

A novel “smart” separator with thermal-triggered flame-retardant properties for lithium-ion batteries to improve their safety.
Keywords: Lithium ion batteries, flame-retardant, smart separator, thermal responsive
Abstract
Although the energy densities of batteries continue to increase, safety problems (for example, fires and explosions) associated with the use of highly flammable liquid organic electrolytes remain a big issue, significantly hindering further practical applications of the next generation of high-energy batteries. We have fabricated a novel “smart” nonwoven electrospun separator with thermal-triggered flame-retardant properties for lithium-ion batteries. The encapsulation of a flame retardant inside a protective polymer shell has prevented direct dissolution of the retardant agent into the electrolyte, which would otherwise have negative effects on battery performance. During thermal runaway of the lithium-ion battery, the protective polymer shell would melt, triggered by the increased temperature, and the flame retardant would be released, thus effectively suppressing the combustion of the highly flammable electrolytes.
INTRODUCTION
Lithium-ion batteries are considered to be one of the most promising power sources of electric vehicles because of their high specific energy densities, stable cycling performance, and other related qualities (1–4). Although the energy densities of batteries continue to increase, safety problems remain a big issue, significantly hindering their further practical applications (5–7). It has been generally recognized that the safety of lithium-ion batteries is closely associated with the highly flammable liquid organic electrolytes, for example, ethylene carbonate (EC) and diethyl carbonate (DEC) (5–8). In the case of internal or external short circuits, undesirable exothermic reactions may lead to a rapid rise in internal temperature and to thermal runaway. The flammable liquid electrolytes would be ignited, eventually leading to fire and battery explosion. With the next generation of high-capacity electrode materials for high-energy batteries (9–11), the safety issue becomes even more critical. Considerable efforts have been devoted to solving this problem, such as by replacing the existing flammable electrolyte with nonflammable ones (12–19) or using flame-retardant separators (20–22), detecting the dendrite via a smart separator for early warning (23), coating the separator with a ceramic layer (24–26), thermal-switching the current collector (27), and autonomic shutdown of lithium-ion batteries using thermoresponsive microspheres, among others (28, 29). However, the risk of battery fire still exists, particularly in cases of local heat spot generation, severe battery extrusion, and other causes. Moreover, battery performance is usually sacrificed in terms of decreased ionic conductivity and energy density. Another straightforward method to reducing the risk of fire and explosion is to add flame-retardant additives into the existing electrolytes (30–36). These additives are generally phosphorus- or halogen-based molecules, which show flame retardancy via either a physical isolation mechanism or a chemical free radical scavenging process (37). However, to achieve considerable nonflammability, a large amount of flame retardant is generally added into the electrolytes, which consequently decreases the ionic conductivity of the electrolytes and significantly deteriorates the electrochemical performance of lithium-ion batteries.
Here, we have fabricated a novel electrospun core-shell microfiber separator with thermal-triggered flame-retardant properties for lithium-ion batteries. As shown in Fig. 1, a free-standing separator was composed of microfibers fabricated by electrospinning. The microfibers exhibit a core-shell structure, where the triphenyl phosphate (TPP), a popular organophosphorus-based flame retardant, is the core and poly(vinylidene fluoride–hexafluoropropylene) (PVDF-HFP) is the shell. The encapsulation of TPP inside the PVDF-HFP protective polymer shell (TPP@PVDF-HFP) has prevented direct exposure of the flame retardant to the electrolyte and has largely slowed down its dissolution, preventing negative effects from the retardants on the electrochemical performance of the battery (Fig. 1A). Moreover, if thermal runaway of the lithium-ion battery happens, the PVDF-HFP polymer shell will melt as temperature increases and then the encapsulated TPP flame retardant will be released into the electrolyte, thus effectively suppressing the combustion of the highly flammable electrolytes (Fig. 1B). We chose PVDF-HFP as the protective shell on the basis of the following considerations: (i) It is insoluble in common electrolytes for lithium-ion batteries, for example, EC/DEC; thus, the polymer protective shell would not dissolve when the normal battery is running; (ii) PVDF-HFP exhibits a relatively low melting point (~160°C), such that it can be melted before or at the early stage of combustion; and (iii) it is inert and stable within the reductive/oxidative electrochemical environment inside the battery. The TPP was chosen as the flame retardant because it is a popularly used (not to mention cheap and efficient) phosphorus-based flame retardant. Moreover, the air pollution level after combustion is much lower than that when halogen-based flame retardants are used. With this smart and adaptive material (38) in the battery, we do not have to make a trade-off between the electrolyte nonflammability and the electrochemical performance of the battery.
Fig. 1. Schematic of the “smart” electrospun separator with thermal-triggered flame-retardant properties for lithium-ion batteries.
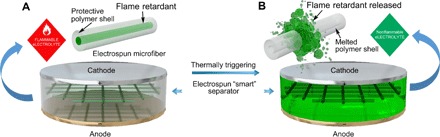
(A) The free-standing separator is composed of microfibers with a core-shell structure, where the flame retardant is the core and the polymer is the shell. The encapsulation of the flame retardant inside the protective polymer shell has prevented direct exposure and dissolution of the flame retardant into the electrolyte, preventing their negative effects on the electrochemical performance of the battery. (B) Upon thermal triggering, the polymer shell would melt and then the encapsulated flame retardant would be released into the electrolyte, thus effectively suppressing the ignition and burning of the electrolytes.
RESULTS
The effect of TPP on battery performances
The efficiency of TPP in suppressing the flammability of the electrolyte was studied first. The electrolyte studied here was 1.0 M LiPF6 in EC/DEC (1:1, w/w), which is commonly used in lithium-ion batteries. As shown in Fig. 2A and movie S1, the EC/DEC electrolyte is highly flammable. It is easily ignited and then combusts violently. However, in the presence of TPP with a concentration of 40 weight % (wt %) in EC/DEC, the flame quickly self-extinguishes, as shown in Fig. 2B and movie S2. To more quantitatively study the flame-retardant property of TPP, we measured the self-extinguishing time (SET) (18) of the electrolytes, which was obtained by normalizing the flame combustion time against the electrolyte mass. As shown in Fig. 2C, the pristine electrolyte, that is, 1 M LiPF6 in EC/DEC (1:1, w/w), is highly flammable with a SET of ~100 s/g. As TPP is added into the electrolyte, the SET of the electrolyte gradually decreases, indicating that the flammability of the electrolyte is drastically reduced as the concentration of TPP increases. The SET value even decreased to near zero when the concentration of TPP increased to 40 wt %. The free radical scavenging mechanism has mainly been suggested as the mechanism for the flame-retardant property of TPP (37). TPP can generate phosphorus-containing free radicals, for example, PO• and PO2•, which can actively capture the H• and HO• radicals emitted by the burning electrolyte so that it can weaken or terminate combustion chain branching reactions, therefore retarding the combustion.
Fig. 2. The influence of TPP on the flammability of the electrolyte and the electrochemical performance of the graphite anode.
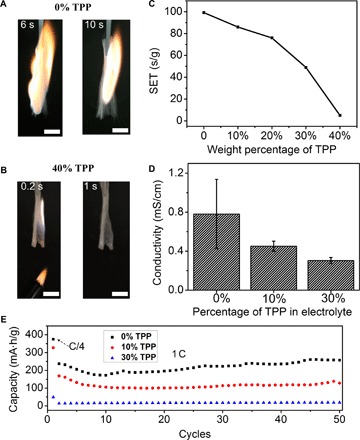
The electrolyte was 1.0 M LiPF6 in EC/DEC (1:1, w/w). Photographs recording the burning of the electrolyte in the presence of (A) 0 wt % and (B) 40 wt % TPP. The respective times, counted from the time when the electrolyte started to burn, are indicated in each picture. Scale bars, 1 cm. (C) SET and (D) ionic conductivities of the electrolyte with different concentrations of TPP. (E) Delithiation capacity of the graphite anode during galvanostatic cycling between 0.01 and 1.5 V. The rate was 0.25 C for the first cycle and 1 C for subsequent cycles. The electrolyte was 1.0 M LiPF6 in EC/DEC (1:1, w/w) in the presence of different concentrations of TPP, which are indicated in the figure.
Although it is efficient in reducing the flammability of the electrolytes, the direct addition of TPP into the electrolyte has severe negative effects on ionic conductivity and battery performance. As the concentration of TPP increases, the ionic conductivity of the electrolyte significantly decreases (Fig. 2D), possibly because of increased viscosity (35). The effects of TPP on the performance of the graphite anode, a popularly used anode in commercial lithium-ion batteries, were tested in a coin cell, where the graphite was used as the working electrode and Li metal was used as both the counter electrode and the reference electrode. In the electrolyte [1 M LiPF6 in EC/DEC (1:1, w/w)], the graphite shows a specific capacity of ~218 mA·hour/g in the first 50 cycles at a galvanostatic charging/discharging rate of 1 C (1 C = charge/discharge in 1 hour). However, as the concentration of TPP in the electrolyte increases, the specific capacity of graphite decreases accordingly, as indicated in Fig. 2E. For example, when the concentration of TPP is 10 wt %, the specific capacity decreased to ~115 mA·hour/g. As the concentration increased to 30 wt %, the specific capacity further decreased to ~17 mA·hour/g. Considering that TPP is electrochemically stable on graphite (fig. S1), the decreased specific capacity should be ascribed to the lowered ionic conductivity of the electrolytes in the presence of TPP. Thus, it is highly desirable and necessary to encapsulate the flame retardant TPP into a protective polymer shell to avoid its negative effects on the battery.
The fabrication and characterization of TPP@PVDF-HFP fibers
To fabricate the desired TPP@PVDF-HFP fiber, TPP and PVDF-HFP were dissolved with a weight ratio of 1:1 in a solvent mixture of dimethylacetamide and acetone (3:7, w/w). Then, the solution was placed into a syringe and used directly for electrospinning (Fig. 3A). Microfibers were successfully obtained, as indicated by the scanning electron microscopy (SEM) image shown in Fig. 3B. To determine the chemical compositions of the microfibers, we carried out energy-dispersive x-ray (EDX) spectrum characterization. As shown in Fig. 3C, the peaks corresponding to the C, O, F, and P elements can be identified, respectively, indicating the coexistence of PVDF-HFP and TPP in the microfibers. Thermogravimetric analysis (TGA) was used to obtain further quantitative information about the composition of the microfiber, as shown in Fig. 3D. The TGA curves reveal that the microfibers show a substantial weight loss starting at ~200°C and ending at ~330°C. For comparison, TGA of TPP alone was also conducted under the same heating condition, as indicated by the blue dashed line in Fig. 3D. The weight loss starts at ~150°C and ends at ~260°C. It should be noted that the weight loss process of TPP in the TPP@PVDF-HFP composite microfiber shows some hysteresis compared with that of the pure TPP, as reflected from the higher starting and ending temperatures. It indicates that the TPP has been encapsulated inside the PVDF-HFP polymer shell; as a result of which the diffusion and evaporation of TPP become sluggish. Further heating leads to a second weight loss starting at ~400°C, which is the same with the TGA curve of pure PVDF-HFP (red dotted line in Fig. 3D). The first and second weight loss account for ~50% of the total sample. This suggests that the weight ratio of TPP in the microfiber is 50%, in accordance with the 1:1 weight ratio of TPP and PVDF-HFP in the starting solution for electrospinning.
Fig. 3. The fabrication and characterization of the TPP@PVDF-HFP microfibers.
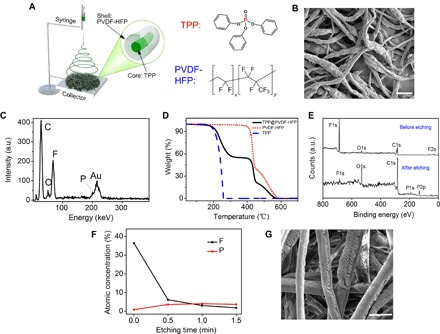
(A) Schematic illustration for the fabrication of the microfibers by electrospinning. (B) SEM image. Scale bar, 5 μm. (C) EDX and (D) TGA measurements of the as-spun fibers. A thin conducting layer of gold was coated onto the fibers for SEM observations. The Au peak in EDX was attributed to the gold layer on the sample. a.u., arbitrary units. (E) The XPS data of the fiber before and after etching (0.5 min). (F) AES sputter depth profiling of the fiber with different etching times. (G) SEM image of the TPP@PVDF-HFP microfibers after etching clearly shows their core-shell structure. Scale bar, 5 μm.
X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) sputter depth profiling were used to further study the microstructure of the microfibers. As shown in Fig. 3E, strong peaks corresponding to elements F and C can be detected, indicating the existence of PVDF-HFP on the surface of the fibers. No peaks corresponding to P were observed, indicating that the amount of TPP on the surface of the microfibers is negligible. The existence of the weak O1s peak is probably attributed to the presence of trace moisture and other oxides in the sample. However, after sputter etching for 0.5 min, the peaks corresponding to P1s and P2p appear, which suggests that the TPP molecules are embedded inside the PVDF-HFP shell. The relative atomic concentrations of elements F and P were then plotted against the sputter etching time. On the surface of the pristine fiber before sputtering, the atomic concentration of F is measured to be 36%, whereas the value sharply decreases to below 5% after sputtering. In contrast, the atomic concentration of P increases from ~0 to ~4% after sputtering, as shown in Fig. 3F. The data clearly indicate that the TPP molecules are encapsulated inside a PVDF-HFP shell of the microfibers rather than exposed on its surface. The sputter etching process on the fibers allows us to directly observe their inner structure, as can be seen in the SEM image in Fig. 3G. Nanoflakes stacking inside the fibers can be clearly observed. The nanoflakes should be formed by the TPP molecules, which are prone to form flake-like crystals (fig. S2). Moreover, there is a thin coating layer on the surface of fibers, which acts as the shell protecting the nanoflakes. The formation of the TPP@PVDF-HFP core-shell microstructure during electrospinning should be ascribed to the following: (i) The solubility difference between PVDF-HFP and TPP in the solvent, in which the TPP shows much higher solubility compared with PVDF-HFP [the saturated concentration of TPP in the mixed solvent, that is, dimethylacetamide and acetone (3:7, w/w), is ~2.9 g/ml, whereas the value is only 0.2 g/ml for PVDF-HFP]. Thus, as the solvent gradually evaporates during electrospinning, the PVDF-HFP could precipitate much earlier than TPP, staying at the surface of the microfiber as the shell, whereas the TPP is still soluble in the remaining solvent and left in the core of the fibers. (ii) In the Taylor cone during electrospinning, the strong electronic field may induce the migration of high polar PVDF-HFP polymer chains toward the interface between the liquid solution and air (39). (iii) The low surface energy of PVDF-HFP and phase separation may cause the formation of a shell layer composed of PVDF-HFP (40). The effects of the thickness of the PVDF-HFP shell and the diameter for the individual fibers within the TPP@PVDF-HFP membrane are studied and discussed in the Supplementary Materials (see fig. S3 and related discussion).
Mechanical property and electrochemical cycling with the TPP@PVDF-HFP separator
Good separator flexibility is critical in battery manufacturing, which requires flexibility for either folding or rolling processes in both pouch and cylindrical cell configurations. The as-spun TPP@PVDF-HFP fiber mat is highly flexible. As shown in fig. S4A, the membrane is coiled around a metal rod to illustrate good flexibility. Also, the membrane shows great bendability (fig. S4B), and there are no cracks formed after bending. In addition, the TPP@PVDF-HFP membrane is quite strong, as indicated by the stress-strain curve in fig. S5. Thus, the TPP@PVDF-HFP membrane is suitable for use as a separator of batteries.
The successful encapsulation of TPP inside the PVDF-HFP polymer shell has significantly decreased the negative effects of TPP on the performance of the graphite anodes. The TPP@PVDF-HFP membrane was pouched into a free-standing round-shaped membrane (Fig. 4B and fig. S6), the thickness of which was measured to be ~40 μm (fig. S7). The TPP@PVDF-HFP fiber membrane was then incorporated into coin cells as the separator, and graphite was used as the working electrode and Li metal was used as the counter electrode and reference electrode. The electrolyte was 1 M LiPF6 in EC/DEC (1:1, w/w). Upon cycling, the graphite anode exhibited a high specific capacity of ~212 mA·hour/g, on average, in the first 70 cycles at a galvanostatic charging/discharging rate of 1 C, which is similar to that of the batteries using commercial polyethylene (PE) separators (~233 mA·hour/g), as shown in Fig. 4A. Assuming that all of the TPP molecules encapsulated inside the separator are dissolved in the electrolyte, the TPP concentration should be ~30 wt % (see Materials and Methods). In such a high concentration, TPP would severely deteriorate the performance of the graphite anode, as has been discussed in Fig. 2E and shown in Fig. 4A. However, the successful encapsulation of TPP inside the polymer shell is efficient in avoiding its negative effects on the graphite anodes. In addition, the voltage profiles exhibited the typical electrochemical features of graphite (Fig. 4, C and D). The shape of the profile does not change when PE separators (Fig. 4C) are replaced with TPP@PVDF-HFP separators (Fig. 4D and fig. S8). In contrast, 30 wt % TPP in the electrolyte will result in a large overpotential and a much lower capacity (Fig. 4E). In addition, the electrospun PVDF-HFP separator was also used as a control, and we directly dissolved the TPP into the EC/DEC electrolyte to a concentration of 30 wt %. Under this condition, the delithiation capacities of the graphite anode during galvanostatic cycling are compared in fig. S9. In great contrast to the TPP@PVDF-HFP separator where TPP was encapsulated inside the PVDF-HFP shell, the TPP directly dissolved in the electrolyte severely deteriorated the performance of the graphite anode. Thus, the TPP@PVDF-HFP separator did not change the electrochemical behaviors of the graphite anode; and so, the encapsulation of TPP inside the PVDF-HFP polymer shell is an efficient way to eliminate its negative effect on the graphite anode.
Fig. 4. Electrochemical performances of the graphite anode using different combinations of separators and electrolytes.
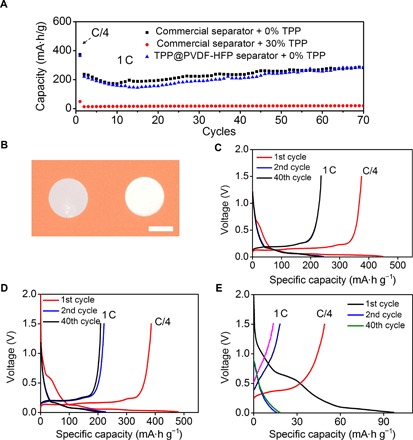
(A) Delithiation capacities of the graphite anode during galvanostatic cycling between 0.01 and 1.5 V. The rate was 0.25 C for the first cycle and 1 C for subsequent cycles. (B) The digital photographs of the commercial PE separator (left) and the free-standing TPP@PVDF-HFP separator (right). Scale bar, 1 cm. Galvanostatic charge/discharge voltage profiles for the graphite anode plotted for the 1st, 2nd, and 40th cycles. Different combinations of electrolytes and separators were used in (C), (D), and (E): (C) pristine EC/DEC electrolyte + PE separator, (D) pristine EC/DEC electrolyte + TPP@PVDF-HFP separator, and (E) electrolyte containing 30 wt % TPP + PE separator. The rate was 0.25 C for the first cycle and 1 C for subsequent cycles.
It should be noted that the PVDF-HFP shell can absorb the electrolyte and swell a little bit during long-term cycling, causing a small amount of TPP to gradually diffuse out of the fibers and dissolve into the electrolyte. However, this has not significantly affected the electrochemical performance of batteries because of the low concentration of TPP in the electrolyte (for detailed discussions, see fig. S10).
It is equally important to show the voltage stability of the separators at the high voltages by which the typical cathodes operate. As shown in fig. S11, the electrochemical high voltage (up to 4.5 V) stability of the TPP@PVDF-HFP separator was confirmed by cyclic voltammetry (CV), indicating the good stability of the TPP@PVDF-HFP separator toward typical cathode materials. In addition, we also tested the electrochemical cycling stability and voltage profiles of a typical cathode, LiCoO2 (LCO) half cells, using the TPP@PVDF-HFP separator. For comparison, we also tested the cells using the commercial PE separator. As shown in fig. S12, the electrochemical behaviors of the LCO are not affected by the TPP@PVDF-HFP separator, indicating the good voltage stability of the separator toward the cathode.
Flame-retardant property study
The TPP@PVDF-HFP separator has significantly improved the flame-retardant property of the electrolyte. The response of the TPP@PVDF-HFP separator upon thermal stimuli was first studied by differential scanning calorimetry (DSC). As shown in Fig. 5A, there are two endothermic peaks located at ~50° and ~150°C, corresponding to the melting points (Tm) of TPP and PVDF-HFP, respectively (fig. S13). Above the melting point of the polymer shell, the fibers are melted, and the encapsulated TPP is released and exposed, as indicated by the SEM observation shown in fig. S14. We further used ultraviolet-visible (UV/Vis) absorbance spectrum to quantitatively monitor the TPP release behavior upon thermal triggering. When dissolved in the electrolyte, TPP exhibits three explicit absorption bands whose peaks are located at 266, 260, and 255 nm. This facilitates us to quantitatively estimate the amount of TPP that has been released into the electrolyte. As shown in Fig. 5B, when the TPP@PVDF-HFP fiber was soaked in the EC/DEC electrolyte and stored at room temperature (~25°C), the release of TPP into the electrolyte was only ~4%. However, upon heating up to 160°C, above the melting point of PVDF-HFP, all of the encapsulated TPP (~100%) was abruptly released into the electrolyte (Fig. 5C). Thus, during thermal triggering, the PVDF-HFP polymer shell of the microfibers melted as the temperature increased above its melting point, facilitating the flame retardant to be released into the electrolyte and retarding/extinguishing the combustion. To demonstrate the flame-retardant property upon thermal triggering, we tested the flammability of the EC/DEC electrolytes in the presence of the TPP@PVDF-HFP separator. In doing so, the TPP@PVDF-HFP separator was wetted by 100 μl of the pristine EC/DEC electrolyte, mounted vertically, and ignited by a direct flame of a lighter. As shown in Fig. 5 (D to F) and movie S3, the flames of the electrolyte diminished rapidly and were completely extinguished within 0.4 s. The SET value of the electrolyte was calculated to be only ~3 s/g. Thus, after thermal stimuli, the separator fabricated by TPP@PVDF-HFP fibers is effective in suppressing the flammability of the EC/DEC electrolytes.
Fig. 5. Study on the flame-retardant property of TPP.
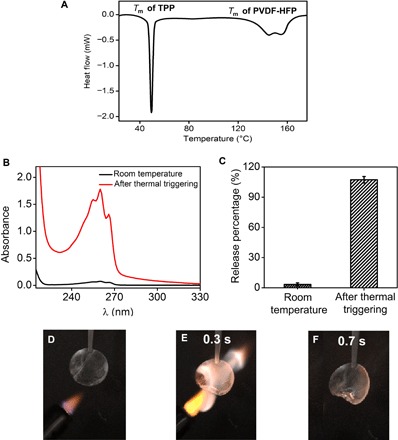
(A) DSC of the TPP@PVDF-HFP separator. (B) The UV/Vis spectrum of TPP that has been dissolved in the EC/DEC electrolyte before and after the TPP@PVDF-HFP was heated up to 160°C. (C) The percentage of TPP being released into the electrolyte before and after thermal triggering at 160°C. The calculated percentage after thermal triggering is slightly above 100%, possibly because of the evaporation of solvent during heating. (D to F) Digital photographs showing the flammability of the TPP@PVDF-HFP separator wetted by the electrolyte. The respective times, counted from the time when the electrolyte started to burn, are indicated in each picture. The diameter of the separator is 1.6 cm.
DISCUSSION
We have fabricated a novel smart electrospun separator with thermal-triggered flame-retardant properties for lithium-ion batteries. The encapsulation of TPP inside a protective polymer shell has prevented direct dissolution of the retardant agent into the electrolyte, which would otherwise have negative effects on battery performance. Thermally triggered melting of the PVDF-HFP polymer shell would release the flame retardant, thus effectively suppressing the combustion of the highly flammable electrolytes under thermal runaway conditions of the lithium-ion battery. It is anticipated that this type of smart separator can be used in other high-energy storage devices, which may encounter thermal runaway safety issues. In the future, mechanical (nail penetration test or crush test) or electrical abuse (overcharge or overdischarge) tests involving large-format cells will be needed for further practical applications.
MATERIALS AND METHODS
Materials synthesis and fabrication
To fabricate the TPP@PVDF-HFP fiber by electrospinning, TPP and PVDF-HFP (Mw = 455,000) were dissolved with a weight ratio of 1:1 in a solvent mixture of dimethylacetamide and acetone (3:7, w/w). A transparent solution was obtained. The concentrations of TPP and PVDF-HFP were both 16 wt %. Then, the solution was placed into a syringe with a stainless steel needle. We used a commercial high-voltage source (ES-30P-5W, Gamma High Voltage Research) for electrospinning. A voltage of 13 kV was applied to the solution to start the spinning process, and the electrospun fibers were collected in a random mat of approximately 10 cm × 10 cm.
Electrochemistry
To make the graphite electrode, a slurry method was used. Graphite powders were mixed with carbon black and PVDF with a ratio of 8:1:1. Then, N-methylpyrrolidone was added as solvent, and stirring was performed overnight. Next, the slurry was cast on copper foil, dried at room temperature, and punched into 1-cm2 electrodes. To guarantee that the electrodes were fully dried, they were kept in a vacuum oven at 100°C for 4 to 5 hours and then kept in an argon-filled glove box for 1 day. Coin cells (2032) were assembled for electrochemical testing. Li metal foil was used as the counter electrode and reference electrode. The rate capability was calculated on the basis of the theoretical capacity of graphite (1 C = 0.372 mA/mg), whereas the specific capacity was calculated on the basis of the mass of graphite. Separators (Celgard 2325) soaked with EC/DEC electrolytes (BASF Selectilyte LP40) were sandwiched by stainless steel electrodes in 2032-type coin cells. The LCO cathode was purchased from MTI and used as received. The battery assembling process is similar to that of graphite. CV measurements were carried out on a BioLogic VMP3 system. For the graphite anode in different electrolytes, the CV was scanned from 2.0 to 0.01 V versus Li/Li+ at a scan rate of 0.05 mV/s. For the CV testing of the electrochemical stability of the TPP@PVDF-HFP separator, Li metal foil was used as the counter electrode and reference electrode. Stainless steel was used as the working electrode. The electrolyte used was 1.0 M LiPF6 in EC/DEC (1:1, w/w).
For the batteries using the TPP@PVDF-HFP separator, the electrospun TPP@PVDF-HFP mat was punched into a round shape with a diameter of 1.6 cm. The mass of each separator was ~40 mg, and ~50 μl of electrolyte was used to wet the separator for each battery.
Characterization
SEM images were taken on an FEI XL30 Sirion. UV/Vis spectroscopy was measured using a Cary 6000i UV-Vis-NIR spectrometer. XPS was carried out on an SSI S-Probe Monochromatized XPS spectrometer with Al Kα radiation at 1486 eV. TGA was performed on a TA Instrument Q500 with a heating rate of 5°C/min. The measurement was performed under simulated air atmosphere (20% O2 + 80% Ar). DSC was measured on a TA Instrument Q2000 with a heating rate of 5°C/min under nitrogen.
The swelling extent of the separator in the standard EC/DEC electrolyte was characterized by the swelling ratio (Q). The separator (original weight, m0) was soaked in the electrolyte [1.0 M LiPF6 in EC/DEC (1:1, w/w)] overnight. The separator became transparent. Then, the separator was taken out from the electrolyte solution; the additional solution on the surface and inside the holes was removed by squeezing the separator and by paper absorption until the weight of the separator (m1) did not change. The swelling ratio was calculated as follows: Q(wt %) = (m1/m0 − 1) × 100%.
The SET was used to quantitatively estimate the flammability of the electrolytes. It was obtained by igniting the preweighed electrolyte (~0.1 g) soaked in a wick fabricated by glass fibers. The electrolyte was exposed to a direct flame from a lighter. After the electrolyte was ignited, the lighter was removed. Then, the time for the flame to self-extinguish was recorded and then normalized by the electrolyte mass, obtaining the SET of the electrolyte. For the testing of the SET in Fig. 5, the TPP@PVDF-HFP separator was wetted by the preweighed electrolyte (~0.1 g), vertically mounted, and then ignited using a lighter.
The TPP release behavior upon thermal triggering was monitored by UV/Vis spectroscopy. TPP@PVDF-HFP fibers (2 mg) were soaked in the 1.0 M LiPF6 in EC/DEC (1:1, w/w) electrolyte (2 ml) in a vial. Then, the vial was heated up to 160°C with a heating rate of 20°C/min and rested for 1 min, after which the vial was taken out. After the solution was cooled down to room temperature, it was used for UV/Vis spectroscopy measurement.
Supplementary Material
Acknowledgments
Funding: We acknowledge the support from the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies, Battery Materials Research Program of the U.S. Department of Energy. Author contributions: K.L. and Y.C. conceived the idea and designed the experiments. K.L. and W.L. synthesized the materials and performed electrochemical tests. Y.Q. and other authors participated in materials characterization. K.L. and Y.C. co-wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1601978/DC1
fig. S1. Cyclic voltammogram for the graphite anode in different electrolytes.
fig. S2. A digital photograph showing that the TPP molecules are flake-like crystals.
fig. S3. SEM image of the TPP@PVDF-HFP fibers.
fig. S4. Digital pictures showing the (A) flexibility and (B) bendability of the TPP@PVDF-HFP membranes.
fig. S5. The stress-strain curve of the TPP@PVDF-HFP membrane.
fig. S6. SEM image of the TPP@PVDF-HFP membrane.
fig. S7. SEM cross-sectional image of the TPP@PVDF-HFP separator.
fig. S8. Voltage profiles of the graphite anode using the PE (black curve) separator and the TPP@PVDF-HFP separator (blue curve).
fig. S9. Electrochemical performances of the graphite anode using TPP@PVDF-HFP separators and PE separators with different electrolytes.
fig. S10. TGA measurements of the TPP@PVDF-HFP separator after long term cycling and the electrochemical performance of related battery.
fig. S11. Electrochemical stability of the TPP@PVDF-HFP separator.
fig. S12. The electrochemical stability of the TPP@PVDF-HFP separator towards LCO cathode.
fig. S13. The measurements on the Tm of TPP and PVDF-HFP.
fig. S14. SEM image of the TPP@PVDF-HFP after heat treatment.
movie S1. The combustion of the EC/DEC electrolyte.
movie S2. The combustion of the EC/DEC electrolyte with 40 wt % TPP.
movie S3. The combustion of the TPP@PVDF-HFP separator wetted by the EC/DEC electrolyte.
REFERENCES AND NOTES
- 1.Tarascon J.-M., Armand M., Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Goodenough J. B., Kim Y., Challenges for rechargeable Li batteries. Chem. Mater. 22, 587–603 (2010). [Google Scholar]
- 3.Armand M., Tarascon J.-M., Building better batteries. Nature 45, 652–657 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Choi J. W., Aurbach D., Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 1, 16013 (2016). [Google Scholar]
- 5.Lu L., Han X., Li J., Hua J., Ouyang M., A review on the key issues for lithium-ion battery management in electric vehicles. J. Power Sources 226, 272–288 (2013). [Google Scholar]
- 6.Balakrishnan P. G., Ramesh R., Prem Kumar T., Safety mechanisms in lithium-ion batteries. J. Power Sources 155, 401–414 (2006). [Google Scholar]
- 7.Jansen A. N., Kahaian A. J., Kepler K. D., Nelson P. A., Amine K., Dees D. W., Vissers D. R., Thackeray M. M., Development of a high-power lithium-ion battery. J. Power Sources 81–82, 902–905 (1999). [Google Scholar]
- 8.Wang Q., Ping P., Zhao X., Chu G., Sun J., Chen C., Thermal runaway caused fire and explosion of lithium ion battery. J. Power Sources 208, 210–224 (2012). [Google Scholar]
- 9.Sun Y. M., Liu N., Cui Y., Promises and challenges of nanomaterials for lithium-based rechargeable batteries. Nat. Energy 1, 16071 (2016). [Google Scholar]
- 10.Yang Y., Zheng G. Y., Cui Y., Nanostructured sulfur cathodes. Chem. Soc. Rev. 42, 3018–3032 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Bruce P. G., Freunberger S. A., Hardwick L. J., Tarascon J.-M., Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Xu K., Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303−4417 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Zhang S. S., A review on electrolyte additives for lithium-ion batteries. J. Power Sources 162, 1379–1394 (2006). [Google Scholar]
- 14.Nakagawa H., Fujino Y., Kozono S., Katayama Y., Nukuda T., Sakaebe H., Matsumoto H., Tatsumi K., Application of nonflammable electrolyte with room temperature ionic liquids (RTILs) for lithium-ion cells. J. Power Sources 174, 1021–1026 (2007). [Google Scholar]
- 15.Wang J. H., Yamada Y., Sodeyama K., Chiang C. H., Tateyama Y., Yamada A., Superconcentrated electrolytes for a high-voltage lithium-ion battery. Nat. Commun. 7, 12032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J., Lin F., Jia H., Yang J., Monroe C. W., NuLi Y., Towards a safe lithium-sulfur battery with a flame-inhibiting electrolyte and a sulfur-based composite cathode. Angew. Chem. Int. Ed. 53, 10099–10104 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Arai J., A novel non-flammable electrolyte containing methyl nonafluorobutyl ether for lithium secondary batteries. J. Appl. Electrochem. 32, 1071–1079 (2002). [Google Scholar]
- 18.Xu K., Ding M. S., Zhang S. S., Allen J. L., Jow T. R., An attempt to formulate nonflammable lithium ion electrolytes with alkyl phosphates and phosphazenes. J. Electrochem. Soc. 149, A622–A626 (2002). [Google Scholar]
- 19.Wonga D. H. C., Thelen J. L., Fu Y. B., Devaux D., Pandya A. A., Battaglia V. S., Balsara N. P., DeSimone J. M., Nonflammable perfluoropolyether-based electrolytes for lithium batteries. Proc. Natl. Acad. Sci. U.S.A. 111, 3327–3331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J., Yue L., Kong Q., Liu Z., Zhou X., Zhang C., Xu Q., Zhang B., Ding G., Qin B., Duan Y., Wang Q., Yao J., Cui G., Chen L., Sustainable, heat-resistant and flame-retardant cellulose-based composite separator for high-performance lithium ion battery. Sci. Rep. 4, 3935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeon D., Lee Y. J., Ryou M.-H., Lee Y. M., New flame-retardant composite separators based on metal hydroxides for lithium-ion batteries. Electrochim. Acta 157, 282–289 (2015). [Google Scholar]
- 22.Arora P., Zhang Z. M., Battery separators. Chem. Rev. 104, 4419–4462 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Wu H., Zhuo D., Kong D. S., Cui Y., Improving battery safety by early detection of internal shorting with a bifunctional separator. Nat. Commun. 5, 5193 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Kim J.-H., Kim J.-H., Kim J.-M., Lee Y.-G., Lee S.-Y., Superlattice crystals–mimic, flexible/functional ceramic membranes: Beyond polymeric battery separators. Adv. Energy Mater. 5, 1500954 (2015). [Google Scholar]
- 25.Park J.-H., Cho J.-H., Park W., Ryoo D., Yoon S.-J., Kim J. H., Jeong Y. U., Lee S.-Y., Close-packed SiO2/poly(methyl methacrylate) binary nanoparticles-coated polyethylene separators for lithium-ion batteries. J. Power Sources 195, 8306–8310 (2010). [Google Scholar]
- 26.Kang S. M., Ryou M. H., Choi J. W., Lee H., Mussel- and diatom-inspired silica coating on separators yields improved power and safety in Li-ion batteries. Chem. Mater. 24, 3481–3485 (2012). [Google Scholar]
- 27.Chen Z., Hsu P.-C., Lopez J., Li Y., To J. W. F., Liu N., Wang C., Andrews S. C., Liu J., Cui Y., Bao Z., Fast and reversible thermoresponsive polymer switching materials for safer batteries. Nat. Energy 1, 15009 (2016). [Google Scholar]
- 28.Baginska M., Blaiszik B. J., Merriman R. J., Sottos N. R., Moore J. S., White S. R., Autonomic shutdown of lithium-ion batteries using thermoresponsive microspheres authors. Adv. Energy Mater. 2, 583–590 (2012). [Google Scholar]
- 29.Shi C., Zhang P., Huang S., He X., Yang P., Wu D. Z., Sun D., Zhao J., Functional separator consisted of polyimide nonwoven fabrics and polyethylene coating layer for lithium-ion batteries. J. Power Sources 298, 158–165 (2015). [Google Scholar]
- 30.Doughty D. H., Rotha E. P., Craftsa C. C., Nagasubramaniana G., Henriksenb G., Amine K., Effects of additives on thermal stability of Li ion cells. J. Power Sources 146, 116–120 (2005). [Google Scholar]
- 31.Hyung Y. E., Vissers D. R., Amine K., Flame-retardant additives for lithium-ion batteries. J. Power Sources 119–121, 383–387 (2003). [Google Scholar]
- 32.Yao X. L., Xie S., Chen C. H., Wang Q. S., Sun J. H., Li Y. L., Lu S. X., Comparative study of trimethyl phosphite and trimethyl phosphate as electrolyte additives in lithium ion batteries. J. Power Sources 144, 170–175 (2005). [Google Scholar]
- 33.Izquierdo-Gonzales S., Li W., Lucht B. L., Hexamethylphosphoramide as a flame retarding additive for lithium-ion battery electrolytes. J. Power Sources 135, 291–296 (2004). [Google Scholar]
- 34.Ota H., Kominato A., Chun W. J., Yasukawa E., Kasuya S., Effect of cyclic phosphate additive in non-flammable electrolyte. J. Power Sources 119–121, 393–398 (2003). [Google Scholar]
- 35.Shim E.-G., Nam T.-H., Kim J.-G., Kim H.-S., Moon S.-I., Electrochemical performance of lithium-ion batteries with triphenylphosphate as a flame-retardant additive. J. Power Sources 172, 919–924 (2007). [Google Scholar]
- 36.Wang X., Yasukawa E., Kasuya S., Nonflammable trimethyl phosphate solvent-containing electrolytes for lithium-ion batteries: I. Fundamental properties. J. Electrochem. Soc. 148, A1058–A1065 (2001). [Google Scholar]
- 37.Granzow A., Flame retardation by phosphorus compounds. Acc. Chem. Res. 11, 177–183 (1978). [Google Scholar]
- 38.Liu K., Kang Y., Wang Z., Zhang X., 25th anniversary article: Reversible and adaptive functional supramolecular materials: “Noncovalent interaction” matters. Adv. Mater. 25, 5530–5548 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Mu X., Liu Y., Fang D., Wang Z., Nie J., Ma G., Electric field induced phase separation on electrospinning polyelectrolyte based core-shell nanofibers. Carbohydr. Polym. 90, 1582–1586 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Wang M., Fang D., Wang N., Jiang S., Nie J., Yu Q., Ma G.. Preparation of PVDF/PVP core-shell nanofibers mats via homogeneous electrospinning. Polymer 55, 2188–2196 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1601978/DC1
fig. S1. Cyclic voltammogram for the graphite anode in different electrolytes.
fig. S2. A digital photograph showing that the TPP molecules are flake-like crystals.
fig. S3. SEM image of the TPP@PVDF-HFP fibers.
fig. S4. Digital pictures showing the (A) flexibility and (B) bendability of the TPP@PVDF-HFP membranes.
fig. S5. The stress-strain curve of the TPP@PVDF-HFP membrane.
fig. S6. SEM image of the TPP@PVDF-HFP membrane.
fig. S7. SEM cross-sectional image of the TPP@PVDF-HFP separator.
fig. S8. Voltage profiles of the graphite anode using the PE (black curve) separator and the TPP@PVDF-HFP separator (blue curve).
fig. S9. Electrochemical performances of the graphite anode using TPP@PVDF-HFP separators and PE separators with different electrolytes.
fig. S10. TGA measurements of the TPP@PVDF-HFP separator after long term cycling and the electrochemical performance of related battery.
fig. S11. Electrochemical stability of the TPP@PVDF-HFP separator.
fig. S12. The electrochemical stability of the TPP@PVDF-HFP separator towards LCO cathode.
fig. S13. The measurements on the Tm of TPP and PVDF-HFP.
fig. S14. SEM image of the TPP@PVDF-HFP after heat treatment.
movie S1. The combustion of the EC/DEC electrolyte.
movie S2. The combustion of the EC/DEC electrolyte with 40 wt % TPP.
movie S3. The combustion of the TPP@PVDF-HFP separator wetted by the EC/DEC electrolyte.