

Abstract
Background
Constitutive activation of the PI3K-AKT-mTOR pathway (mTOR pathway) underlies megalencephaly in many patients. Yet, prevalence of the involvement of the PI3K-AKT-mTOR pathway in patients with megalencephaly remains to be elucidated, and molecular diagnosis is challenging. Here, we have successfully established a combination of genetic and biochemical methods for diagnosis of mTOR pathway-associated megalencephaly, and have attempted to delineate the clinical characteristics of the disorder.
Methods
Thirteen patients with an increased head circumference and neurological symptoms participated in the study. To evaluate the activation of the mTOR pathway, we performed western blot analysis to determine the expression levels of phosphorylated S6 ribosomal protein (phospho-S6 protein) in lymphoblastoid cell lines from 12 patients. Multiplex targeted sequencing analysis for 15 genes involved in the mTOR pathway was performed on 12 patients, and whole-exome sequencing was performed on one additional patient. Clinical features and MRI findings were also investigated.
Results
We identified pathogenic mutations in six (AKT3, 1 patient; PIK3R2, 2 patients; PTEN, 3 patients) of the 13 patients. Increased expression of phospho-S6 protein was demonstrated in all five mutation-positive patients in whom western blotting was performed, as well as in three mutation-negative patients. Developmental delay, dysmorphic facial features were observed in almost all patients. Syndactyly/polydactyly and capillary malformations were not observed, even in patients with AKT3 or PIK3R2 mutations. There were no common phenotypes or MRI findings among these patients.
Conclusions
A combination of genetic and biochemical methods successfully identified mTOR pathway involvement in nine of 13 (approximately 70%) patients with megalencephaly, indicating a major contribution of the pathway to the pathogenesis of megalencephaly. Our combined approach could be useful to identify patients who are suitable for future clinical trials using an mTOR inhibitor.
Keywords: AKT3, MCAP, MPPH, Multiplex targeted sequencing, Phosphorylated S6 ribosomal protein, PIK3R2, PTEN
Background
Megalencephaly is accompanied by hyperplasia of the brain parenchyma, and is defined by a head circumference greater than +2 SD from the mean of the general population. Various diseases involve development of the condition, including metabolic diseases, such as Alexander disease and Canavan disease, and syndromes, such as Sotos syndrome and Noonan syndrome [1].
In recent years, both megalencephaly-capillary malformation syndrome (MCAP, OMIM 602501) and megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome (MPPH, OMIM 603387) have been shown to result from gain-of-function mutations in the PI3K-AKT-mTOR pathway (mTOR-pathway) [2]. MCAP and MPPH show very similar symptoms; the main symptoms are progressive megalencephaly, polymicrogyria, capillary malformations, syndactyly, and connective tissue dysplasia in the former [3–5], and progressive megalencephaly, polymicrogyria, and polydactyly in the latter [5–7]. Riviere et al. performed whole exome sequencing (WES) with a next-generation sequencer and identified germline mutations in AKT3 and PIK3R2 and a postzygotic mutation in PIK3CA in patients with MCAP, MPPH, and overlapping phenotypes of MCAP and MPPH [2]. Thereafter, Mirzaa et al. identified a germline mutation in CCND2 in patients with MPPH [8]. Moreover, a loss-of-function mutation in PTEN, which suppresses the mTOR pathway, has been identified in autistic patients with macrocephaly [9, 10]. However, the prevalence of mTOR pathway involvement in patients with megalencephaly remains to be elucidated.
The mTOR pathway is involved in various functions including protein synthesis, lipid synthesis, autophagy, and energy metabolism, and is fundamental to essential biological functions. mTOR was originally discovered as a target protein for the immunosuppressant rapamycin [11, 12]. Rapamycin is used to treat tuberous sclerosis, a disease caused by mutations in either of the genes TSC1 or TSC2, both of which are involved in the regulation of the mTOR pathway [13, 14]. Elucidation of the molecular mechanisms underlying megalencephaly is crucial to determine the value of investigating therapeutic agents, such as rapamycin, in the context of mTOR pathway-associated megalencephaly.
In this study, we conducted genetic and biochemical analyses in 13 patients with increased head circumference and neurological symptoms such as developmental delay and epilepsy, and investigated clinical features and imaging of mTOR pathway-associated megalencephaly.
Methods
Study subjects
We analyzed 13 patients with increased head circumference (>2 SD) and neurological symptoms such as developmental delay or epilepsy. These patients were included in this study, with a possibility of mTOR involvement after other diseases had been ruled out. All patients’ disease was sporadic. Clinical examination failed to make a specific diagnosis for each patient. Experimental protocols were approved by the Ethical Committee for the Study of Human Gene Analysis at Nagoya City University Graduate School of Medical Sciences (approval number 164). Written informed consent was obtained from all patients or their parents.
Whole-exome sequencing
We performed WES in one parent-patient (Patient 1) trio because she had participated in another study of WES on brain malformation. To do this, genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Midi Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Genomic DNA was captured using the SureSelect XT Human All Exon V5 capture library (Agilent Technologies, Santa Clara, CA, USA), and sequenced using the Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) with 100 bp paired-end reads. Exome data processing, variant calling and variant annotation were performed as described previously [15].
Multiplex targeted sequencing
We performed multiplex targeted sequencing in 12 patients. Amplicon libraries of the target gene exons from 15 genes involved in the mTOR pathway were prepared with an Ion AmpliSeq Custom Panel (Thermo Fisher Scientific, Waltham, MA, USA). The number of exons, amplicons, and total targeted bases were 300, 412, and 43216 bases, respectively. This panel allowed theoretical coverage of 97.4% of the targeted sequences (Table 1). The library was prepared using the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific), according to the manufacturer’s instructions. Emulsion PCR was performed using the Ion OneTouch system (Thermo Fisher Scientific) with the Ion OneTouch 200 Template Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Multiplex targeted sequencing was performed with an Ion Torrent Personal Genome Machine (PGM) system using an Ion PGM 200 Sequencing Kit and an Ion 316 or 318 Chip (Thermo Fisher Scientific) according to the manufacturer’s instructions. Sequence data was analyzed using a CLC Genomic Workbench 7.0 (CLC bio, Aarhus, Denmark) and default settings.
Table 1.
The panel of targeted genes involved in mTOR pathway
| Gene | Chr | Number of exons | Number of amplicons | Total Bases | Overall coverage (%) |
|---|---|---|---|---|---|
| PIK3CA | 3 | 20 | 34 | 3407 | 98.3 |
| PIK3CB | 3 | 22 | 30 | 3433 | 98.7 |
| PIK3CD | 1 | 22 | 37 | 3355 | 97.6 |
| PIK3R1 | 5 | 18 | 23 | 2506 | 100 |
| PIK3R2 | 19 | 15 | 19 | 2337 | 79.2 |
| PIK3R3 | 1 | 10 | 14 | 1486 | 100 |
| PTEN | 10 | 9 | 12 | 1302 | 97.6 |
| PDPK1 | 16 | 14 | 19 | 1811 | 95.3 |
| AKT1 | 14 | 13 | 22 | 1573 | 97.5 |
| AKT2 | 19 | 14 | 18 | 1687 | 98.9 |
| AKT3 | 1 | 14 | 17 | 1624 | 98.8 |
| RHEB | 7 | 8 | 8 | 635 | 95.1 |
| MTOR | 1 | 57 | 69 | 8220 | 99.7 |
| TSC1 | 9 | 22 | 29 | 3834 | 100 |
| TSC2 | 6 | 42 | 61 | 6006 | 96.5 |
| Total | 300 | 412 | 43216 | 97.4 |
Chr chromosome
Overall coverage means percent of coverage in target sequence
Validation analysis by Sanger sequencing
We performed conventional Sanger sequencing to validate candidate mutations. We amplified the genomic regions by PCR (primer sequences are available on request), and directly sequenced using an ABI 310 Genetic Analyzer (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Mutation analysis of CCND2
We further sequenced the final exon of CCND2 in patients with no candidate mutation by Sanger sequencing.
Lymphoblastoid cell lines
Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines (LCLs) were established from patients’ peripheral blood using a standard method. LCLs were cultured at 5% CO2 in RPMI medium (Sigma-Aldrich, Tokyo, Japan) with 10% FCS, L-glutamine and an antibiotic.
Western blot analysis
Equal amounts of protein were boiled with SDS sample buffer (45 mmol/L Tris–HCl, pH 6.8, 10% glycerol, 1% SDS, 0.01% bromophenol blue, 50 mmol/L DTT). Proteins in the lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, Japan). Membranes were blocked for 1 h with 5% dried skimmed milk in PBS with 0.1% Tween-20 (Sigma-Aldrich). Membranes were then incubated overnight with primary antibodies against phosphorylated S6 ribosomal protein (phospho-S6) (Ser240/244; diluted 1:1,000; Cell Signaling Technology, Danvers, MA, USA), and GAPDH (diluted 1:10,000; Cell Signaling Technology), followed by 1 h incubation with horseradish peroxidase–conjugated secondary antibody (GE Healthcare, Little Chalfont, UK). Bands were quantified using ImageJ, and the intensity of phospho-S6 was normalized with that of GAPDH. We obtained ratios to the normal control values. Five normal controls showed the ratio of 1.02 ± 0.17. Thus, we adopted 5xSD as cut-off and considered those of 2 or greater to be significantly increased and positive in this study.
Results
WES of patient 1 generated 5.86 giga bases of nucleotide sequence. The average read depth of on-target regions was 69.3. This analysis identified a de novo AKT3 heterozygous mutation [c.686A > G; p.(N229S)] (Table 2) that has been reported previously [2, 16–18], and is considered pathogenic.
Table 2.
Genetic data, clinical features, and MRI findings of patients in this study
| Patient1 | Patient2 | Patient3 | Patient4 | Patient5 | Patient6 | Patient7 | Patient8 | Patient9 | Patient10 | Patient11 | Patien12 | Patient13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 3y0m | 2y3m | 6y8 m | 4y2m | 4y9m | 4y6m | 16y | 4y8 m | 7y4m | 9y | 2y10m | 3y6m | 2y0m |
| Sex | F | M | M | F | F | F | M | M | M | M | M | M | M |
| Elevated p-S6 protein (Relative density) | + (3.4) | + (3.0) | + (3.8) | + (3.6) | ND | + (2.1) | + (2.1) | + (2.8) | + (2.6) | - (0.8) | - (0.9) | - (1.1) | - (1.0) |
| Gene | AKT3 | PIK3R2 | PIK3R2 | PTEN | PTEN | PTEN | - | - | - | - | - | - | - |
| Mutation | c.686A > G p.(N229S) | c.1117G > A p.(G373R) | c.1117G > A p.(G373R) | c.640C > T p.(Q214*) | c.740 T > C p.(L247S) | c.1006C > G p.(Y336*) | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| Inheritance | de novo | de novo | de novo | Mother negative | de novo | de novo | - | - | - | - | - | - | - |
| Clinical features | |||||||||||||
| Gestational age | 36w5d | 36w6d | 40w1d | 37w4d | 38w6d | 39w5d | 36w6d | 36w5d | 41w0d | 38w1d | 39w | 38w6d | 38w3d |
| Birth weight g (SD)a | 2942 (+1.4) | 3450 (+2.7) | 3532 (+1.2) | 2536 (−0.3) | 2854 (+0) | 3200 (+0.7) | 2915 (+1.0) | 3490 (+3.0) | 2464 (−2.3) | 3885 (+3.3) | 3830 (+2.6) | 4120 (+3.5) | 3866 (+3.0) |
| Birth length cm (SD)a | 47.5 (+0.3) | 49.2 (+1.0) | 52.5 (+1.4) | 47 (−0.4) | 50 (+0.7) | 51 (+1.0) | 48 (+0.4) | 48.8 (+0.9) | 47 (−1.6) | 50.6 (+1.3) | NA | NA | 48.4 (+0) |
| Birth OFC cm (SD)a | 39 (+4.9) | 36 (+2.5) | 34 (+0.4) | 33 (+0.1) | 33 (−0.1) | 34 (+0.5) | 35 (+1.8) | 37 (+3.3) | 30 (−2.0) | 41 (+6.4) | 36.5 (+3.0) | 37 (+3.0) | 39 (+4.6) |
| Birth OFC percentile a | >99 | >99 | 67 | 53 | 44 | 70 | 96 | >99 | 2 | >99 | >99 | >99 | >99 |
| Last weight SDb | −1.9 | 0 | −2.3 | +0.4 | −0.7 | +0.8 | −2.6 | −0.2 | −1.2 | −0.2 | +2.8 | +2.2 | +1.4 |
| Last length SDb | −2.0 | −0.2 | −2.8 | −0.4 | −1.9 | +0.2 | −3.3 | −1.3 | −2.7 | 1.1 | +0.7 | −1.8 | +0.2 |
| Last OFC SDc | +6.3 | +4.6 | +3.0 | +4.5 | +4.3 | +3.5 | +4.9 | +4.6 | +2.0 | +8.5 | +5.4 | +3.8 | +4.5 |
| Last OFC percentile c | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | 98 | >99 | >99 | >99 | >99 |
| Overgrowth /Asymmetry | - | + | - | - | - | - | - | - | - | + | - | + | - |
| Vascular malformations | - | - | - | - | - | - | - | - | - | + | - | - | + |
| Syndactyly | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Polydactyly | - | - | - | - | - | - | + | - | - | + | - | + | - |
| Connective tissue dysplasia | - | + | - | - | - | - | - | - | + | - | - | - | - |
| Dysmorphic features | + | + | + | + | + | + | + | + | + | + | + | + | + |
| DQ (assessed method) age | DD | DQ 42 (Denver) 10 m | DD | DQ 76 (KIDS) 2y5m | DQ 85 (KIDS) 4y6m | DQ 59 (K-test) 4y1m | DQ 12 (KIDS) 6y6m | DQ 71 (K-test) 6y8 m | DQ 35 (KIDS) 5y9m | DD | DQ 72 (K-test) 3y0m | DQ 46 (K-test) 1y7m | NA |
| Meaningful words | - | - | - | 1y6m | 8 m | 2y6m | + | 2y | - | + | 2y | - | - |
| Walking alone | - | - | - | 2y4m | 2y2m | 1y6m | + | 1y11m | 1y11m | 4y6m | 1y6m | 2y3m | 1y3m |
| Hypotonia | + | + | + | - | + | - | + | + | + | - | - | + | + |
| Seizure | + | - | + | - | - | - | + | - | - | - | - | - | - |
| MRI findings | |||||||||||||
| Ventriculomegaly | + | + | + | - | - | - | + | + | - | + | + | + | + |
| Polymicrogyria | + | + | + | - | - | - | + | - | - | + | - | + | - |
| Cerebellar tonsillar ectopia | - | - | - | - | - | - | - | + | - | + | - | + | - |
| White matter abnormalities | + | + | + | - | - | - | - | - | - | + | - | + | - |
aSD and percentile were determined on the basis of the national data reported by the Ministry of Health, Welfare, and Labor in Japan in 2010
bSD was determined on the basis of the national data reported by the Ministry of Health, Welfare, and Labor in Japan in 2000
cSD and percentile were determined on the basis of CDC growth Charts for the United States in 2000
DD apparently developmentally delayed but not scored by a standardized method, Denver Denver Developmental Screening Test, DQ developmental quotient, F female, KIDS Kinder Infant Development Scale, K-test the revised version of Kyoto Scale of Psychological Development, M male, NA not available, OFC occipitofrontal circumference, p-S6 phosphorylated S6 protein, SD, standard deviation
Accession number.: AKT3, NM_005465.4; PIK3R2, NM_005027.3; PTEN, NM_000314.5
Multiplex target next-generation sequencing was performed for another 12 patients. The median number of total sequenced bases per patient, of mapped reads, and of mean read length were 84.6 mega bases, 527 k reads, and 160 bases, respectively. The average read depth of the on-target regions was 1002-fold; 93.6% of the target regions had above 100-fold coverage. Using this multiplex targeted sequencing, a de novo PIK3R2 heterozygous mutation [c.1117G > A; p.(G373R)] was identified in two patients, and PTEN heterozygous mutations [c.640C > T; p.(Q214*), c.740 T > C; p.(L247S), c.1006C > G; p.(Y336*)] were identified in three patients (Table 2). Five mutations in patients for whom both parents DNA were available for testing were confirmed to be de novo. Only a mother was available for testing one of the PTEN mutations [c.640C > T; p.(Q214*)], and was found to be negative (Table 2). The missense mutation in PTEN [c.740 T > C; p.(L247S)] was not reported previously. This de novo mutation was located in the C2 domain that is involved in binding to phospholipids in biological membranes [19], and was indicated to be “deleterious” and “possibly damaging” by in silico analysis with SIFT and PolyPhen-2, respectively [20, 21]. Thus, we considered it pathogenic according to the American College of Medical Genetics and Genomics interpretation guidelines [22]. Other mutations were identical to previously reported mutations, and were considered pathogenic [2, 23–25]. As shown in Table 3, the allelic frequency of the mutated allele detected by multiplex target next-generation sequencing was approximately 50%. Hence, all mutations were considered to be germline mutations.
Table 3.
Mutant allele frequency in multiplex target next-generation sequencing
| Subject | Gene | Mutation | Mutant allele | Total allele | % |
|---|---|---|---|---|---|
| Patient 1a | AKT3 | p.(N229S) | 43 | 78 | 55.1 |
| Patient 2 | PIK3R2 | p.(G373R) | 847 | 1828 | 46.3 |
| Patient 3 | PIK3R2 | p.(G373R) | 343 | 689 | 47.8 |
| Patient 4 | PTEN | p.(Q214*) | 1497 | 2922 | 51.2 |
| Patient 5 | PTEN | p.(L247S) | 238 | 500 | 47.6 |
| Patient 6 | PTEN | p.(Y336*) | 96 | 214 | 44.9 |
aThe mutation, which had previously been identified by WES, was reconfirmed by target next-generation sequencing
For the remaining seven patients in whom we did not detect any mutations by multiplex targeted sequencing, the last exon of CCND2 was analyzed in addition, but no mutations were detected. CCND2 is a recently described new MPPH gene [8], and the last exon corresponds to a mutational hotspot. CCND2 was not included in the original targeted sequencing panel because it was reported after our panel was created. It is true that Sanger sequencing has limitations in the identification of somatic mutations. However, all previous CCND2 mutations are considered to be heterozygous germline mutations, and thus at least major germline mutations in CCND2 were excluded in our patients.
Next, we analyzed the expression level of phospho-S6 protein in LCLs available from 12 patients by western blot analysis. Phospho-S6 protein lies downstream of the mTOR pathway, and is a marker of pathway activation [26, 27]. Of six patients for whom pathogenic mutations were identified, LCLs were established from five (LCL was not established for patient 5). All five patients showed an apparent increase in phospho-S6 protein expression. In addition, the expression level was also elevated in three of seven patients for whom no pathogenic mutations were identified (Fig. 1). Patients’ phenotypes are shown in Table 2. While all patients showed +2 SD or larger in head circumference, the head circumference at birth was not necessarily significantly large. Developmental delay, dysmorphic facial features including prominent forehead, long head, and ocular hypertelorism were observed in almost all patients. Syndactyly/polydactyly and capillary malformations, which are considered main symptoms of MCAP and MPPH, were not observed even in patients with AKT3 or PIK3R2 mutations. Regarding brain MRI findings, while ventriculomegaly, polymicrogyria and white matter abnormalities were observed in all patients with AKT3 and PIK3R2 mutations, no obvious abnormalities were found in patients with PTEN mutations (Fig. 2, Table 2). Phenotypes and MRI findings varied among the three mutation-negative patients who showed increased expression of phospho-S6 protein by western blot analysis. Moreover, no apparent phenotypic difference was noted between these patients and the patients with neither a pathogenic mutation, nor an increase in phospho-S6 expression.
Fig. 1.
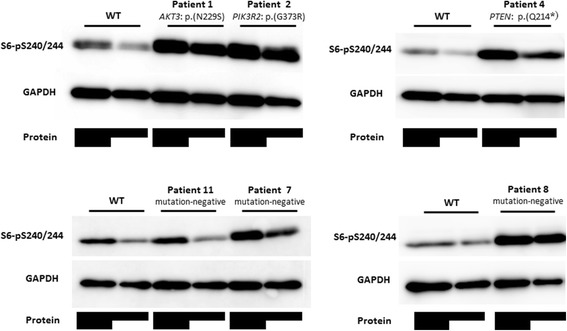
Representative western blot analysis of phospho-S6 protein levels in control and patient LCLs. Expression of phospho S6 (Ser 240/244) and GAPDH in variable amounts of protein extract (total protein indicated by shaded bars) are depicted in the upper and lower panel, respectively. After normalisation with GAPDH, there was increased expression of phospho-S6 in patients with mutations (Patient 1, 2, 4) and without mutations (Patient 7, 8), compared to wild type (WT) and a patient without mutations (Patient 11)
Fig. 2.
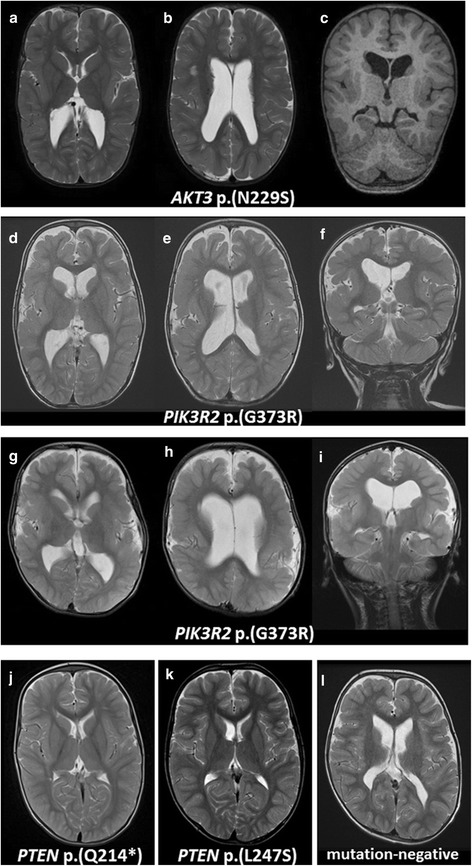
MRI findings in PI3K-AKT-mTOR pathway-associated megalencephaly. T2-weighted axial images (a, b, d, e, g, h, j-l), T1-weighted coronal image (c), T2-weighted images (f, i) of patients (a-c, Patient 1 with an AKT3 mutation [p.(N229S)] at 2 years of age; d-f, Patient 2 with a PIK3R2 mutation [p.(G373R)] at 2 years; g-i, Patient 3 with a PIK3R2 mutation [p.(G373R)] at 6 years; j, Patient 4 with a PTEN mutation [p.(Q214*)] at 2 years; k, Patient 5 with a PTEN mutation [p.(L247S)] at 1 year and 9 months; l, Patient 8 without mutation at 4 years. In Patient 1–3, ventriculomegaly (b, e, h), bilateral polymicrogyria that appears to be most severe in the perisylvian regions but is also present in other regions (a, c, d, f, g, i), and an abnormally high intensity signal from white matter (b, e, h) were observed. The patient with PTEN mutations showed no abnormalities (j, k). Patient 8, without mutation, showed only mild ventriculomegaly (l)
Discussion
In this study, we identified constitutive activation of the mTOR pathway in nine of 13 patients with megalencephaly of unknown etiology, indicating a significant role for the mTOR pathway in pathogenesis of genetic megalencephaly syndromes. We identified pathogenic mutations through multiplex targeted sequencing or WES in six patients. Of note is that all five mutation-positive patients in whom western blot analysis was performed showed abnormal activation of the mTOR pathway. Accordingly, we did not detect pathogenic mutations in four patients who did not show abnormal activation of the mTOR pathway. Thus, western blot analysis of phospho-S6 protein could be used as a biochemical marker to suggest megalencephaly associated with the constitutional activation of the mTOR pathway. It can also aid in confirming the results of molecular genetics analysis. Recently, Loconte et al. described three patients with PIK3CA-related overgrowth syndrome, including one patient with MCAP, and demonstrated the usefulness of a combination of genetic and biochemical methods [28]. They used skin fibroblasts for biochemical investigation, partially because some of their patients showed somatic mosaicism involving only limited parts of the limbs. Skin biopsy would be an important alternative method, particularly for patients with mosaicism, but it is not easily applied as an initial investigation. Our study demonstrated that blood could be used as a starting material to find possible involvement of the mTOR pathway.
In a report from Riviere et al., a PIK3R2 mutation found at the same site [c.1117G > A; p.(G373R)] always presented clinical symptoms of MPPH. PIK3CA mutations were identified at multiple sites, and all but one case showed MCAP. AKT3 mutations were found in patients with overlapping phenotypes or MPPH [2]. Thus, genotype and phenotype correlate considerably with each other. Nevertheless, syndactyly/polydactyly and capillary malformations, which are considered to be the main symptoms of both MCAP and MPPH, were not observed in patients with AKT3 or PIK3R2 mutations in our study. Mutations in PTEN, which suppresses the mTOR pathway, are also found in PTEN hamartoma syndrome, such as Cowden disease, Bannayan syndrome, and Proteus syndrome [19]. However, there is no evident genotype–phenotype association. The PTEN mutation [c.640C > T; p.(Q214*)] detected in our study has also been previously reported in Bannayan syndrome [23], but hamartomatous gastrointestinal polyposis or pigmented patches on the penis was not noted in our patient at the time of final evaluation. Another patient with a PTEN mutation also had no major anomaly other than megalencephaly and dysmorphic facial features. Neither patient had complications with tumors. While all patients showed a +2 SD or larger head circumference, the head circumference at birth was not necessarily large, indicating that megalencephaly in our patients was progressive. While developmental delays were observed in all patients, patients with AKT3 or PIK3R2 mutations had no meaningful words and had a more severe disease phenotype than those with PTEN mutations. Overall, no apparent differences in phenotype could be identified between patients with or without the involvement of the mTOR pathway and between patients with or without identified pathogenic mutations, suggesting genetic heterogeneity of the disorder.
Regarding head MRI findings, enlargement of the ventricle, polymicrogyria, and white matter abnormalities were observed in all patients with AKT3 or PIK3R2 mutations, and seemed indicative of mTOR pathway involvement. One mutation-negative patient, however, did exhibit these features, and thus these findings are not sufficiently specific to indicate all underlying pathogenesis. No abnormal MRI findings were observed in patients with PTEN mutations. The lack of a cortical anomaly has been reported for PTEN macrocephaly [10], and it appears to be an indicator of PTEN involvement.
In our study, target sequencing was performed primarily on 15 genes involved in the mTOR pathway. Therefore, the first conceivable limitation is the possibility of mutations in other genes that were not covered by our gene panel. In addition, our gene panel do not cover the entire exon region (the coverage ratio of PIK3R2 in particular is low at 79.2%), and the Ion PGM method offers only limited indel detection. Nevertheless, our target sequencing is useful in early diagnosis in clinical settings due to the lower costs and shorter turn-around time than WES. Secondly, our strategy is limited when analyzing cases of mosaicism. Indeed, Riviere et al. performed targeted ultra-deep sequencing (coverage of more than 10,000 reads) of the PIK3CA mutation sites in mutation-negative affected individuals, as well as in known mutation carriers and control individuals [2]. This experiment detected additional low-level mosaic mutations missed by Sanger sequencing. Although a 100-fold or greater depth was obtained for about 93% of the target area with our target sequencing method, it is still possible that low-level mosaics were not detectable using this approach. Ultra-deep sequencing could be performed to identify such low-level mosaicism. Our biochemical analysis also used materials from peripheral blood, and thus it could not identify activation of the mTOR pathway in patients with mosaicism. Riviere et al. reported that mTOR pathway activation was also confirmed by means of western blot analysis using LCLs with low-level mosaics of a mutated allele frequency of 16% in the peripheral blood [2]. However, it is unknown whether similar results could be obtained with our protocol. It is also unknown whether similar results can be obtained in patients with lower allelic frequencies of the mutated allele. In rare cases, low-level mosaic mutations are detected only in the saliva, buccal mucosa, and LCLs. Therefore, affected tissues need to be used for molecular diagnosis of patients with mosaicism. The final limitation of this study is the small sample size. Further studies with larger sample sizes are warranted to perform WES or ultra-deep sequencing in patients who are positive with western blot analysis but negative with genetic analysis.
mTOR inhibitors are effective treatment for not only cancer, but for the brain and renal tumors of tuberous sclerosis [14]. They are also effective for epilepsy [13]. Moreover, their effects on intellectual disability and autism have also been demonstrated in experiments using model animals [29, 30]. Thus, the mTOR pathway associated-megalencephaly, a disease for which there is no curative treatment available, could also be a therapeutic target. Seizure related death was reported in a patient with mTOR pathway-associated megalencephaly [7], and thus development of a specific therapeutic intervention is crucial.
Conclusions
A combination of genetic and biochemical methods was able to identify the involvement of the mTOR pathway in approximately 70% of patients with megalencephaly of an unknown etiology. Our combined approach could be useful to identify patients who are suitable for future clinical trials using an mTOR inhibitor.
Acknowledgements
We wish to acknowledge Core Laboratory, Nagoya City University Graduate School of Medical Sciences. We wish to thank all members in the laboratories of Department of Pediatrics and Neonatology, Nagoya City University Graduate School of Medical Sciences, for helpful assistance.
Funding
This study was supported in part by a grant for Research on Applying Health Technology from the Ministry of Health, Labour and Welfare of Japan to F. M., N. O., M. K., M. Y., Y. K., K. K., and S. S.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the ClinVar repository (http://www.ncbi.nlm.nih.gov/clinvar/).
Authors’ contributions
YN performed all experiments except whole-exome sequencing. FM performed whole-exome sequencing and the analysis. MK, NO, TT, MY, YK, KK, and SS participated in whole-exome sequencing. AH, NA, SF, SM, AU, and YK contributed to analyzing patients’ clinical features. YJ, MNakanishi performed western blotting. MNakagawa interpreted MRI findings. IH, TT, KA, and KO performed multiplex targeted sequencing. NY and SS wrote the manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this report and any accompanying images.
Ethics approval and consent to participate
Experimental protocols were approved by the Ethical Committee for the Study of Human Gene Analysis at Nagoya City University Graduate School of Medical Sciences (approval number 164). Written informed consent for analyses was obtained from all patients or their parents.
Abbreviations
- LCL
Lymphoblastoid cell line
- MCAP
Megalencephaly-capillary malformation syndrome
- MPPH
Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome
- MRI
Magnetic resonance imaging
- mTOR
Mechanistic target of rapamycin
- SD
Standard deviation
- WES
Whole exome sequencing
Contributor Information
Yutaka Negishi, Email: negishi@med.nagoya-cu.ac.jp.
Fuyuki Miya, Email: miya@src.riken.jp.
Ayako Hattori, Email: aykhat@med.nagoya-cu.ac.jp.
Yoshikazu Johmura, Email: johmuray@med.nagoya-cu.ac.jp.
Motoo Nakagawa, Email: lmloltlolol@gmail.com.
Naoki Ando, Email: nando@med.nagoya-cu.ac.jp.
Ikumi Hori, Email: ikumil93@med.nagoya-cu.ac.jp.
Takao Togawa, Email: t.togawa@med.nagoya-cu.ac.jp.
Kohei Aoyama, Email: kohei-a@med.nagoya-cu.ac.jp.
Kei Ohashi, Email: k.ohashi@med.nagoya-cu.ac.jp.
Shinobu Fukumura, Email: fukumura@sapmed.ac.jp.
Seiji Mizuno, Email: seiji_mizuno@aichi-colony.jp.
Ayako Umemura, Email: ayako5346@yahoo.co.jp.
Yoko Kishimoto, Email: y.kishimoto@shimada-ryoiku.or.jp.
Nobuhiko Okamoto, okamoto@osaka.email.ne.jp.
Mitsuhiro Kato, Email: ktmt-hro@umin.ac.jp.
Tatsuhiko Tsunoda, Email: tsunoda@src.riken.jp.
Mami Yamasaki, Email: myamasaki@ajk.takatsuki-hp.or.jp.
Yonehiro Kanemura, Email: kanemura@onh.go.jp.
Kenjiro Kosaki, Email: kkosaki@z3.keio.jp.
Makoto Nakanishi, Email: mkt-naka@med.nagoya-cu.ac.jp.
Shinji Saitoh, Phone: 81-52-853-8246, Email: ss11@med.nagoya-cu.ac.jp.
References
- 1.Williams CA, Dagli A, Battaglia A. Genetic disorders associated with macrocephaly. Am J Med Genet A. 2008;146A(15):2023–2037. doi: 10.1002/ajmg.a.32434. [DOI] [PubMed] [Google Scholar]
- 2.Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, Hennekam RC, Mueller RF, Brueton L, Super M, Steen-Johnsen J, et al. Macrocephaly with cutis marmorata, haemangioma and syndactyly--a distinctive overgrowth syndrome. Clin Dysmorphol. 1997;6(4):291–302. doi: 10.1097/00019605-199710000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, Higgins JV, Stevens CA, Twersky S, Weksberg R, et al. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. 1997;70(1):67–73. doi: 10.1002/(SICI)1096-8628(19970502)70:1<67::AID-AJMG13>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 5.Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM, Jr, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A(2):269–291. doi: 10.1002/ajmg.a.34402. [DOI] [PubMed] [Google Scholar]
- 6.Garavelli L, Leask K, Zanacca C, Pedori S, Albertini G, Della Giustina E, Croci GF, Magnani C, Banchini G, Clayton-Smith J, et al. MRI and neurological findings in macrocephaly-cutis marmorata telangiectatica congenita syndrome: report of ten cases and review of the literature. Genet Couns. 2005;16(2):117–128. [PubMed] [Google Scholar]
- 7.Conway RL, Pressman BD, Dobyns WB, Danielpour M, Lee J, Sanchez-Lara PA, Butler MG, Zackai E, Campbell L, Saitta SC, et al. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A. 2007;143A(24):2981–3008. doi: 10.1002/ajmg.a.32040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirzaa GM, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet. 2014;46(5):510–515. doi: 10.1038/ng.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11(2):111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- 10.McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3(3):137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 11.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. doi: 10.3389/fnmol.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51(1):27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson JR. Therapeutic targeting of mTOR in tuberous sclerosis. Biochem Soc Trans. 2009;37(Pt 1):259–264. doi: 10.1042/BST0370259. [DOI] [PubMed] [Google Scholar]
- 15.Negishi Y, Miya F, Hattori A, Mizuno K, Hori I, Ando N, Okamoto N, Kato M, Tsunoda T, Yamasaki M, et al. Truncating mutation in NFIA causes brain malformation and urinary tract defects. Human Genome Variation. 2015;2:15007. doi: 10.1038/hgv.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nellist M, Schot R, Hoogeveen-Westerveld M, Neuteboom RF, van der Louw EJ, Lequin MH, Bindels-de Heus K, Sibbles BJ, de Coo R, Brooks A, et al. Germline activating AKT3 mutation associated with megalencephaly, polymicrogyria, epilepsy and hypoglycemia. Mol Genet Metab. 2015;114(3):467–473. doi: 10.1016/j.ymgme.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Harada A, Miya F, Utsunomiya H, Kato M, Yamanaka T, Tsunoda T, Kosaki K, Kanemura Y, Yamasaki M. Sudden death in a case of megalencephaly capillary malformation associated with a de novo mutation in AKT3. Childs Nerv Syst. 2015;31(3):465–471. doi: 10.1007/s00381-014-2589-y. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura K, Kato M, Tohyama J, Shiohama T, Hayasaka K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, et al. AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin Genet. 2014;85(4):396–398. doi: 10.1111/cge.12188. [DOI] [PubMed] [Google Scholar]
- 19.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 20.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Longy M, Coulon V, Duboue B, David A, Larregue M, Eng C, Amati P, Kraimps JL, Bottani A, Lacombe D, et al. Mutations of PTEN in patients with bannayan-riley-ruvalcaba phenotype. J Med Genet. 1998;35(11):886–889. doi: 10.1136/jmg.35.11.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tapper WJ, Foulds N, Cross NC, Aranaz P, Score J, Hidalgo-Curtis C, Robinson DO, Gibson J, Ennis S, Temple IK, et al. Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PLoS One. 2014;9(1):e86940. doi: 10.1371/journal.pone.0086940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 27.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips WA, Loconte DC, Grossi V, Bozzao C, Forte G, Bagnulo R, Stella A, Lastella P, Cutrone M, Benedicenti F, et al. Molecular and functional characterization of three different postzygotic mutations in PIK3CA-related overgrowth spectrum (PROS) patients: effects on PI3K/AKT/mTOR signaling and sensitivity to PIK3 inhibitors. PLoS One. 2015;10(4):e0123092. doi: 10.1371/journal.pone.0120762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292. doi: 10.1038/ncomms2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the ClinVar repository (http://www.ncbi.nlm.nih.gov/clinvar/).