substrate-induced activation of dienelactone hydrolase: an enzyme with a naturally occurring Cys-His-Asp triad. Protein Eng 6: 575–583 [DOI] [PubMed] [Google Scholar]
- Clemens S (2001) Molecular mechanisms of plant metal tolerance and homeostasis. Planta 212: 475–486 [DOI] [PubMed] [Google Scholar]
- Clemens S, Kim EJ, Neumann D, Schroeder JI (1999) Tolerance to toxic metals by a gene family of phytochelatin synthases from plants and yeast. EMBO J 18: 3325–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens S, Schroeder JI, Degenkolb T (2001) Caenorhabditis expresses a functional phytochelatin synythase. Eur J Biochem 268: 3640–3643 [DOI] [PubMed] [Google Scholar]
- Cobbett CS (2000) Phytochelatins and their roles in heavy metal detoxification. Plant Physiol 123: 825–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbett C, Goldsbrough P (2002) Phytochelatins and metallothioneins: roles in heavy metal detoxification and homeostasis. Annu Rev Plant Biol 53: 159–182 [DOI] [PubMed] [Google Scholar]
- Elmsley J (2001) Nature's Building Blocks. An A-Z Guide to the Elements. Oxford University Press, Oxford, UK
- Freedman JH, Ciriolo MR, Peisach J (1989) The role of glutathione in copper metabolism and toxicity. J Biol Chem 264: 5598–5605 [PubMed] [Google Scholar]
- Ghosh M, Shen J, Rosen BP (1999) Pathways of As(III) detoxification in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 96: 5001–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill E, Löffler S, Winnacker E-L, Zenk MH (1989) Phytochelatins, the heavy metal-binding peptides of plants are synthesized from glutathione by a specific γ-glutamylcysteine dipeptidyl transpeptidase (phytochelatin synthase). Proc Natl Acad Sci USA 86: 6838–6842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S-B, Smith AP, Howden R, Dietrich WM, Bugg S, O'Connell MJ, Goldsbrough PB, Cobbett CS (1999) Phytochelatin synthase genes from Arabidopsis and the yeast Schizosaccharomyces pombe. Plant Cell 11: 1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC (1984) Iron and free radical reactions: two aspects of antioxidant protection. Trends Biochem Sci 11: 372–375 [Google Scholar]
- Howden R, Goldsbrough PB, Andersen CR, Cobbett CS (1995) Cadmium-sensitive, cad1 mutants of Arabidopsis thaliana are phytochelatin deficient. Plant Physiol 107: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humm A, Fritsche E, Mann K, Gohl M, Huber R (1997. a) Recombinant expression and isolation of human L-arginine:glycine amidinotranferase and identification of its active site cysteine residue. Biochem J 322: 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humm A, Fritsche E, Steinbacher S, Huber R (1997. b) Crystal structure and mechanism of human L-arginine:glycine amidinotranferase: a mitochondrial enzyme involved in creatine biosynthesis. EMBO J 16: 3373–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Hiratake J, Suzuki H, Kumagai H, Sakata K (2000) Identification of catalytic nucleophile of Escherichia coli γ-glutamyltranspeptidase by γ-monofluorophosphono derivative of glutamic acid: N-terminal thr-391 in small subunit is the nucleophile. Biochemistry 39: 7764–7771 [DOI] [PubMed] [Google Scholar]
- Inzé D, Van Montagu M (1995) Oxidative stress in plants. Curr Opin Biotechnol 6: 153–158 [Google Scholar]
- Kamphuis IG, Drenth J, Baker EN (1985) Thiol proteases. Comparative studies based on the high-resolution structures of papain and actinidin, and on amino acid sequence information for cathepsins B and H, and stem bromelain. J Mol Biol 182: 317–329 [DOI] [PubMed] [Google Scholar]
- Kondo N, Imai K, Isobe M, Goto T, Murasugi A, Wada-Nakagawa C, Hayashi Y (1984) Cadystin A and B, major unit peptides comprising cadmium binding peptides induced in a fission yeast—separation, revision of structures and synthesis. Tetrahedron Lett 25: 3869–3872 [Google Scholar]
- Kraut J (1977) Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem 46: 331–358 [DOI] [PubMed] [Google Scholar]
- Lane TW, Morel FM (2000) A biological function for cadmium in marine diatoms. Proc Natl Acad Sci USA 97: 4627–4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z-S, Lu Y-P, Thiele DJ, Rea PA (1997) A new pathway for vacuolar cadmium sequestration in Saccharomyces cerevisiae: YCF1-mediated transport of bis(glutathionato)cadmium. Proc Natl Acad Sci USA 94: 42–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler S, Hochberger A, Grill E, Winnacker E-L, Zenk MH (1989) Termination of the phytochelatin synthase reaction through sequestration of heavy metals by the reaction product. FEBS Lett 258: 42–46 [Google Scholar]
- Maier T, Yu C, Kullertz G, Clemens S (2003) Localization and functional characterization of phytochelatin synthases. Planta 218: 300–308 [DOI] [PubMed] [Google Scholar]
- Maser P, Thomine S, Schroeder JI, Ward JM, Hirschi K, Sze H, Talke IN, Amtmann A, Maathuis FM, Sanders D, et al (2001) Phylogenetic relationships within cation transporter families of Arabidopsis. Plant Physiol 126: 1646–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz DF, Kreppel L, Speiser DM, Scheel G, MacDonald G, Ow DW (1992) Heavy metal tolerance in the fission yeast requires an ATP-binding cassette-type vacuolar membrane transporter. EMBO J 11: 3491–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz DF, Ruscitti T, McCue KF, Ow DW (1995) Transport of metal-binding peptides by HMT1, a fission yeast ABC-type vacuolar membrane protein. J Biol Chem 270: 4721–4728 [DOI] [PubMed] [Google Scholar]
- Oven M, Page JE, Zenk MH, Kutchan TM (2002) Molecular characterization of the homo-phytochelatin synthase of soybean Glycine max. Relation to phytochelatin synthase. J Biol Chem 277: 4747–4754 [DOI] [PubMed] [Google Scholar]
- Rabenstein DL (1989) Metal complexes of glutathione and their biological significance. In D Dolphin, R Poulson, O Avramovic, eds, Glutathione: Chemical, Biochemical and Medical Aspects. John Wiley & Sons, New York, pp 147–186
- Rauser WE (1999) Structure and function of metal chelators produced by plants: the case for organic acids, amino acids, phytin and metallothioneins. Cell Biochem Biophys 31: 19–48 [DOI] [PubMed] [Google Scholar]
- Ruotolo R, Peracchi A, Bolchi A, Infusini G, Amoresano A, Ottonello S (2004) Domain organization of phytochelatin synthase. Functional properties of truncated enzyme species identified by limited proteolysis. J Biol Chem 279: 14686–14693 [DOI] [PubMed] [Google Scholar]
- Rychlewski L, Jaroszewski L, Li W, Godzik A (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9: 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satofuka H, Fukui T, Takagi M, Atomi H, Imanaka T (2001) Metal-binding properties of phytochelatin-related peptides. J Inorg Biochem 86: 595–602 [DOI] [PubMed] [Google Scholar]
- Stadtmann ER (1993) Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem 62: 797–821 [DOI] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18: 321–336 [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Wemmie JA, Moye-Rowley WS, Thiele DJ (1994) A yeast metal resistance protein similar to human cystic fibrosis transmembrane conductance regulator (CFTR) and multidrug resistance-associated protein. J Biol Chem 269: 22853–22857 [PubMed] [Google Scholar]
- Tsuji N, Nishikori S, Iwabe O, Shiraki K, Miyasaka H, Takagi M, Hirata K, Miyamoto K (2004) Characterization of phytochelatin synthase-like protein encoded by alr0975 from a prokaryote, Nostoc sp. PCC 7120. Biochem Biophys Res Commun 315: 751–755 [DOI] [PubMed] [Google Scholar]
- Vatamaniuk OK, Bucher EA, Rea PA (2002) Worms take the ‘phyto’ out of ‘phytochelatins’. Trends Biotechnol 20: 61–64 [DOI] [PubMed] [Google Scholar]
- Vatamaniuk OK, Bucher EA, Ward JT, Rea PA (2001) A new pathway for heavy metal detoxification in animals: phytochelatin synthase is required for cadmium tolerance in Caenorhabditis elegans. J Biol Chem 276: 20817–20820 [DOI] [PubMed] [Google Scholar]
- Vatamaniuk OK, Mari S, Lang A, Chalasani S, Demkiv L, Rea PA (2004) Phytochelatin synthase, a dipeptidyltransferase that undergoes multisite acylation with γ-glutamycysteine during catalysis. Stoichiometric and site-directed mutagenic analysis of Arabidopsis thaliana PCS1-catalyzed phytochelatin synythesis. J Biol Chem 279: 22449–22460 [DOI] [PubMed] [Google Scholar]
- Vatamaniuk OK, Mari S, Lu Y-P, Rea PA (1999) AtPCS1, a phytochelatin synthase from Arabidopsis thaliana: isolation and in vitro reconstitution. Proc Natl Acad Sci USA 96: 7110–7115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatamaniuk OK, Mari S, Lu Y-P, Rea PA (2000) Mechanism of heavy metal activation of phytochelatin (PC) synthase: blocked thiols are sufficient for PC synthase-catalyzed transpeptidation of glutathione and related thiol peptides. J Biol Chem 275: 31451–31459 [DOI] [PubMed] [Google Scholar]
- Voet D, Voet JG (2004) Biochemistry, Ed 3. John Wiley & Sons, New York
- Zenk MH (1996) Heavy metal detoxification in higher plants: a review. Gene 179: 21–30 [DOI] [PubMed] [Google Scholar]