

ABSTRACT
Streptococcus pneumoniae is an ovoid-shaped Gram-positive bacterium that grows by carrying out peripheral and septal peptidoglycan (PG) synthesis, analogous to model bacilli, such as Escherichia coli and Bacillus subtilis. In the model bacilli, FtsZ and FtsA proteins assemble into a ring at midcell and are dedicated to septal PG synthesis but not peripheral PG synthesis; hence, inactivation of FtsZ or FtsA results in long filamentous cells unable to divide. Here, we demonstrate that FtsA and FtsZ colocalize at midcell in S. pneumoniae and that partial depletion of FtsA perturbs septum synthesis, resulting in elongated cells with multiple FtsZ rings that fail to complete septation. Unexpectedly, complete depletion of FtsA resulted in the delocalization of FtsZ rings and ultimately cell ballooning and lysis. In contrast, depletion or deletion of gpsB and sepF, which in B. subtilis are synthetically lethal with ftsA, resulted in enlarged and elongated cells with multiple FtsZ rings, with deletion of sepF mimicking partial depletion of FtsA. Notably, cell ballooning was not observed, consistent with later recruitment of these proteins to midcell after Z-ring assembly. The overproduction of FtsA stimulates septation and suppresses the cell division defects caused by the deletion of sepF and gpsB under some conditions, supporting the notion that FtsA shares overlapping functions with GpsB and SepF at later steps in the division process. Our results indicate that, in S. pneumoniae, both GpsB and SepF are involved in septal PG synthesis, whereas FtsA and FtsZ coordinate both peripheral and septal PG synthesis and are codependent for localization at midcell.
IMPORTANCE Streptococcus pneumoniae (pneumococcus) is a clinically important human pathogen for which more therapies against unexploited essential targets, like cell growth and division proteins, are needed. Pneumococcus is an ovoid-shaped Gram-positive bacterium with cell growth and division properties that have important distinctions from those of rod-shaped bacteria. Gaining insights into these processes can thus provide valuable information to develop novel antimicrobials. Whereas rods use distinctly localized protein machines at different cellular locations to synthesize peripheral and septal peptidoglycans, we present evidence that S. pneumoniae organizes these two machines at a single location in the middle of dividing cells. Here, we focus on the properties of the actin-like protein FtsA as an essential orchestrator of peripheral and septal growth in this bacterium.
KEYWORDS: FtsA, Gram-positive cocci, Streptococcus pneumoniae, cell division
INTRODUCTION
Bacterial cell division starts with the localization of the tubulin-like FtsZ at midcell (1), and FtsZ is the most widespread cell division protein conserved in almost all bacterial species (recently reviewed in reference 2). Upon GTP binding, FtsZ polymerizes into protofilaments (3, 4), and its attachment to the cell membrane is crucial for its assembly into a ring-like structure, known as the Z ring (5, 6). Subsequently, additional proteins are assembled at midcell to form the cell division machinery, a multiprotein complex known as the divisome (reviewed in references 7 and 8).
Among these proteins, actin-like FtsA (9, 10) is needed both to tether FtsZ to the cell membrane and to recruit the late-assembling cell division proteins required to complete the process (10–15). Although it is less conserved than FtsZ, FtsA is also widespread in many bacterial species (2). FtsA is essential in Escherichia coli, and its inactivation results in filamentous cells that are unable to divide (16, 17). The ftsA gene can be deleted in Bacillus subtilis, although at high temperatures, ftsA-null cells filament, exhibiting reduced viability and defective sporulation (18, 19). FtsA seems to be essential in Streptococcus pneumoniae, as no transformants were obtained by insertion-deletion mutagenesis of ftsA (20).
FtsA is recruited to midcell at the earliest stage of the division process. In E. coli, its correct localization depends on the previous localization of FtsZ (21), whereas FtsZ localization does not depend on the localization of FtsA (22). However, localization of FtsZ in the absence of FtsA may be mediated by another FtsZ membrane anchor, ZipA (5, 11, 23, 24). FtsA and ZipA are the only two anchors for FtsZ in E. coli, and in the absence of both, Z rings do not form (5). E. coli FtsA variants able to suppress the lack of ZipA support the idea that these proteins have a redundant function in stabilizing the Z ring (12, 25, 26). However, unlike FtsA, ZipA is absent in Gram-positive bacteria, where the mechanism of FtsZ tethering to the membrane remains to be clarified; other FtsZ-interacting proteins, like SepF (27, 28) or EzrA (29, 30), may fulfill this function (31).
In this study, we investigated the role of FtsA in S. pneumoniae (pneumococcus), a clinically relevant human pathogen and a reference species to study cell division in Gram-positive ovococci that have an ovoid-shaped morphology. Similar to the model rod-shaped E. coli and B. subtilis, S. pneumoniae grows and divides by carrying out peripheral peptidoglycan (PG) synthesis with septal PG synthesis (32–36). Pneumococcal cell division initiates with the localization of FtsZ and FtsA to midcell, and later-division proteins, such as StkP, penicillin-binding protein 2x (PBP2x), PBP1a, GpsB, and DivIVA, localize only after the Z ring has been assembled (20, 37–41). However, genetic evidence for the essential role of the Fts proteins involved in the initial steps of cell division is presently lacking. In particular, it is not yet known how the PG synthetic complex involved in septation (the divisome) is assembled and coordinated with the protein complex involved in peripheral elongation (the elongasome) (7, 42), which in S. pneumoniae is also located at midcell (35, 36, 43–45).
Here, we confirm that FtsA is essential in S. pneumoniae and show that depletion of FtsA initially inhibits septation and ultimately results in cell lysis. This is distinct from the role of FtsA in model rod-shaped bacteria and suggests that FtsA is required for both peripheral wall and septal wall synthesis in this organism. We also demonstrate that the depletion of FtsA results in delocalization of Z rings in S. pneumoniae, implicating FtsA as a dominant membrane tether and/or stabilizer of the Z ring. This behavior, along with the ability of increased levels of FtsA to stimulate septation and to suppress septation defects of other cell division mutants, distinguishes S. pneumoniae FtsA from FtsA proteins of model rod-shaped bacteria. Overall, our results support the idea that both the elongation and septation synthesis machines act in complexes at midcell that include FtsA as an essential organizing component.
RESULTS
FtsA colocalizes with FtsZ early in the division cycle and relocalizes to new sites prior to septum completion.
FtsZ and FtsA localize to midcell early in the division cycle of S. pneumoniae (20), as in model rod-shaped bacteria. Consistently, green fluorescent protein (GFP)-FtsA was found to localize at the division site before StkP, DivIVA, PBP1a, and PBP2x (39, 41) but after LocZ (46). Given the localization profile and that S. pneumoniae FtsA also interacts directly with FtsZ, it was hypothesized that the two proteins arrive at midcell at the same time and colocalize during the cell cycle (35).
To test this, we used an ftsZ-mCherry-erm construct (47) that when introduced via transformation into the Rx1strain, integrated at the ftsZ native locus, resulting in the expression of ftsZ-mCherry from its own promoter as the only source of FtsZ in the cells. We then introduced the ftsZ-mCherry fusion into the Rx1 strain expressing both native ftsA and gfp-ftsA, which is fully functional and able to complement an ftsA deletion (see below), and we observed cells at six different stages of cell division, from newborn (stage 1) to completion of division near to cell separation (stage 6). The results clearly show that localization of GFP-FtsA and FtsZ-mCherry overlapped completely at all stages (Fig. 1), supporting a model in which FtsA and FtsZ are in a complex, as previously suggested by immunofluorescence and immunoelectron microscopy (20). The clearest example of colocalization is at stage 4, where both proteins can be seen in the process of disassembling from the active division septum to new division sites (Fig. 1, arrows). At stage 5, there is no detectable FtsA or FtsZ remaining at midcell, suggesting that unlike in E. coli, where FtsA lags significantly behind FtsZ (48), the two proteins move in concert in S. pneumoniae.
FIG 1.
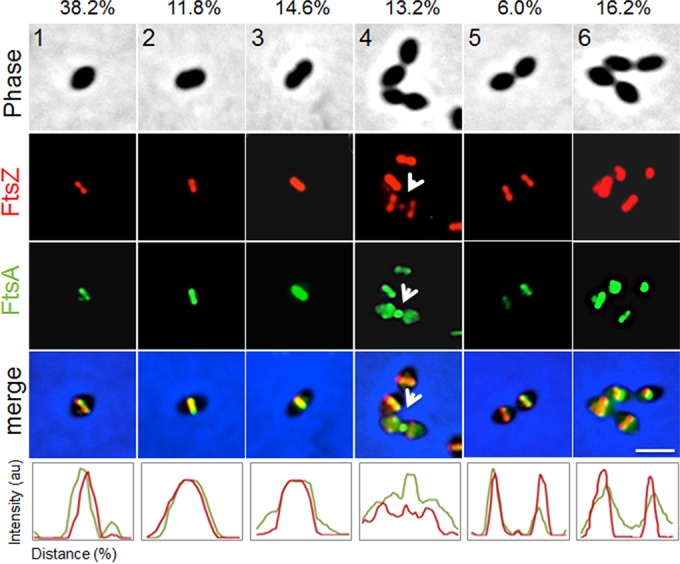
FtsA colocalizes with FtsZ throughout the cell division cycle. For representative stages of the cell division cycle 1 to 6 (top row), Rx1 cells producing FtsZ-mCherry and GFP-FtsA were imaged by phase-contrast and fluorescence microscopy. Arrows highlight FtsZ and FtsA at a deeply constricting septum upon relocalization of some FtsZ and FtsA for the next round of division. Percentages of cells at the indicated stage are listed above the representative images. Fluorescence intensity plots for each stage are shown at the bottom. au, arbitrary units. Scale bar, 1 μm.
To explore the nature of FtsA/FtsZ colocalization further, we determined whether the two proteins also colocalize throughout the cell cycle in an unencapsulated derivative (Δcps mutant) of serotype 2 strain D39, which is the progenitor strain of laboratory strain Rx1 (see Table S1 in the supplemental material) (49; B. E. Rued, J. J. Zheng, A. Mura, H.-C. T. Tsui, M. J. Boersma, J. L. Mazny, F. Corona, A. J. Perez, D. Fadda, L. Doubravová, K. Buriánková, P. Branny, O. Massidda, and M. E. Winkler, unpublished data). In these experiments, markerless FLAG-FtsA and FtsZ-Myc fusions were expressed in the same cell as their native chromosomal locations. Control experiments showed that cells expressing the epitope-tagged proteins grew with doubling times similar to the D39 Δcps parent strain and produced similar amounts of total FtsA and FtsZ proteins (Fig. S3A). Other controls described in Materials and Methods ruled out antibody cross-reactivity artifacts. In agreement with the Rx1 results (Fig. 1), two-dimensional immunofluorescence microscopy (2D-IFM) of D39 Δcps cells showed that FLAG-FtsA and FtsZ-Myc colocalize at all stages of cell division (Fig. 2A), where cells in images were retrospectively binned into four division stages for image averaging (Fig. S3B) (40). The widths of equatorial and septal bands of FLAG-FtsA and FtsZ-Myc in D39 Δcps cells at three division stages were averaged and statistically compared in the 2D-IFM images (Fig. 2B). At all division stages, the width of the FtsA band was greater than that of the FtsZ band, supporting the model of FtsA acting as a membrane anchor for FtsZ in S. pneumoniae. High-resolution three-dimensional structured illumination microscopy (3D-SIM) IFM confirmed the conclusion that FLAG-FtsA and FtsZ-Myc colocalize at all stages of pneumococcal cell division (Fig. 2C). Again, the diameters of the FtsA bands appeared to be slightly greater than those of the FtsZ bands in most cells (Fig. 2C), consistent with the 2D-IFM measurements that included a much larger number of cells (Fig. 2B). Rotated 3D-SIM IFM images showed discrete patches of FtsA and FtsZ in both unconstricted and deeply constricted septal rings (Fig. 2C), similar to the patchy patterns observed in other bacteria (50–53). The signals from FtsA and FtsZ appear to not always coincide in some 3D-SIM images (Fig. 2C); however, given the resolution distortion inherent in 3D-SIM image rotation, a conclusion that FtsA and FtsZ occasionally occupy different segments of division rings requires corroboration by other high-resolution microscopy techniques.
FIG 2.

Two-dimensional- and 3D-SIM IFM of FtsA and FtsZ in strain D39 Δcps reveals colocalization of FtsA in a ring wider than the FtsZ ring. Strain IU10304 (D39 Δcps FLAG-ftsA ftsZ-Myc) was grown to mid-exponential phase in BHI broth and processed for dual-protein 2D- and 3D-SIM IFM with DAPI labeling of DNA, as described in Materials and Methods. (A) Representative fields of 2D-IFM images showing FtsA and FtsZ colocalization during the cell cycle. (B) Top, schematic diagram showing the FtsZ ring versus FtsA ring measurements. Bottom, scatter plot of the paired widths from the same cells of FtsZ and FtsA fluorescently immunolabeled regions at midcell equators and septa of cells at division stages 1 to 3. Width measurements and plotting were done using the IMA-GUI program (see Materials and Methods). The dotted reference line intercepts the origin with a slope of 1. Differences between the paired widths were calculated for each cell in each divisional stage, and the null hypothesis that the mean difference was 0 was tested by 1-sample Student's t test, where *** represents a P value of <0.001 (44). Septal widths of stage 4 cells were not analyzed, because FtsZ and/or FtsA may have been already absent from midcell and not completely assembled yet at future division sites. (C) Representative 3D-SIM IFM and DAPI images obtained of strain IU10304 at different division stages. Each image (i to v) is a different cell in which one or more divisome rings are rotated along the axis indicated on the left of the panel. DNA (DAPI-stained image) is pseudocolored white or blue in column 1 or 5, respectively. FtsA and FtsZ are pseudocolored red and green, respectively. The first row of each image represents images captured in the xy plane, while second-row images were obtained by rotating a section of the cell around the x or y axis. Individual rotated daughter and/or septal rings are indicated by the corresponding arrows of the nonrotated cells. (i) Postdivisional pneumococci (two stage 1 cells) showing that both daughter cells contain FtsZ and FtsA ring colocalization at future division sites. (ii) Predivisional cell showing FtsZ and FtsA ring septal colocalization. (iii) Early divisional cell showing FtsZ and FtsA rings at the equator of one of the two future dividing daughter cells. (iv) Middivision cell showing constricting FtsZ and FtsA rings at actively dividing septa and newly formed FtsZ and FtsA rings at equators of future dividing daughter cells. (v) Late-division pneumococci in which FtsA and FtsZ rings are located at equators of future dividing daughter cells and only a dot of both FtsA and FtsZ is present at the former active division septum. Images are representative of >50 3D-reconstructed cells in different division stages from two independent experiments. Scale bars in panels A and C, 1 µm.
Partial depletion of FtsA inhibits septation, and more complete depletion causes isotropic cell expansion and lysis.
We next wanted to test whether FtsA is essential for S. pneumoniae cell division and to pinpoint the consequences of FtsA inactivation. We took advantage of the Zn2+-inducible system that has been used for depletion of essential genes, including those involved in cell division (41, 54). For this purpose, the Rx1 ftsA+//PZngfp-ftsA+ merodiploid strain, where the double slash indicates carriage of an additional copy of the ftsA gene at the bgaA locus and expression of GFP-FtsA upon induction with ZnCl2 (Table S1), was transformed with a construct containing an ftsA gene deletion and a promoterless chloramphenicol marker (ftsA::Plesscat) to replace the native ftsA gene by insertion/deletion mutagenesis to obtain an Rx1 ΔftsA//PZngfp-ftsA+ conditional null strain (Fig. S1A and Table S1). Transformants were selected on tryptone soya blood agar (TSBA) plates containing chloramphenicol and ZnCl2, and correct insertion of the fragment at the native locus was confirmed by PCR. No transformants were obtained in TSBA plates without ZnCl2, in agreement with a previous report (20).
The functionality of the GFP-FtsA protein in the absence of the native gene was confirmed by the inability of transformants to grow, in TSB or TSBA, at ZnCl2 concentrations of <0.075 mM, whereas they grew at ≥0.075 mM ZnCl2, and normal colony growth was observed at ≥0.45 mM ZnCl2 (Fig. 3A to C). The growth kinetics and morphology of cells grown in the presence of 0.45 to 0.65 mM ZnCl2 were similar to those of the parental Rx1 and Rx1 ftsA+//PZngfp-ftsA+ strains (Fig. 3A, and data not shown), whereas growth was impaired at lower ZnCl2 concentrations, with a rapid decrease in cell density observed at ZnCl2 concentrations of <0.075 mM. Consistent with these results, immunoblotting with anti-FtsA antibodies showed that cellular levels of GFP-FtsA in the ftsA conditional null mutant were proportional to the levels of ZnCl2 added to the cultures, indicating that depletion of GFP-FtsA occurred efficiently and rapidly. The GFP-FtsA levels were already lower (∼10% less) than the levels of native FtsA at the highest ZnCl2 concentrations, under these conditions, and became significantly more depleted at ZnCl2 concentrations of <0.15 mM (Fig. 3B). GFP-FtsA levels at 0.075 mM ZnCl2 were ∼35% of the native FtsA protein levels. GFP-FtsA became undetectable at ZnCl2 concentrations of ≤0.0375 mM, which correlated well with the largest decreases in optical density (Fig. 3A), due to cell lysis. Consistently, morphological changes were detectable after 30 to 120 min upon depletion, with cells that elongated, enlarged, and bulged (not shown). As depletion continued, the phenotype deteriorated, resulting in highly heterogeneous morphologies after 4 h (Fig. 3C), similar to those described for the depletion of PBP2x (41). At ZnCl2 concentrations of ≤0.0375 mM (undetectable GFP-FtsA levels), cells started ballooning after 120 min and then lysed soon after. At ZnCl2 concentrations of 0.075 to 0.15 mM (GFP-FtsA levels at ∼35 to 70% of the native FtsA protein levels), cells grew in chains of irregular elongated cells and then lysed. Finally, at concentrations of 0.45 to 0.65 mM ZnCl2 (GFP-FtsA levels at ∼90% of the native FtsA protein levels), cells grew well and showed an overall wild-type morphology, indicating complementation. Similar results were obtained when 0.01 mM MnCl2 was added to the TSB medium in addition to ZnCl2 to counteract the potential toxicity of ZnCl2 (not shown).
FIG 3.

Depletion of FtsA inhibits septation and results in cell ballooning and lysis. (A) Growth curves of cells depleted of FtsA in TSB at 37°C using different levels of ZnCl2 induction to express GFP-FtsA in the Rx1 ΔftsA//PZngfp-ftsA+ strain. (B) Immunoblots showing native FtsA or GFP-FtsA in uninduced samples (0 mM ZnCl2) of Rx1 and its merodiploid derivative Rx1 ftsA+//PZngfp-ftsA+ and in uninduced (0 mM ZnCl2) or induced samples of Rx1 ΔftsA//PZngfp-ftsA+ (upper image). An anti-FtsZ immunoblot for the same samples was used as a loading control (lower image). (C) Phase-contrast micrographs of representative fields of Rx1 ftsA+//PZngfp-ftsA+ or Rx1 ΔftsA//PZngfp-ftsA+ cells grown in TSB at 37°C in the absence or in the presence of different ZnCl2 concentrations (0, 0.075, 0.15, and 0.65 mM), taken 4 h from the start of FtsA depletion (upper images); growth on TSBA plates (Rx1 ftsA+//PZngfp-ftsA+ control strain on the left side and Rx1 ΔftsA//PZngfp-ftsA+ on the right side) with the same ZnCl2 concentration is shown in the lower images. O/N, overnight. (D) Combined phase-contrast and fluorescence micrographs of Rx1 ΔftsA//PZngfp-ftsA+ cells grown in C+Y medium at 37°C with or without different ZnCl2 concentrations (0, 0.05, 0.10, and 0.15 mM), taken 5 h from the start of FtsA depletion; GFP-FtsA localization to multiple bands in filamentous cells is highlighted by arrows. Average cell length and width at each stage are shown below the corresponding micrograph. Scale bars, 1 μm.
To understand the consequences of FtsA depletion in more detail, we also examined cell morphology and localization of GFP-FtsA at different levels of GFP-FtsA induction in the ΔftsA mutant background after growth under depletion conditions in C+Y medium (Fig. 3D). At concentrations of 0.15 mM ZnCl2 (corresponding approximately to 0.65 mM ZnCl2 in TSB), cells showed a wild-type morphology and size, although they were slightly smaller, with GFP-FtsA properly localized at midcell and future division sites. Similarly, at 0.10 mM ZnCl2, cells still showed a normal morphology, size, and GFP-FtsA localization but formed more chains (Fig. 3D) and started lysing before the beginning of stationary phase. At 0.05 mM ZnCl2, cells elongated significantly, and GFP-FtsA continued to localize at potential division sites in the filamenting cells. However, maximum depletion of FtsA (ZnCl2 levels <0.05 mM or with no ZnCl2) resulted in the isotropic expansion of cells and complete delocalization of GFP-FtsA until the cells lysed (Fig. 3D and data not shown).
Similar FtsA depletion experiments were performed using a D39 Δcps ΔftsA//PZnftsA+ conditional null strain (Fig. S1B and Table S1). Growth of this strain was also Zn dependent, with a normal growth curve and cell morphology at 37°C in brain heart infusion (BHI) broth containing 0.5 mM ZnCl2 plus 0.05 mM MnSO4, which, like MnCl2, was added to counteract potential toxicity from ZnCl2. At low ZnCl2/MnSO4 concentrations (<0.125/0.0125 mM), cultures failed to grow after ∼3 h, at which time cells became spherical, contained ∼18% FtsA compared to Zn-replete cultures, and started to lyse (data not shown). Upon partial ZnCl2/MnSO4 depletion (0.25/0.025 mM), cells grew to a lower final density, contained decreased (∼57%) FtsA protein, and formed a mixture of cell shapes (normal, enlarged, elongated, and irregular) compared to those under the zinc-replete conditions or to the parent strain (data not shown). At 37°C in BHI broth, the D39 Δcps ΔftsA//PZnftsA+ conditional null strain formed <5% elongated cells, unlike the Rx1 ΔftsA//PZngfp-ftsA+ conditional null strain (Fig. 3C and D and 4; also, data not shown). However, elongated D39 Δcps ΔftsA//PZnftsA+ cells were readily observed (>50% of the cell population) when cells were depleted for FtsA at 30°C in BHI (not shown). We concluded that the depletion of FtsA causes a block in cell septation leading to cell elongation in different pneumococcal strains, which can be influenced by temperature and growth conditions.
FIG 4.

Cell division septa are abnormal in filamentous cells after moderate FtsA depletion. Representative micrographs of ultrathin cell sections showing morphological defects of cells depleted of FtsA in TSB at 37°C. Samples for microscopy were taken 4 h after the start of FtsA depletion (at the indicated levels of ZnCl2) and washed, fixed, and vitrified, as described in Materials and Methods. Septation defects characteristic of each depletion condition are indicated by arrows. Scale bars, 0.2 μm.
Depletion of FtsA causes rapid defects in septal morphology.
Cell filamentation during partial FtsA depletion indicated that normal septation was inhibited. To take a closer look at the effects of FtsA depletion on cell division septa, we imaged Rx1 cells under various depletion conditions by transmission electron microscopy (EM). After depleting FtsA for ∼4 h in TSB medium at 37°C, division septa exhibited significant defects, which were more severe at lower concentrations of ZnCl2 (Fig. 4). At 0.075 mM ZnCl2, cells displayed altered morphology and defective septa, with accumulation of cell wall/membrane material similar to what was reported for S. aureus depleted of FtsZ (55), suggesting that cell wall synthesis may be dispersed. At 0.15 mM ZnCl2, cell division was largely restored, but morphology and septa were still aberrant. At this inducer concentration, 75.4% (49/65) of the septa were either incipient or incomplete, whereas only 24.6% (16/65) were closed, some of which were constricting, consistent with the ability of some of cells to grow and divide. Finally, at 0.65 mM ZnCl2, cell morphology and division were restored in the majority (>90%) of cells, consistent with this level of GFP-FtsA being able to complement the ftsA-null phenotype.
FtsA depletion causes dispersion of PG synthesis.
Given the possibility that PG synthesis is dispersed upon depletion of FtsA, we compared the sites of active PG synthesis in Rx1 ΔftsA//PZngfp-ftsA+ cells by staining with HADA (HCC-amino–d-alanine), a fluorescent hydroxy-coumarin derivative of d-alanine (56). Short pulses of HADA incorporate into PG in regions of active penicillin-binding protein transpeptidase activity (56, 57). Rx1 ΔftsA//PZngfp-ftsA+ cells were grown in the absence or presence of different ZnCl2 concentrations and observed by fluorescence microscopy (Fig. 5). Similar to what we observed for Rx1, HADA staining of the Rx1 ΔftsA//PZngfp-ftsA+ cells grown in ZnCl2 concentrations of 0.15 or 0.10 mM revealed a pattern of new PG synthesis at division sites that colocalized with GFP-FtsA for most of the cell cycle (83.3% and 82.7%, respectively). This pattern is consistent with the localization profile and timing reported for FtsZ, FtsA and the respective PBPs involved in peripheral and septal PG synthesis (20, 33, 37, 40, 41, 44, 45). In cells grown in 0.05 mM ZnCl2, 71.4% of the division sites still showed colocalization of GFP-FtsA and HADA signals. On the contrary, at ZnCl2 concentrations lower than 0.05 mM, the HADA fluorescent signal progressively dispersed throughout the surface of the isotropically expanded cells (Fig. 5 and data not shown). This HADA labeling pattern strongly suggests that PG synthesis becomes delocalized when FtsA is severely depleted.
FIG 5.
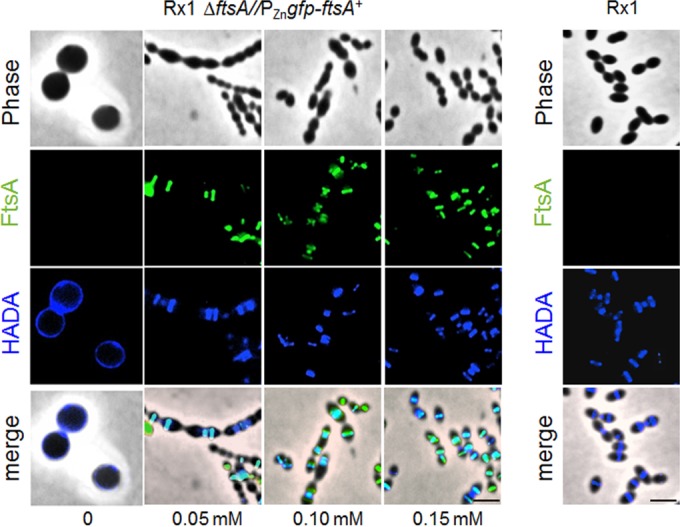
Depletion of FtsA causes dispersion of PG synthesis. Fluorescence micrographs showing PG synthesis in S. pneumoniae cells depleted of FtsA. Rx1 and Rx1 ΔftsA//PZngfp-ftsA+ cells were grown for 5 h in C+Y medium in the absence or in the presence of ZnCl2, and the incorporation of PG was detected by pulse-labeling with HADA, as described in Materials and Methods. The green fluorescent signal corresponds to GFP-FtsA, while the blue fluorescent signal corresponds to incorporation of HADA and indicates newly synthesized PG. Cyan staining signifies overlap between green and blue. Scale bars, 1.5 μm.
FtsA is required for efficient midcell localization of FtsZ.
Because localization of FtsA is dependent on FtsZ in other species and they mostly colocalize in the cell, we asked whether FtsZ rings were still present in FtsA-depleted cells. We used the Rx1 ΔftsA mutant strain producing both GFP-FtsA and FtsZ-mCherry fusions described above to test whether FtsZ-mCherry was localized in S. pneumoniae under FtsA depletion conditions. At 0.65 mM ZnCl2 with levels of GFP-FtsA at ∼90% of the native FtsA protein levels (Fig. 3B), FtsZ-mCherry was properly localized both at growing septa and at future division sites. Cells were slightly elongated with respect to the Rx1 ΔftsA//PZngfp-ftsA+ parent carrying native FtsZ (with lengths and widths of 1.98 ± 1.01 μm by 0.64 ± 0.08 μm [means ± standard deviations; n = 224] and 1.68 ± 0.41 μm by 0.64 ± 0.06 μm [n = 240], respectively) and formed more chains (Fig. 6A); this effect may be due to a decreased ability of FtsZ-mCherry to interact with GFP-FtsA compared with native FtsZ. Under stronger depletion conditions (0.075 or 0.15 mM ZnCl2), at levels of GFP-FtsA ∼35% and ∼70% of the native FtsA protein, cells elongated and enlarged (maximal average length and width, 2.11 ± 1.11 μm by 0.68 ± 0.08 μm). Under these conditions, some FtsZ-mCherry rings remained, but the fusion protein became noticeably dispersed in expanded cells. These results suggest that mild depletion of FtsA in S. pneumoniae prevents FtsZ rings from activating septation, and further depletion prevents the localization of FtsZ to midcell rings. To exclude the possibility that the delocalization of FtsZ-mCherry in the absence of FtsA was due to the mCherry tag, the results were confirmed by immunoelectron microscopy using anti-FtsA or anti-FtsZ in the Rx1 ΔftsA//PZngfp-ftsA+ strain carrying native FtsZ (Table 1).
FIG 6.

Depletion of FtsA results in delocalization of FtsZ. (A) Rx1 ΔftsA//PZngfp-ftsA+ and Rx1 ftsA+//PZngfp-ftsA+ cells expressing ftsZ-mCherry were subjected to different concentrations of ZnCl2 for 5 h in TSB at 28°C and imaged by fluorescence and phase-contrast microscopy, as described in Materials and Methods. Scale bars, 1 μm. (B) Individual time course of growing Rx1 ftsA+//PZngfp-ftsA+ cells expressing ftsZ-mCherry (upper images) and Rx1 ΔftsA//PZngfp-ftsA+ cells expressing ftsZ-mCherry (lower images), showing how FtsA depletion leads to perturbation of FtsZ-mCherry rings and cell elongation, followed by cell expansion and lysis. The number in the upper right corner of each frame indicates minutes after ZnCl2 removal. Two prominent FtsZ-mCherry rings that eventually delocalize upon ftsA depletion are highlighted by white and yellow arrows. Scale bars, 1 μm.
TABLE 1.
Localization of FtsA and FtsZ upon depletion of FtsA
| Strain | ZnCl2 concn (mM) | FtsA molecules/cella | % midcell localizationb |
|
|---|---|---|---|---|
| FtsA | FtsZ | |||
| Rx1 ftsA+//PZngfp-ftsA+ | 0 | 2,200 | 67 | 70 |
| Rx1 ΔftsA//PZngfp-ftsA+ | 0.65 | 1,980 | 62 | 68 |
| 0.15 | 1,540 | 40 | 48 | |
| 0.075 | 770 | 12 | 18 | |
Estimated number of molecules/cell of native FtsA (or GFP-FtsA) with respect to the number reported in reference 20.
Percentage of immunogold particles at the division site (midcell and future division sites).
To determine how rapidly FtsZ delocalized from midcell during FtsA depletion, we monitored FtsZ-mCherry localization in individual Rx1 ΔftsA//PZngfp-ftsA+ ftsZ-mCherry cells over time in comparison with those of the Rx1 ftsA+//PZngfp-ftsA+ ftsZ-mCherry parent (Fig. 6B and Movie S1). Although FtsZ-mCherry localized to distinct bands in cells at early time points, by 120 min after FtsA depletion, many cells became significantly elongated and had lost FtsZ-mCherry bands (Fig. 6B and Movie S2). In addition, most FtsZ-mCherry rings that were present were not able to complete septation (Fig. 6B, arrows), probably because they formed the aberrant septa shown in Fig. 4. Several cells at this time point started to expand, and the expansion correlated with significantly diffuse FtsZ-mCherry localization. By 270 min, all cells were abnormally shaped, and many of them lysed; importantly, FtsZ-mCherry was completely delocalized from midcell in the remaining intact cells, despite some areas of strong fluorescence.
Excess FtsA stimulates production of smaller cells.
The effects of the depletion of FtsA prompted us to check whether overproduction of FtsA might also inhibit cell division, as it does in E. coli (58, 59), or instead promote division. We induced overproduction of FtsA in Rx1 ftsA+//PZngfp-ftsA+ cells grown with different levels of ZnCl2, followed by phase-contrast and fluorescence microscopy. As the inducer concentration increased from 0.05 to 0.15 mM ZnCl2 and GFP-FtsA increased concomitantly, GFP-FtsA continued to localize to division septa (Fig. 7A and B). Notably, the population of cells became both shorter and narrower (Fig. 7C), changing from an average length and width of 1.53 ± 0.29 by 0.64 ± 0.05 μm (n = 540) with no inducer to 1.37 ± 0.27 by 0.60 ± 0.05 μm (n = 224) (Student's t test, P < 0.001) at full induction, a statistically significant 10% decrease in length and 6% decrease in width. As growth rates were not significantly changed at the different inducer concentrations (data not shown), the data suggest that increased amounts of FtsA accelerate the septation process. In addition, we scored the percentage of Rx1 ftsA+//PZngfp-ftsA+ ftsZ-mCherry cells with FtsZ-mCherry that had a deeply constricted FtsZ ring plus two new rings at future division sites. In support of our hypothesis, the percentage was 3.4% (14/409) at 0 mM ZnCl2 but decreased to 1.7% (8/461) and 1.6% (12/759) at 0.45 and 0.65 mM ZnCl2, respectively. The lower percentage of deeply constricting cells with excess FtsA is consistent with the idea that more rapidly dividing cells will spend less time in any specific stage of division.
FIG 7.

Excess FtsA levels stimulate production of smaller cells. (A) Fluorescence micrographs of Rx1 cells expressing PZngfp-ftsA, grown in prewarmed C+Y medium with 0, 0.05, 0.10, and 0.15 mM ZnCl2 at 28°C until the cells reached an OD of ∼0.3. Scale bar, 1 μm. (B) Anti-FtsA immunoblot showing the amounts of native FtsA and GFP-FtsA in uninduced or induced samples. (C) Cell length and width distributions for each induction condition are shown, overlaid onto the distribution profile (in black) from the no-ZnCl2 control culture.
We also tested the effects of FtsA overexpression in the D39 Δcps ftsA+//PZnftsA+ merodiploid strain at 37°C in BHI broth with 0.5 mM ZnCl2 plus 0.05 mM MnSO4. A slight, but statistically marginal, decrease in median cell length of the D39 Δcps strain was routinely detected upon overexpression of FtsA (data not shown), in contrast to the greater decrease observed for the Rx1 strain growing in C+Y medium (Fig. 7). Consistent with this weaker effect, Western blot analysis showed that FtsA was overproduced by only about 1.7× in the D39 Δcps strain grown in BHI broth (data not shown) compared to the greater overproduction (>3×) of GFP-FtsA attained in the Rx1 strain grown in C+Y medium (Fig. 7B). As similar results were obtained for Rx1 ftsA+//PZngfp-ftsA grown in TSB or BHI with 0.65 mM ZnCl2 (data not shown), this is likely the result of differences in Zn-dependent gene expression in the different growth media.
Excess FtsA suppresses late septation defects in cells lacking division protein SepF or GpsB.
In B. subtilis, SepF has been shown to share some properties with FtsA, including the ability to tether FtsZ to the membrane (27, 28, 31), and to permit cell division in the absence of FtsA when overproduced (28). Deletion of the gene encoding SepF, previously called ylmF, in S. pneumoniae is not lethal but results in significant defects in septation as well as other morphological abnormalities (60). These septation defects are not caused by FtsZ ring mislocalization, as FtsZ-mCherry localized to many potential septation sites in elongated ΔsepF mutant cells (Fig. S4A), and there were multiple bands of HADA staining within the elongated cell regions (Fig. S4B). Two-dimensional- and 3D-SIM IFM of D39 Δcps ΔsepF mutants expressing FLAG-FtsA and FtsZ-Myc confirmed that ΔsepF mutants form elongated cells containing multiple distinct unconstricted septal bands of colocalized FtsA and FtsZ surrounding dispersed DNA nucleoids (Fig. 8A and B). Despite their morphological defects, D39 Δcps ΔsepF mutants grow similarly to their wild-type parent in BHI broth at 37°C (Fig. 8C).
FIG 8.

FtsA and FtsZ remain colocalized in a ΔsepF mutant. Strain IU10632 (D39 Δcps FLAG-ftsA ftsZ-Myc ΔsepF) was grown to mid-exponential phase in BHI broth and processed for dual-protein 2D- and 3D-SIM IFM with DAPI labeling of DNA, as described in Materials and Methods. (A) Representative field of phase-contrast and 2D-IFM images. Cultures of ΔsepF mutants are characterized by mixture of cells of relatively normal morphology (long arrows) and elongated and slightly wider cells without septal constriction (shorter arrows). FtsA and FtsZ are colocalized in both cell types. Images are representative of 2 independent experiments. (B) 3D-SIM IFM images. The first row represents an image captured in the xy plane, while the second-row images were obtained by rotating the chain of cells to show the FtsA and FtsZ rings. Scale bars, 1 μm. Images are representative of >50 3D-reconstructed cells in different division stages from two independent experiments. (C) Representative growth curves of the wild-type isogenic D39 Δcps ftsA+ ftsZ+ sepF+ parent (IU1824), ftsA+ ftsZ+ ΔsepF mutant strain (IU8039), and FLAG-ftsA ftsZ-Myc ΔsepF strain (IU10632). ΔsepF mutant strains with the ftsA+ ftsZ+ or the FLAG-ftsA ftsZ-Myc background grew similarly (with growth rates similar to but growth yields lower than those of the sepF+ parent).
Based on these observations and the ability of excess FtsA to stimulate septation, we reasoned that excess FtsA might be able to repair the septation defect of ΔsepF mutant cells in S. pneumoniae. To test this possibility, we introduced the Zn2+-inducible gfp-ftsA construct into Rx1 ΔsepF mutant cells and examined cells grown under different inducer concentrations. As expected, cells grown with no inducer were elongated relative to wild-type cells, and some abnormally small or misshapen cells were also observed (Fig. 9A). Cells grown with inducer levels as low as 0.05 mM, which represents an ∼2× increase in the total FtsA plus GFP-FtsA content, were already less elongated, and some of them showed clear constricting GFP-FtsA rings at midcell and relocalized GFP-FtsA rings at future division sites. Increased levels of induction, similar to those that stimulated septation in wild-type cells described above, resulted in even shorter cells, again with GFP-FtsA at the division septa, although even at the highest levels of GFP-FtsA induction, cells remained enlarged compared to wild-type cells. In the D39 Δcps ΔsepF mutants, overexpression of FtsA also restored cell lengths of nonseptating and septating cells to those of the sepF+ parent strain (Fig. S5). We concluded that the ΔsepF septation defect is repaired by excess FtsA.
FIG 9.

Reversal of septation defects in Rx1 cells lacking either SepF or GpsB by excess FtsA. (A) Phase-contrast or fluorescence micrographs of Rx1 ΔsepF cells or Rx1 ΔsepF cells expressing gfp-ftsA at various induction levels to provide an excess of FtsA (immunoblot panel is on the right). The arrow highlights a dividing cell with normal morphology and FtsA ring pattern. Scale bars, 1 μm. (B) Phase-contrast or fluorescence micrographs of Rx1 ΔgpsB sup4 cells or Rx1 ΔgpsB sup4 cells expressing gfp-ftsA at various induction levels to provide an excess of FtsA (immunoblot panel is on the right). Scale bars, 1 μm. See the text for additional details.
GpsB is another cell division protein in B. subtilis that has been functionally linked to FtsA. B. subtilis cells lacking GpsB do not show any distinctive phenotype but cannot grow when FtsA is simultaneously inactivated (61). In S. pneumoniae, GpsB, like FtsA, is essential in the progenitor D39 genetic background (40, 62), whereas Rx1 ΔgpsB mutants are viable because of the presence of preexisting suppressors in the laboratory strain genetic background but that grow very slowly (Rued et al., unpublished data). Attempts to transform a ΔgpsB deletion amplicon into a D39 Δcps ftsA+//PZnftsA+ merodiploid strain overexpressing FtsA in the presence of 0.5 mM ZnCl2 plus 0.05 mM MnSO4 were unsuccessful (data not shown), implying that overproduction of FtsA alone does not suppress the requirement for GpsB in this strain. Likewise, FtsA overproduction did not improve the poor growth of an Rx1 ΔgpsB ftsA+//PZnftsA+ merodiploid strain. However, the Rx1 ΔgpsB mutant spontaneously accumulated another suppressor mutation, designated sup4, whose identity is reported elsewhere (Rued et al., unpublished data). The Rx1 ΔgpsB sup4 strain grows much faster than the Rx1 ΔgpsB mutant but still forms elongated cells with distinct multiple FtsZ rings and bands of HADA labeling (Fig. 9B and S4A and B), indicative of a defect in a late step of septum formation or closure.
As with ΔsepF mutant cells, we asked whether Rx1 ΔgpsB sup4 cells could be rescued by overproduction of GFP-FtsA. Without inducer, many ΔgpsB sup4 mutant cells were significantly elongated (Fig. 9B), whereas even with the lowest level of inducer (0.05 mM ZnCl2), ΔgpsB sup4 mutant cells were already shorter and displayed clear GFP-FtsA rings at the division septa. Higher levels of GFP-FtsA induction had similar effects. We concluded that in some genetic backgrounds, such as Rx1 ΔgpsB sup4, the ΔgpsB mutant septation defects can be completely corrected by extra FtsA.
The presence of multiple rings of FtsZ and zones of PG synthesis in elongated D39 and Rx1 ΔsepF mutants and Rx1 ΔgpsB sup4 cells (Fig. 9 and S4) suggests that the cell division defects occur at a late stage of septation. Previous data indicated that localization of GpsB lags behind FtsZ during the division process (40), consistent with the late septation defect, so we tested whether SepF localized like GpsB. As we did with the colocalization of FtsZ and FtsA, GFP fusions of either SepF (GFP-SepF) or GpsB (GFP-GpsB) were induced in cells producing FtsZ-mCherry, and micrographs were used to sort cells into six stages of S. pneumoniae cell division. As can be seen in Fig. 10, both GFP-SepF and GFP-GpsB colocalize with FtsZ-mCherry in stages 1 to 3, but in stage 4, FtsZ-mCherry has already relocalized from the midcell septum to new division sites, while both GFP-GpsB (as expected) and GFP-SepF clearly lag behind at the midcell septum. These results are consistent with the idea that a later stage of septation organized by FtsA is blocked by the inactivation of SepF and GpsB, such that FtsA overexpression can rescue ΔsepF and ΔgpsB deletion phenotypes, with the ΔgpsB deletion phenotype in selected genetic backgrounds.
FIG 10.

SepF localizes to the septum later than FtsZ and disassembles later than FtsZ, similar to GpsB. (A) Representative phase-contrast and fluorescence micrographs of Rx1 cells expressing ftsZ-mCherry and gfp-sepF at each of the six stages of cell division are shown. Arrows highlight the lag of GFP-SepF with respect to FtsZ-mCherry in stage 4. (B) Representative phase-contrast and fluorescence micrographs of Rx1 cells expressing ftsZ-mCherry and gfp-gpsB at each of the six stages of cell division are shown. Arrows highlight the lag of GFP-GpsB with respect to FtsZ-mCherry in stage 4. Percentages of cells at the indicated stage are listed above the representative images. Fluorescence intensity plots for each stage are shown at the bottom. au, arbitrary units. Scale bars, 1 μm.
DISCUSSION
Here, we show that in contrast to the model rod-shaped bacteria E. coli and B. subtilis, the inactivation of FtsA in S. pneumoniae results in cell lysis rather than cell filamentation. The results confirm that ftsA is essential in S. pneumoniae (20) and rule out the possibility of a polar effect on the downstream ftsZ gene. Such an effect was postulated to explain ftsA essentiality in S. pneumoniae (28), because B. subtilis ftsA is not essential (18, 19) unless sepF (28) or gpsB (61) is also inactivated.
S. pneumoniae, like E. coli and B. subtilis, grows and divides by carrying out coordinated peripheral and septal PG synthesis (32–36). Such a mechanism predicts that in ovoid-shaped cocci, like S. pneumoniae, even an early block in cell division, such as inactivation of FtsA, would cause cells to elongate into rods rather than expand and lyse (35, 63). The cell lysis observed upon depletion of FtsA can be rationalized by considering at least two characteristics that distinguish ovoid-shaped streptococci from their rod-shaped counterparts, despite the presence of two PG biosynthetic complexes. First, proteins within peripheral and septal PG synthesis complexes all localize to midcell (33–36). Second, inactivation or depletion of proteins that function late in cell division (e.g., for septum remodeling or closure) are associated with a variety of phenotypes but not cell lysis (39, 40, 47, 60, 62, 64), whereas those involved in peripheral or septal PG synthesis result in cell lysis, independently of the respective protein's role in cell elongation or cell division (40, 41, 44, 65, 66).
This correlation underscores the intimate coordination and chronology between the two PG biosynthetic machineries (34–36, 43). This connection is best illustrated by S. pneumoniae PBP2x (the E. coli PBP3 homolog), which is the essential transpeptidase required to build the cell division septum. Selective inhibition of PBP2x by methicillin results in cell elongation, while its depletion results in cell lysis (40, 41, 44, 66). Here, the depletion of FtsA shows similar behavior, with partial depletion resulting in cell elongation and inhibition of septum closure, and full depletion resulting in cell enlargement and lysis. This two-stage progression is particularly evident in time-lapse studies (Fig. 6B, bottom panels; see also Movie S2 in the supplemental material).
In filaments of an ftsA temperature-sensitive (Ts) E. coli mutant at 42°C, FtsZ can localize at cell division sites (22), indicating that FtsZ assembly at midcell can occur independently of FtsA. In contrast, FtsZ rapidly delocalizes after depletion of FtsA in S. pneumoniae, indicating that FtsA is required for efficient assembly of Z rings at midcell in this organism. This supports the notion that FtsA is a key membrane anchor for FtsZ in Gram-positive organisms that lack a ZipA homolog (19, 67), although EzrA and SepF both bind the membrane and FtsZ and thus also have the potential to be membrane tethers for the Z ring (29–31). Of these two proteins, EzrA is essential in S. pneumoniae (35; A. J. Perez, unpublished data), whereas SepF is not (60; this work). Another distinction from the E. coli division apparatus is that whereas FtsZ disassembles from the septum before FtsA in that species (48, 68), FtsA and FtsZ colocalize at all stages of cell division in S. pneumoniae, with no significantly detectable lag by FtsA. This is consistent with a complete dependence of FtsZ localization on functional FtsA in S. pneumoniae.
Why is FtsA essential in S. pneumoniae? In B. subtilis, the lack of essentiality of FtsA has been attributed to the presence of SepF, which was proposed also to anchor FtsZ to the membrane and thus support growth in the absence of FtsA (31). In line with this notion, knockouts of sepF and ftsA are synthetically lethal in B. subtilis, and overproduction of SepF can compensate in part for the cell division defects caused by the absence of FtsA (28). Nevertheless, although a B. subtilis ftsA-null mutant is viable, it shows a severe Ts phenotype at 45°C, growing as filaments prone to lysis and in which FtsZ is somewhat dispersed (18, 19). FtsZ is localized at regular intervals along the filaments, but the majority of the Z rings are abnormal (19), suggesting that under these conditions, a functional FtsA is still required for proper Z-ring activity despite the presence of SepF. In S. pneumoniae, the deletion of sepF results in a highly heterogeneous population of elongated and enlarged cells clearly impaired in cell division (60), but Z rings and bands of HADA staining are still present in the elongated cells (Fig. 8 and S4). This result strongly suggests that SepF is not a key membrane anchor for the Z ring, but rather that SepF, as originally proposed and consistent with its name (27), plays a later role in cell division, likely in septal ring progression or closure, along with GpsB and PBP2x (40). The ability of extra FtsA to correct defects of ΔsepF mutants or ΔgpsB mutants in some genetic backgrounds suggests that FtsA has a direct or indirect role in promoting septum closure, likely activating PBP2x. Because FtsA is also crucial for maintaining a coherent Z ring in S. pneumoniae, it is possible that SepF has a similarly early stage role as well. Overall, our results suggest that FtsA is critical for both early and late stages in septum formation. Consistent with this idea, new results suggest that EzrA could mediate early and late stages of septum formation by forming complexes with Z-ring components and with GpsB, which forms complexes with PBPs and is required for PBP2x migration to the center of division septa (44; Rued et al., unpublished).
Indeed, one surprising finding in this study is that depletion of FtsA in S. pneumoniae leads to cell lysis. This indicates that in the absence of FtsA, cells can neither elongate nor divide, strongly implicating a role for FtsA in both activities and further supporting the idea that the activities of the biosynthetic components that catalyze pneumococcal peripheral and septal PG synthesis are coordinated throughout the cell cycle, even as PBP2x separates to the centers of septa in middle-to-late-division stages (35, 45, 62). Consistently, PG synthesis is dispersed in the absence of FtsA. As cell expansion and lysis are preceded by cell elongation and FtsZ delocalization, one model would be that cell lysis is a direct consequence of the loss of Z rings and that the role of FtsA is mainly to anchor FtsZ to the membrane to maintain Z-ring integrity. In this model, FtsZ directs the synthesis of both the peripheral wall and the septum from midcell.
In a second model, FtsA itself is a key orchestrator and coordinator of septal and peripheral PG synthesis through interactions with additional proteins. In this model, FtsA would organize the PG synthesis machineries at midcell to build both the peripheral wall and the septum, possibly by partly assuming functions played by MreB, which is present in model rod-shaped bacteria (69) but absent in S. pneumoniae (33, 65). FtsA and MreB are both structural homologs of actin (70), and both have the ability to interact with FtsZ (71). Moreover, FtsA and MreB have been proposed to share an evolutionary origin that subsequently diverged to perform parallel functions within the divisome and the elongasome, respectively (72). It is therefore tempting to speculate that in S. pneumoniae, which compared to bacilli has a different and less sophisticated elongation scheme, a single actin-like protein, FtsA, could perform both roles, taking over the functions of MreB, as seems to occur in preseptal PG synthesis in E. coli (71, 73–75). Some features of FtsA in S. pneumoniae, such as its higher concentration and FtsA-to-FtsZ ratio (20), are consistent with this replacement role. The ability of extra FtsA to generate cells that are both shorter and narrower also suggests that FtsA may control a rate-limiting step in septal as well as peripheral PG synthesis. Finally, there is a strong precedent from Gram-negative bacteria that FtsA not only anchors FtsZ to cell membranes but plays regulatory roles in divisome assembly and activation (2).
FtsA attaches to the cell membrane by its C-terminal membrane-targeting sequence (76) and, accordingly, we have observed that FtsZ cannot be efficiently maintained at midcell in the absence of FtsA. However, because FtsZ is similarly needed for FtsA to find the septum, further work will be necessary to separate any direct role for FtsA in directing both modes of PG synthesis from effects on FtsZ assembly itself. Nevertheless, our results illustrate how integration of the elongation and septation machineries at a single midcell location in S. pneumoniae leads to key differences in cell growth patterns of ovoid-shaped cocci compared to rods. Moreover, although FtsZ and FtsA colocalize in midcell ring complexes throughout division, it remains to be determined whether the organization of FtsZ and FtsA is uniform within these rings or whether the organization of these two essential proteins is dynamic within rings, thereby allowing interactions with additional proteins at different stages of the cell cycle.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. S. pneumoniae strains were derived from Rx1, an unencapsulated laboratory strain (77), and from an unencapsulated derivative (Δcps) of serotype 2 strain D39, which was the progenitor of Rx1 (49). Rx1 strains were routinely grown on Columbia agar (CA; Sigma-Aldrich) or tryptone soya agar (TSA; Oxoid) supplemented with 5% defibrinated sheep blood (Columbia blood agar [CBA] or tryptone soya blood agar [TSBA]) or tryptone soya broth (TSB; Oxoid) at 37°C or 28°C statically, in an atmosphere of 5% CO2, when available (38, 60). Rx1 strains were also grown in the semisynthetic C medium (78) supplemented with 0.1% yeast extract (C+Y). In particular, this medium was used in gene expression experiments from the PczcD (PZn) promoter to allow more precise control of ZnCl2 induction, as well as better visualization of the GFP fluorescence (39, 54). D39 Δcps strains were grown statically in brain heart infusion broth (BHI) or on TSBA plates at 37°C or 30°C in an atmosphere of 5% CO2, as described previously (40, 44, 45). When appropriate, chloramphenicol (4.5 μg/ml), tetracycline (2.5 μg/ml), and/or erythromycin (0.25 μg/ml) were added to the growth medium. For induction of the PZn promoter in Rx1 strains, ZnCl2 (Sigma-Aldrich) was added to liquid medium and blood agar plates at different concentrations, depending on the medium used (39). The PZn promoter was induced in D39 Δcps strains by the addition of different concentrations of ZnCl2 and 0.1× the concentration of MnSO4 (45). Competent S. pneumoniae cells were prepared and transformed by the addition of competence-inducing peptide 1 (CSP-1), as previously described (40, 60).
E. coli DH5α (79) was used as a host for cloning and plasmid propagation. Strains were grown in Luria-Bertani (LB) broth and agar at 37°C and supplemented with ampicillin (150 μg/ml) when required.
Growth curves and microscopy.
For physiological and morphological analyses of Rx1 strains, cells were inoculated from frozen glycerol stocks or starters (optical density [OD], 0.3) into prewarmed TSB or C+Y medium and incubated at the appropriate temperature. Growth was monitored turbidimetrically every 30 min with an Ultrospec 3100 (Amersham Pharmacia Biotech) or with a DU 730 spectrophotometer (Beckman Coulter) at 650 nm (TSB) or 600 nm (C+Y). Growth determinations were performed for D39 Δcps strains, as described previously (40, 44, 45).
For microscopic analysis of Rx1 strains, samples were taken every 30 min and observed immediately under the microscope. Alternatively, samples (1 ml) were transferred to Eppendorf tubes, centrifuged, resuspended, and fixed in 50 to 100 μl of a 4% paraformaldehyde solution (Immunofix; Bio-Optica). Aliquots of 2 to 10 μl were transferred to poly-l-lysine-coated slides and examined using a Zeiss Axioskop HBO 50 or an Olympus CellR IX 81 microscope equipped with 100× phase-contrast and fluorescence objectives. Photographs were taken with a Zeiss MC100 Spot camera or with an Olympus FV2T digital black-and-white (B/W) FireWire camera. Sizes (in micrometers) of at least 100 cells for each sample were measured using automated MicrobeTracker software and the results further analyzed in Microsoft Excel. For fluorescence microscopy, 2 μl was spotted onto a microscope slide, covered with a 1% phosphate-buffered saline (PBS)–agarose slab, and observed using an Olympus CellR IX 81 microscope equipped with an Olympus FV2T digital B/W FireWire camera.
Time-lapse microscopy was essentially carried out according to the method previously reported (39). Briefly, cells were grown in TSBA plates for 14 h at 28°C. Cells were then scraped, rinsed once in 1× PBS, and suspended in prewarmed TSB, without or with the appropriate concentration of ZnCl2, to an OD at 650 nm (OD650) of 0.05 to 0.1. The suspension (2 μl) was attached to a thin 1% low-melting-point agarose–TSB matrix. The microscope slide was incubated at 28°C in a temperature-controlled chamber of an Olympus CellR IX 81 microscope. Phase-contrast and fluorescence images were taken every 30 min. Movies were assembled using the ImageJ software.
D39 Δcps strains expressing epitope-tagged proteins from native chromosomal loci were constructed using methods described previously (40, 44, 45), as outlined in Table S1 using PCR amplicons listed in Table S2 (Fig. S1B and S2). Two-dimensional epifluorescence microscopy (EFM), 2D immunofluorescence microscopy (IFM), and 3D-SIM IFM of D39 Δcps strains expressing epitope-tagged proteins were performed as described previously (40, 44, 45). The primary antibodies used were anti-FLAG rabbit polyclonal antibody (1:100 dilution; catalog no. F7425; Sigma) and mouse monoclonal antibody against c-Myc (1:100 dilution; catalog no. R950-250; Life Technologies). The secondary antibodies used were 1:100 dilutions of Alexa Fluor 488–highly cross-absorbed goat anti-mouse antibody and 1:100 dilutions of Alexa Fluor 568–highly cross-absorbed goat anti-rabbit antibody (catalog no. A11031; Life Technologies). Cells were mounted in SlowFade gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (catalog no. S36936; Thermo Fisher/Life Technologies). DNA in nucleoids was stained using mounting medium SlowFade gold antifade reagent with DAPI (catalog no. S36936; Life Technologies). Control experiments showed that labeling of single-tagged strains with the double-labeling procedure containing both sets of primary and secondary antibodies produced signal only in the expected fluorescence channel (data not shown), indicating that there was no detectable nonspecific binding of the primary or secondary antibodies to untagged proteins, that nonspecific binding of secondary antibodies did not occur to the primary antibodies of a different animal, and that the excitation and emission wavelength cutoffs of the fluorescence filter blocks allowed the detection of fluorescence signals from only Alexa Fluor 488 or Alexa Fluor 568, depending on the setting. Cells in 2D-EFM and 2D-IFM images were binned into division stages, and cell dimensions and protein locations were determined and quantitated using a MATLAB GUI program reported before (40).
Plasmid construction and DNA manipulation.
Standard protocols for molecular cloning, transformation, and DNA analysis were used (80). Oligonucleotides were obtained from Sigma-Aldrich. Restriction enzymes, DNA polymerase, and T4 ligase were purchased from Roche Diagnostics or New England BioLabs. The plasmids, constructs, and primers used in this study are listed in Tables S1 and S2.
Construction of an ftsA conditional knockout mutant and depletion of FtsA.
To delete ftsA in Rx1, we first constructed a merodiploid strain by transforming the PvuI-digested pJWV25-PZngfp-ftsA plasmid (39) into Rx1 and verified the correct insertion of the PZngfp-ftsA construct at the bgaA locus by PCR using the primers listed in Table S2. The resulting merodiploid strain, Rx1 ftsA+//PZngfp-ftsA+, carries an additional copy of the ftsA gene at the bgaA locus and expresses GFP-FtsA upon induction with ZnCl2. The construct for the ftsA knockout was obtained by a two-step PCR procedure (20). Three sets of primers, AKOF1/AKOR2, AKOF3/AKOR4, and AKOF5/AKOR6 (Table S2), were used to generate an ftsA::Plesscat construct containing a promoterless cat gene (Fig. S1A). The first PCR-derived fragments, obtained using Rx1 chromosomal and pR326 DNA as the templates, were purified using a QIAquick PCR purification kit (Qiagen), mixed at a ratio of 1:1:1, and reamplified in a second PCR, using the AKOF1/AKOR6 primers. The construct was then transformed into the S. pneumoniae Rx1 ftsA+//PZngfp-ftsA+ merodiploid parent strain, and transformants were selected on TSBA plates containing chloramphenicol (4.5 μg/ml) and 0.45 mM ZnCl2 after 24 h of incubation at 37°C. Transformants were isolated in single colonies on TSBA plates containing tetracycline (2.5 μg/ml), chloramphenicol (4.5 μg/ml), and 0.45 or 0.65 mM ZnCl2 and stored as frozen glycerol stocks. The correct insertion of the ftsA::cat construct at the native locus was confirmed by PCR using primers external to the region of integration (Fig. S1A and Table S2). Zinc-dependent growth of the Rx1 ΔftsA//PZngfp-ftsA+ mutant was confirmed in TSBA and TSB in the absence or presence of different concentrations of ZnCl2 ± 0.01 mM MnCl2. Frozen glycerol starters (OD, 0.3) were prepared from cells grown in TSB with 0.45 or 0.65 mM ZnCl2 at 37°C.
For depletion experiments, the Rx1 ΔftsA//PZngfp-ftsA+ strain was inoculated from frozen glycerol starters into 10 ml of TSB or C+Y medium containing 0.65 or 0.15 mM ZnCl2, respectively, serially diluted into the same medium, and incubated at 37°C. Overnight cultures with an OD of ≤0.4 were centrifuged (8,000 × g for 5 min at 25°C), and the pellets were washed with 10 ml of the respective medium lacking ZnCl2 and collected again by centrifugation (8,000 × g for 5 min at 25°C). Residual supernatant fractions were removed, and the pellets were resuspended in 10 ml of prewarmed medium and used to inoculate a series of 10-ml cultures in the appropriate medium and the following final concentrations of ZnCl2: 0, 0.075, 0.15, and 0.65 mM for TSB and 0, 0.05, 0.1, and 0.15 for C+Y medium. Alternatively, the Rx1 ΔftsA//PZngfp-ftsA+ strain was inoculated directly from frozen starters (1:50) into 40 ml of prewarmed TSB or C+Y medium without or with the appropriate concentration of ZnCl2. Growth was monitored turbidimetrically every 30 min, and cells were observed at various time points by phase-contrast and fluorescence microscopy. Similar FtsA depletion experiments were performed using a D39 Δcps ΔftsA//PZnftsA+ conditional null strain (Fig. S1B and Table S1). Growth of this strain was also Zn dependent and showed a normal growth curve and cell morphology in BHI broth containing 0.5 mM ZnCl2 plus 0.05 mM MnSO4 at 37°C (data not shown).
Construction of strains expressing other fluorescently tagged cell division proteins.
To obtain S. pneumoniae Rx1 and derivatives expressing gfp-sepF and gfp-gpsB encoding the respective tagged fluorescent proteins, competent cells were transformed with PvuI-digested pJWV25-PZngfp-sepF or pJWV25-PZngfp-gpsB plasmid. Transformants were selected on TSBA or CBA plates containing 2.5 μg/ml tetracycline and verified for correct insertion of the respective PZngfp-sepF and PZngfp-gpsB constructs at the bgaA locus by PCR. To obtain S. pneumoniae Rx1 and derivative strains expressing ftsZ-mCherry, competent cells were transformed with an ftsZ::mCherry-Pc-erm construct (47). Transformants were selected on TSBA or CBA plates containing 0.25 μg/ml erythromycin. Fluorescence microscopy of cells expressing ftsZ-mCherry was performed as described above from cells cultured at 28°C.
Western blot immunodetection and analysis.
Western blotting was performed to evaluate the expression of native and/or fusion proteins in Rx1 strains. Cells were grown in the appropriate medium at the required temperature and monitored turbidimetrically until they reached an OD of 0.3 to 0.4. Cultures (2 ml) were rapidly chilled and microcentrifuged (15,800 × g, 3 min, 4°C). When necessary, different culture volumes were taken to contain approximately the same cellular dry weight, as calculated from the ODs (81). Pellets were resuspended in 40 μl of 1× SEDS lysis buffer (0.02% SDS, 15 mM EDTA, 0.01% deoxycholate, 150 mM NaCl) and incubated for 5 min at 37°C (39). Lysates were then mixed with 40 μl of 2× Laemmli buffer (Bio-Rad) and boiled at 100°C for 10 min. After separation by SDS-PAGE, proteins were transferred to a nitrocellulose (Protran BA83; Schleicher and Schuell) or a polyvinylidene difluoride (PVDF) (Immobilon-P) membrane. After blocking, membranes were incubated with anti-FtsA and anti-FtsZ antibodies (20) or anti-GFP (Santa Cruz Biotechnology, Inc.) polyclonal antibody, at the appropriate dilution, for 1 h at room temperature and, after washing, with horseradish peroxidase (HRP)-conjugated antibodies (Bio-Rad) or A8275 (Sigma). Finally, chemiluminescent bands were detected using the Immun-Star HRP chemiluminescent kit (Bio-Rad) and Kodak BioMax light films or SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and Agfa medical blue X-ray film. After blotting, membranes were stained with 0.4% amido black solution (Lachema N.P., Brno, Czech Republic). Quantitative analysis was performed using the public domain software ImageJ version 1.48v (National Institutes of Health [https://imagej.nih.gov/ij/index.html]).
For Western blot assays of D39 Δcps strains, total cell lysates were prepared using SEDS lysis buffer, as described above, with the following modifications. Two milliliters of cells was collected by centrifugation (5 min at 16,000 × g at 4°C) and resuspended in 1 ml of cold PBS at 4°C. Following another centrifugation, the supernatants were removed, and the pellets were frozen rapidly at −80°C and used within 1 to 24 h. The cell pellets were thawed at 25°C for 10 min, resuspended in 40 μl of 1× SEDS lysis buffer with protease inhibitor (catalog no. 88266; Thermo), and incubated at 37°C for 15 min on a shaking block (600 rpm). The protein concentration of each lysate sample was determined using the DC protein assay (Bio-Rad), and protein levels were normalized for each lane in the same gel. Thirty microliters of lysates was mixed with 30 μl of 2× Laemmli buffer (Bio-Rad) and boiled at 100°C for 10 min. Proteins were separated by SDS-PAGE on 4 to 15% TGX precast gradient gels (Bio-Rad). A PAGE ruler prestained protein standard (catalog no. 10748-010; Life Technologies) was used to determine polypeptide molecular weights. Rabbit anti-FtsZ and rabbit anti-FtsA were used as primary antibodies to determine FtsZ and FtsA amounts, respectively (20), at a 1:10,000 ratio. Secondary incubations were performed using HRP–donkey anti-rabbit antibody for 1 h at a 1:10,000 ratio. Chemiluminescent signal in the protein bands was detected and quantitated as described previously (82).
Electron microscopy.
Transmission electron and immunoelectron microscopy were carried out similarly as previously described (20). S. pneumoniae Rx1 strains were cultured in TSB or C+Y medium at the appropriate temperature, and growth was monitored turbidimetrically every 30 min. At the desired times, cultures were rapidly chilled on ice, and different volumes, corresponding to a total cellular dry weight of 0.580 to 0.600 μg, were taken, centrifuged (4,320 × g, 15 min, 4°C), washed in 10 mM phosphate (pH 7.0), and fixed in 1 ml of 4% paraformaldehyde and 0.05% glutaraldehyde in 1× PBS for 1 h at 4°C. The fixed cells were gently washed with 1× PBS and stored at 4°C until used. Small pellets of fixed cells were cryoprotected with glycerol, applied to small pieces of filter paper, and quickly frozen in liquid ethane. Frozen specimens were transferred to a Reichert-Jung AFS freeze substitution unit (Leica, Vienna, Austria) for 48 h at 90°C in a mixture of methanol and 0.5% (wt/vol) uranyl acetate for complete substitution of the water in the sample. After freeze substitution, samples were infiltrated using Lowicryl K4M (EML Laboratories, Berkshire, United Kingdom) at 30°C and polymerized with UV light. Ultrathin sections were deposited on 200-mesh gold grids covered with Formvar and carbon and processed for immunogold labeling as follows. After 30 min of blocking with TBS-BSA-Tween buffer (30 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% bovine serum albumin, 0.2% Tween 20), sections were incubated for 1 h in TBS-BSA-Tween containing anti-FtsA (1:100) or anti-FtsZ (1:100) antibody, and then the grids were washed with TBS. Next, grids were blocked again for 15 min on TBS-BSA-Tween before a 45-min incubation with 10 nM gold-labeled goat anti-rabbit IgG (1:40) in TBS-BSA-Tween. The grids were then washed three times with TBS and four times with distilled water, for 10 min each. Images were collected with a JEOL JEM1011 transmission electron microscope (Zeiss) and photographed with an 11-megapixel charge-coupled-device Gatan Erlangshen ES1000W camera. The software used for image capture was Gatan digital micrograph 1.8.0, and Adobe Photoshop CS5 was used for processing. Statistical analysis was done as previously described (20).
Short-pulse-labeling with fluorescent d-amino acids.
The fluorescent d-amino acid HADA (HCC-amino–d-alanine), a hydroxy coumarin derivative of d-alanine (56), was purchased from the Department of Chemistry, Indiana University, Bloomington, IN. For HADA short-pulse-labeling, 1 ml of cells, cultured as described above, was taken and aliquoted into Eppendorf tubes. Two microliters of HADA (stock solution 100 mM in dimethyl sulfoxide [DMSO], stored at −80°C) was added, and after gentle mixing, samples were incubated at 37°C for 4 min. Tubes were immediately put on ice for 1 min and then spun down at 16,000 × g for 5 min at 4°C. The supernatants were discarded, and pellets were washed twice with PBS and then resuspended in 40 to 80 μl of PBS. Two microliters of the cell suspensions was spotted onto a microscope slide, covered with a 1% PBS-agarose slab, and immediately observed using an Olympus CellR IX 81 microscope equipped with an Olympus FV2T digital B/W camera. The HADA signal was visualized using a DAPI filter with a 1-s exposure.
Supplementary Material
ACKNOWLEDGMENTS
We thank Josef Čáslavský and Aleš Ulrych for their advice on time-lapse microscopy and colocalization studies. We also thank the Electron Microscopy Facility of CNB-CSIC for their technical support. We thank Michael B. Whalen, Linda Doubravová, Federico Corona, and Jesus Bazan for helpful comments on the work.
Work in the O.M. lab was supported by the Autonomous Region of Sardinia (RAS) LR7/2007 (grant CRP2-401). A.M. was supported by a Ph.D. scholarship from the Sardinia Regional Government (European Social Fund 2007-2013). Work in the P.B. laboratory was supported by the Czech Science Foundation (grant P302/12/0256), by the Ministry of Education, Youth and Sports of the Czech Republic (grant LH 12055), and by Institutional Research Concept (grant RVO 61388971). Work in the M.E.W. lab was supported by grant GM113172 from the National Institutes of Health. The work of D.D. was supported by the Deutsche Forschungsgemeinschaft (grant Ha 1011/13-1). Work in the M.V. lab was funded by the Spanish Ministerio de Ciencia e Innovación (grant BIO2011-28941-C03-01) and by the Spanish Ministerio de Economía y Competitividad (grant BFU2014-52070-C2-1-P). W.M. was supported by grant GM61074 from the National Institutes of Health.
A.M., D.F., A.J.P., M.L.D., H.-C.T.T., A.I.R., M.K., D.D., M.E.W., P.B., M.V., W.M., and O.M participated in the conception or design of the study; A.M., D.F., A.J.P., M.L.D., H.-C.T.T., D.M., A.I.R., M.K., M.E.W., W.M., and O.M. participated in the acquisition, analysis, or interpretation of the data; A.M., W.M., and O.M wrote the manuscript; and D.F., A.I.R., M.K., D.D., H.-C.T.T., P.B., M.V., and M.E.W. made comments on and revisions to the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00608-16.
REFERENCES
- 1.Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 2.Busiek KK, Margolin W. 2015. Bacterial actin and tubulin homologs in cell growth and division. Curr Biol 25:R243–R254. doi: 10.1016/j.cub.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Boer P, Crossley R, Rothfield L. 1992. The essential bacterial cell-division protein FtsZ is a GTPase. Nature 359:254–256. doi: 10.1038/359254a0. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee A, Lutkenhaus J. 1994. Guanine nucleotide-dependent assembly of FtsZ into filaments. J Bacteriol 176:2754–2758. doi: 10.1128/jb.176.9.2754-2758.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J 21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson HP, Anderson DE, Osawa M. 2010. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egan AJF, Vollmer W. 2013. The physiology of bacterial cell division. Ann N Y Acad Sci 1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- 8.Rico AI, Krupka M, Vicente M. 2013. In the beginning, Escherichia coli assembled the proto-ring: an initial phase of division. J Biol Chem 288:20830–20836. doi: 10.1074/jbc.R113.479519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sánchez M, Valencia A, Ferrándiz MJ, Sander C, Vicente M. 1994. Correlation between the structure and biochemical activities of FtsA, an essential cell division protein of the actin family. EMBO J 13:4919–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Ent F, Löwe J. 2000. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J 19:5300–5307. doi: 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 12.Pichoff S, Shen B, Sullivan B, Lutkenhaus J. 2012. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA's self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol 83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma X, Margolin W. 1999. Genetic and functional analyses of the conserved C-terminal core domain of Escherichia coli FtsZ. J Bacteriol 181:7531–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbin BD, Geissler B, Sadasivam M, Margolin W. 2004. Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J Bacteriol 186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rico AI, García-Ovalle M, Mingorance J, Vicente M. 2004. Role of two essential domains of Escherichia coli FtsA in localization and progression of the division ring. Mol Microbiol 53:1359–1371. doi: 10.1111/j.1365-2958.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- 16.Lutkenhaus JF, Donachie WD. 1979. Identification of the ftsA gene product. J Bacteriol 137:1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donachie WD, Begg KJ, Lutkenhaus JF, Salmond GPC, Martínez-Salas E, Vicente M. 1979. Role of the ftsA gene product in control of Escherichia coli cell division. J Bacteriol 140:388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beall B, Lutkenhaus J. 1992. Impaired cell division and sporulation of a Bacillus subtilis strain with the ftsA gene deleted. J Bacteriol 174:2398–2403. doi: 10.1128/jb.174.7.2398-2403.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen SO, Thompson LS, Harry EJ. 2005. Cell division in Bacillus subtilis: FtsZ and FtsA association is Z-ring independent, and FtsA is required for efficient midcell Z-ring assembly. J Bacteriol 187:6536–6544. doi: 10.1128/JB.187.18.6536-6544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. 2005. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Mol Microbiol 55:699–711. [DOI] [PubMed] [Google Scholar]
- 21.Addinall SG, Lutkenhaus J. 1996. FtsA is localized to the septum in an FtsZ-dependent manner. J Bacteriol 178:7167–7172. doi: 10.1128/jb.178.24.7167-7172.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addinall SG, Bi EF, Lutkenhaus J. 1996. FtsZ ring formation in fts mutants. J Bacteriol 178:3877–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale CA, de Boer PA. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185. doi: 10.1016/S0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 24.Shiomi D, Margolin W. 2008. Compensation for the loss of the conserved membrane targeting sequence of FtsA provides new insights into its function. Mol Microbiol 67:558–569. doi: 10.1111/j.1365-2958.2007.06085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geissler B, Elraheb D, Margolin W. 2003. A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A 100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geissler B, Shiomi D, Margolin W. 2007. The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology 153:814–825. doi: 10.1099/mic.0.2006/001834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamoen LW, Meile JC, de Jong W, Noirot P, Errington J. 2006. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol Microbiol 59:989–999. doi: 10.1111/j.1365-2958.2005.04987.x. [DOI] [PubMed] [Google Scholar]
- 28.Ishikawa S, Kawai Y, Hiramatsu K, Kuwano M, Ogasawara N. 2006. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol Microbiol 60:1364–1380. doi: 10.1111/j.1365-2958.2006.05184.x. [DOI] [PubMed] [Google Scholar]
- 29.Levin PA, Kurtser IG, Grossman AD. 1999. Identification and characterization of a negative regulator of FtsZ ring formation in Bacillus subtilis. Proc Natl Acad Sci U S A 96:9642–9647. doi: 10.1073/pnas.96.17.9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cleverley RM, Barrett JR, Baslé A, Bui NK, Hewitt L, Solovyova A, Xu ZQ, Daniel RA, Dixon NE, Harry EJ, Oakley AJ, Vollmer W, Lewis RJ. 2014. Structure and function of a spectrin-like regulator of bacterial cytokinesis. Nat Commun 5:5421. doi: 10.1038/ncomms6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duman R, Ishikawa S, Celik I, Strahl H, Ogasawara N, Troc P, Lowe J, Hamoen LW. 2013. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc Natl Acad Sci U S A 110:E4601–E4610. doi: 10.1073/pnas.1313978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higgins ML, Shockman GD. 1970. Model for cell wall growth of Streptococcus faecalis. J Bacteriol 101:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zapun A, Vernet T, Pinho MG. 2008. The different shapes of cocci. FEMS Microbiol Rev 32:345–360. doi: 10.1111/j.1574-6976.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- 34.Sham LT, Tsui H-CT, Land AD, Barendt SM, Winkler ME. 2012. Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opin Microbiol 15:194–203. doi: 10.1016/j.mib.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massidda O, Nováková L, Vollmer W. 2013. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol 15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- 36.Pinho MG, Kjos M, Veening JW. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 37.Morlot C, Zapun A, Dideberg O, Vernet T. 2003. Growth and division of Streptococcus pneumoniae: localization of the high molecular weight penicillin-binding proteins during the cell cycle. Mol Microbiol 50:845–855. doi: 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- 38.Fadda D, Santona A, D'Ulisse V, Ghelardini P, Ennas MG, Whalen MB, Massidda O. 2007. Streptococcus pneumoniae DivIVA: localization and interactions in a MinCD-free context. J Bacteriol 189:1288–1298. doi: 10.1128/JB.01168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beilharz K, Nováková L, Fadda D, Branny P, Massidda O, Veening JW. 2012. Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Natl Acad Sci U S A 109:E905–E913. doi: 10.1073/pnas.1119172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Land AD, Tsui H-CT, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. 2013. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol 90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. 2014. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Mol Microbiol 92:733–755. doi: 10.1111/mmi.12588. [DOI] [PubMed] [Google Scholar]
- 42.den Blaauwen T, de Pedro MA, Nguyen-Distèche M, Ayala JA. 2008. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev 32:321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 43.Philippe J, Vernet T, Zapun A. 2014. The elongation of ovococci. Microb Drug Resist 20:215–221. doi: 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. 2014. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Mol Microbiol 94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. 2016. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol Microbiol 100:1039–1065. doi: 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holečková N, Doubravová L, Massidda O, Molle V, Buriánková K, Benada O, Kofroňová O, Ulrych A, Branny P. 2015. LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio 6(1):e01700-14. doi: 10.1128/mBio.01700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sham LT, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Natl Acad Sci U S A 108:E1061–E1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Söderström B, Mirzadeh K, Toddo S, von Heijne G, Skoglund U, Daley DO. 2016. Coordinated disassembly of the divisome complex in Escherichia coli. Mol Microbiol 101:425–438. doi: 10.1111/mmi.13400. [DOI] [PubMed] [Google Scholar]
- 49.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strauss MP, Liew AT, Turnbull L, Whitchurch CB, Monahan LG, Harry EJ. 2012. 3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: implications for triggering cytokinesis. PLoS Biol 10:e1001389. doi: 10.1371/journal.pbio.1001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buss J1, Coltharp C, Huang T, Pohlmeyer C, Wang SC, Hatem C, Xiao J. 2013. In vivo organization of the FtsZ-ring by ZapA and ZapB revealed by quantitative super-resolution microscopy. Mol Microbiol 89:1099–1120. doi: 10.1111/mmi.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rowlett VW, Margolin W. 2014. 3D-SIM super-resolution of FtsZ and its membrane tethers in Escherichia coli cells. Biophys J 107:L17–L20. doi: 10.1016/j.bpj.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holden SJ, Pengo T, Meibom KL, Fernandez Fernandez C, Collier J, Manley S. 2014. High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization. Proc Natl Acad Sci U S A 111:4566–4571. doi: 10.1073/pnas.1313368111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eberhardt A, Wu LJ, Errington J, Vollmer W, Veening JW. 2009. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol Microbiol 74:395–408. doi: 10.1111/j.1365-2958.2009.06872.x. [DOI] [PubMed] [Google Scholar]
- 55.Pinho MG, Errington J. 2003. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol Microbiol 50:871–881. doi: 10.1046/j.1365-2958.2003.03719.x. [DOI] [PubMed] [Google Scholar]
- 56.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, VanNieuwenhze MS. 2012. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent d-amino acids. Angew Chem Int Ed Engl 51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boersma MJ, Kuru E, Rittichier JT, VanNieuwenhze MS, Brun YV, Winkler ME. 2015. Minimal peptidoglycan (PG) turnover in wild-type and PG hydrolase and cell division mutants of Streptococcus pneumoniae D39 growing planktonically and in host-relevant biofilms. J Bacteriol 197:3472–3485. doi: 10.1128/JB.00541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai K, Lutkenhaus J. 1992. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J Bacteriol 174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dewar SJ, Begg KJ, Donachie WD. 1992. Inhibition of cell division initiation by an imbalance in the ratio of FtsA to FtsZ. J Bacteriol 174:6314–6316. doi: 10.1128/jb.174.19.6314-6316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fadda D, Pischedda C, Caldara F, Whalen MB, Anderluzzi D, Domenici E, Massidda O. 2003. Characterization of divIVA and other genes located in the chromosomal region downstream of the dcw cluster in Streptococcus pneumoniae. J Bacteriol 185:6209–6214. doi: 10.1128/JB.185.20.6209-6214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tavares JR, de Souza RF, Meira GLS, Gueiros-Filho FJ. 2008. Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisome. J Bacteriol 190:7096–7107. doi: 10.1128/JB.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, Grangeasse C. 2014. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet 10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lleo MM, Canepari P, Satta G. 1990. Bacterial cell shape regulation: testing of additional predictions unique to the two-competing-sites model for peptidoglycan assembly and isolation of conditional rod-shaped mutants from some wild-type cocci. J Bacteriol 172:3758–3771. doi: 10.1128/jb.172.7.3758-3771.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fleurie A, Cluzel C, Guiral S, Freton C, Galisson F, Zanella-Cleon I, Di Guilmi AM, Grangeasse C. 2012. Mutational dissection of the S/T-kinase StkP reveals crucial roles in cell division of Streptococcus pneumoniae. Mol Microbiol 83:746–758. doi: 10.1111/j.1365-2958.2011.07962.x. [DOI] [PubMed] [Google Scholar]
- 65.Land AD, Winkler ME. 2011. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol 193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berg KH, Stamsås GA, Straume D, Håvarstein LS. 2013. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol 195:4342–4354. doi: 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szwedziak P, Wang Q, Freund SMV, Löwe J. 2012. FtsA forms actin-like protofilaments. EMBO J 31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Söderström B, Skoog K, Blom H, Weiss DS, von Heijne G, Daley DO. 2014. Disassembly of the divisome in Escherichia coli: evidence that FtsZ dissociates before compartmentalization. Mol Microbiol 92:1–9. doi: 10.1111/mmi.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carballido-López R, Formstone A. 2007. Shape determination in Bacillus subtilis. Curr Opin Microbiol 10:611–616. doi: 10.1016/j.mib.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 70.van den Ent F, Amos LA, Löwe J. 2001. Prokaryotic origin of the actin cytoskeleton. Nature 413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- 71.Fenton AK, Gerdes K. 2013. Direct interaction of FtsZ and MreB is required for septum synthesis and cell division in Escherichia coli. EMBO J 32:1953–1965. doi: 10.1038/emboj.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szwedziak P, Löwe J. 2013. Do the divisome and elongasome share a common evolutionary past? Curr Opin Microbiol 16:745–751. doi: 10.1016/j.mib.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 73.Aaron M, Charbon G, Lam H, Schwarz H, Vollmer W, Jacobs-Wagner C. 2007. The tubulin homologue FtsZ contributes to cell elongation by guiding cell wall precursor synthesis in Caulobacter crescentus. Mol Microbiol 64:938–952. doi: 10.1111/j.1365-2958.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- 74.Varma A, de Pedro MA, Young KD. 2007. FtsZ directs a second mode of peptidoglycan synthesis in Escherichia coli. J Bacteriol 189:5692–5704. doi: 10.1128/JB.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varma A, Young KD. 2009. In Escherichia coli, MreB and FtsZ direct the synthesis of lateral cell wall via independent pathways that require PBP 2. J Bacteriol 191:3526–3533. doi: 10.1128/JB.01812-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krupka M, Cabré EJ, Jiménez M, Rivas G, Rico AI, Vicente M. 2014. Role of the FtsA C terminus as a switch for polymerization and membrane association. mBio 5(6):e02221-14. doi: 10.1128/mBio.02221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ravin AW. 1959. Reciprocal capsular transformations of pneumococci. J Bacteriol 77:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lacks S, Hotchkiss RD. 1960. A study of the genetic material determining an enzyme in Pneumococcus. Biochim Biophys Acta 39:508–518. doi: 10.1016/0006-3002(60)90205-5. [DOI] [PubMed] [Google Scholar]
- 79.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 80.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 81.Massidda O, Kariyama R, Daneo-Moore L, Shockman GD. 1996. Evidence that the PBP 5 synthesis repressor psr of Enterococcus hirae is also involved in the regulation of cell wall composition and other cell wall-related properties. J Bacteriol 178:5272–5278. doi: 10.1128/jb.178.17.5272-5278.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wayne KJ, Sham LT, Tsui HC, Gutu AD, Barendt SM, Keen SK, Winkler ME. 2010. Localization and cellular amounts of the WalRKJ (VicRKX) two-component regulatory system proteins in serotype 2 Streptococcus pneumoniae. J Bacteriol 192:4388–4394. doi: 10.1128/JB.00578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.